Introduction
Heme oxygenase-1 (HO-1), as an inducible and
cytoprotective enzyme, has a protective effect against cellular
oxidative stress. Studies have confirmed that cholesterol has an
initiating role in several metabolic diseases, including obesity,
diabetes and myocardial infarction (1,2). In
particular, the excessive intake of cholesterol results in lipid
overload, promotes the formation of foam cells, and increases the
production of pro-inflammatory factors, including interleukin
(IL)-6, IL-1, tumor necrosis factor (TNF)-α, reactive oxygen
species (ROS) and cytokines (2,3).
These factors are released into the circulation and cause vascular
endothelial damage.
HO-1, as an important molecule in the antioxidant
defense system, degrades heme into carbon monoxide (CO), biliverdin
and ferrous iron. HO-1 is involved in the inhibition of ROS
production, the consumption of redundant ROS, and the induction of
heme metabolism. Abundant ROS are usually produced under various
pathological statuses, including irradiation, inflammatory
processes, electron transport reactions and lipid peroxidation
(4,5). High levels of ROS disrupt the balance
between ROS and reactive nitrogen species, and further cause
oxidative stress in the organism (6). Several studies have shown that ROS
can induce the proliferation of pre-adipocytes and increase the
size of adipocytes (7). The mass
and volume of adipocytes is increased by the activation of NADPH
oxidases and endoplasmic reticulum (ER) stress, further increasing
the oxidative stress status, which increases the generation of ROS
in adipocytes. The accumulation of ROS can increase the expression
of glucose transporter 1 (GLUT1) by increasing transcription rate
and mRNA stability, which leads to increased expression levels of
GLUT1 and induces the uptake of glucose in cells (8,9).
HO-1 can degrade the p38α mitogen-activated protein
kinase (MAPK) isoform, and then alter the ratio of p38α and p38β to
induce the cytoprotective effect of the HO-1/CO metabolism pathway
against the release of ROS from the mitochondrial and lipid
peroxidation processes (10). CO
can also mimic the effect of HO-1 and degrade the p38α MAPK
isoform, also exerting an antioxidant effect. Biliverdin can
convert into bilirubin, and bilirubin is a potent antioxidant.
HO-1, CO and biliverdin also decrease the intracellular
concentrations of ROS and protect cells from injuries caused by
oxidative stress (11). The
accumulation of low density lipoprotein (LDL) cholesterol can
increase the production of ROS, and ROS can transform LDL to
cytotoxic oxidized-LDL, which results in the release of numerous
pro-inflammatory cytokines by macrophages or T cells (12). An increase in the expression of
TNF-α can decrease adhesion in endothelial cells, facilitating the
transmigration of neutrophils and increase in vascular
permeability, and promoting the formation of intercellular gaps.
IL-6 also can alter the shape of endothelial cells and disrupt the
endothelial cell-cell barrier (13,14).
The stimulation of long-term high levels of lipids induces the
release of inflammatory factors, and an increase in the production
of ROS can finally result in atherosclerosis of the coronary
artery. The rise of ROS caused by high lipid levels may directly
interact with Kelch-like ECH associated protein 1 (Keap1) and cause
the dissociation of the Keap1/nuclear factor erythroid 2-related
factor 2 (Nrf2) complex in the cytoplasm, promoting the
transportation of Nrf2 into the nucleus. Nrf2 can recognize
specific DNA-binding elements of the HO-1 promoter, and then
increase the expression level of HO-1 (11). However, the detailed mechanisms
underlying the effect of HO-1 against cholesterol-induced oxidative
damage in vascular endothelial cells remains to be elucidated.
The present study was performed to investigate the
molecular mechanism through which HO-1 alleviates injury in
endothelial cells caused by cholesterol. It was confirmed that
cholesterol stimulation upregulated the expression of HO-1 in a
time-dependent manner. The upregulated expression of HO-1
alleviated oxidative damage in the vascular endothelial cells by
activation of the MAPK/extracellular signal-regulated kinase (ERK)
pathway and inhibition of the phosphoinositide 3-kinase (PI3K)/AKT
signaling pathway.
Materials and methods
Materials
H-DMEM was obtained from Basalmedia Technologies
Co., Ltd. (Shanghai, China) and FBS was from Bailing Biotechnology
Co., Ltd. (Lanzhou, China). Lipofectamine 3000 transfection reagent
(cat. no. R0531) was from Thermo Fisher Scientific, Inc. (Waltham,
MA, USA). Cholesterol was obtained from Sigma-Aldrich; Merck
Millipore (Darmstadt, Germany). Tin protoporphyrin (SnPP; cat. no.
sc-203452) was from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). The primary rabbit monoclonal anti-c-Myc (cat. no. ab32072),
anti-ERK1+ERK2 (cat. no. ab184699), anti-phosphorylated (p-)
ERK1+p-ERK2 (cat. no. ab76299), anti-nuclear factor (NF)-κB p65
(cat. no. ab7970), anti-HO-1 (cat. no. ab52947), anti-Nrf2 (cat.
no. ab62352), anti-AKT (cat. no. ab179463) and anti-p-AKT (cat. no.
ab81283) antibodies were from Abcam (Cambridge, MA, USA). The cell
membrane permeable calcium fluorescent probe and CellROX Orange
reagent were from Yeasen Biotechnology Co., Ltd. (cat. no.
40704ES50, Shanghai, China). The mitochondrial membrane potential
(ΔΨm) assay kit with JC-1 (cat. no. C2006) was from Beyotime
Institute of Biotechnology (Haimen, China). The anti-rabbit
HRP-labeled secondary antibodies were from KPL, Inc. (Gaithersburg,
MD, USA).
Cell culture
HEK293T and EA.hy926 cells obtained from the
Shanghai Cell Resource Center of the Chinese Academy of Sciences
(Shanghai, China; cat. nos. GNHu17 and GNHu39) were cultured at a
37°C in a humidified 5% CO2 atmosphere in H-DMEM
containing 10% FBS, respectively. According to a previous study
(15), the EA.hy926 cells
(5×105) were seeded into 100 mm plastic dishes and
cultured for 24 h, following which the cells were divided into
three groups (SnPP, HO-1-overexpression and control). Cells in the
SnPP group were treated with 20 mmol/l SnPP at 37°C for 24 h; cells
in the HO-1-overexpression group were transfected using TurboFect™
transfection reagent (cat. no. R0531; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol, with 10 µg
pCDNA3.1-HO-1 vector for 24 h; and cells in the control group
received no treatment. Following treatment, the cells in all groups
were treated with 100 mmol/l cholesterol at 37°C for 0, 12 and 24
h. For all assays, the cells were washed in sterile PBS prior to
removal of the redundant cholesterol.
MTT assay
An MTT assay was performed according to a previous
study (16). Briefly, a
concentration of 1×104 cells was seeded into each well
of a 96-well plate (cat. no. 3599; Corning Incorporated, Corning,
NY, USA). The cells in the three groups were respectively incubated
with 5 mg/ml MTT buffer at 37°C for 3 h, following incubation with
100 mmol/l cholesterol for 0, 12 and 24 h. The optical density at
490 nm was measured using the SpectraMax series microplate reader
(Molecular Devices, LLC, Sunnyvale, CA, USA).
Western blot analysis
EA.hy926 cells were cultured and divided into three
groups as described above. Following treatment with 100 mmol/l
cholesterol for 0, 12 and 24 h, the cells were lysed with RIPA
buffer (50 mmol/l Tris-HCl, 150 mmol/l NaCl, 24 mmol/l sodium
deoxycholate, 3.68 mmol/l SDS, 1% TritonX-100 and cocktail protease
inhibitors) at 4°C. A BCA assay was used to determine the
concentration of protein, and 60 µg of the protein samples were
loaded and separated by 10% SDS-PAGE, followed by transfer onto a
0.22 µm nitrocellulose membrane using a semi-dry electroblotter.
The membranes were respectively incubated with primary antibodies
(1:1,000) overnight at 4°C, followed by incubation with secondary
antibody (1:5,000) for 1 h at room temperature. The quantification
of protein was performed using ECL immunoblotting reagent (KPL,
Inc.). The gray values of bands were quantified using Scion Image
software (version 4.0.3.2; Scion Corporation, Frederick, MD, USA)
and were normalized with β-actin.
Analyses of ΔΨm, ROS and
[Ca2+]i
According to a previous study (17), a total of 1×104 cells
were seeded into a confocal plate (cat. no. 801002; Nest
Scientific, Rahway, NJ, USA). Following treatment with 100 mmol/l
cholesterol for 0, 12 and 24 h, the cells were incubated with 4
µmol/l Fluo-4, 5 µmol/l CellROX Orange reagent and 5 µg/ml JC-1 at
37°C for 30 min following removal of culture medium, respectively.
Following incubation, the cells were washed with PBS three times
and were then visualized using confocal microscopy (×20 objective;
Leica TCS SP8; Leica Microsystems GmbH, Wetzlar, Germany). The
images were analyzed using Image-Pro Plus 6.0 software (Media
Cybernetics, Inc., Bethesda, MD, USA).
Statistical analysis
Data are presented as the mean ± standard error of
the mean of three independent experiments. Multi-way analysis of
variance was performed to evaluate the differences between groups
using GraphPad Prism 6 software (GraphPad Software Inc., La Jolla,
CA, USA) followed by the Tukey-Kramer post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of cholesterol on the growth
of EA.hy926 cells
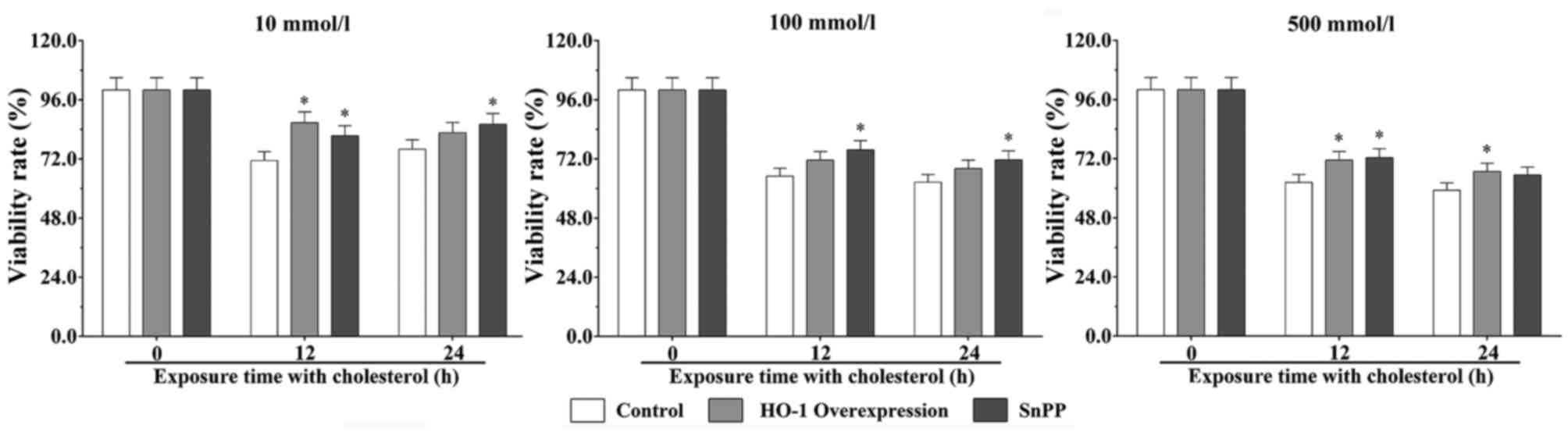
The MTT results showed that cholesterol inhibited
the proliferation and viability of EA.hy926 cells, and exhibited in
a dose- and time-dependent effect (Fig. 1). Following stimulation with 10
mmol/l cholesterol for 12 h, the cell viability rates in the SnPP
treatment group and HO-1 overexpression group were significantly
increased, compared with that in the control group (P<0.05).
Following stimulation with cholesterol for 24 h, the viability of
the HO-1-overexpressing cells treated with 10 mmol/l cholesterol
for 24 h was significantly increased, compared with that of the
control group (P<0.05). Following stimulation with 100 mmol/l
cholesterol for 12 and 24 h, the viability of cells in the HO-1
overexpression group were significantly increased, compared with
that of the control group (P<0.05). Following stimulation with
500 mmol/l cholesterol for 12 h, the viability of cells in the SnPP
treatment and HO-1 overexpression groups were significantly
increased, compared with that in the control group (P<0.05).
Following stimulation with cholesterol for 24 h, the viability of
cells in the SnPP treatment group was significantly increased,
compared with that in the control group (P<0.05).
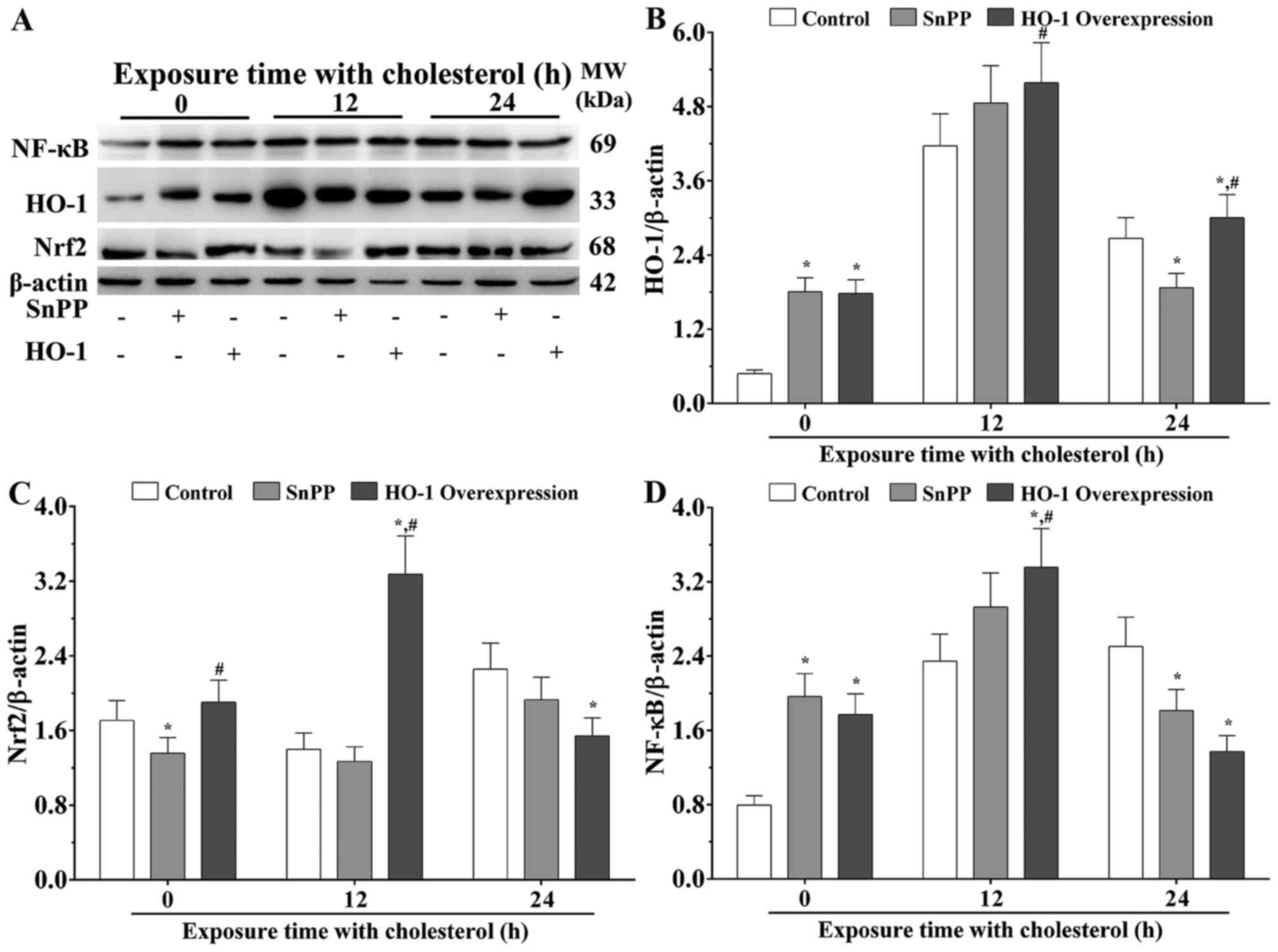
Upregulated expression of HO-1 is
protective during cholesterol stimulation
The present study hypothesized that a high
concentration of cholesterol stimulation results in the
overexpression of HO-1 via activation of the Nrf2 signaling
pathway. Under normal condition, the results of the western blot
analysis showed that, compared with the control group, the
expression levels of HO-1 in the cells with SnPP treatment and HO-1
overexpression were significantly increased (P<0.05). Following
stimulation with cholesterol for 12 h, the expression level of HO-1
was significantly increased in the HO-1 overexpression group,
compared with that in the SnPP treated group (P<0.05). Following
stimulation with cholesterol for 24 h, the expression level of HO-1
was significantly decreased in the SnPP treated group, compared
with that in the control group (P<0.05), and was significantly
increased in the HO-1-overexpressing group, compared with that in
the control group and SnPP treatment group (P<0.05; Fig. 2A and B).
Nrf2 is a type of transcription activator, which can
bind to antioxidant response elements (ARE) in the promoter regions
of downstream target genes, including HO-1. As shown in Fig. 2C, without stimulation with
cholesterol, the expression level of Nrf2 in the SnPP treatment
group was significantly decreased, compared with that in the
control group (P<0.05), and that in the HO-1 overexpression
group was significantly increased, compared with that in the SnPP
treatment group (P<0.05). Following stimulation with cholesterol
for 12 h, the expression level of Nrf2 was significantly increased
in the HO-1 overexpression group, compared with levels in the
control and SnPP treatment groups, respectively (P<0.05).
Following stimulation with cholesterol for 24 h, the expression
level of Nrf2 in the HO-1 overexpression group was significantly
decreased compared with the control group (P<0.05; Fig. 2C).
NF-κB, as a pleiotropic transcription factor, can be
activated by several stimuli associated with a number of biological
processes, including immunity, inflammation, cell growth,
differentiation, apoptosis and tumorigenesis. NF-κB is the endpoint
of several signal transduction events. As shown in Fig. 2D, without cholesterol stimulation,
the expression levels of NF-κB were significantly increased in the
SnPP-treated group and HO-1 overexpression group, compared with
that in the control group (P<0.05). Following stimulation with
cholesterol for 12 h, the expression level of NF-κB was
significantly increased in the HO-1 overexpression group, compared
with the levels in the control and SnPP-treated groups (P<0.05).
Following stimulation with cholesterol for 24 h, the expression
level of NF-κB was significantly decreased in the SnPP-treated
group and the HO-1 overexpression group compared with the control
group (P<0.05; Fig. 2D).
Molecules involved in the regulatory
progresses of HO-1 following cholesterol stimulation
The MAPK/ERK and PI3K/AKT signaling pathways are
associated with the process of ROS metabolism. The results of the
present study confirmed that the ratios of p-AKT/AKT were
significantly decreased in the SnPP treatment group and HO-1
overexpression group, compared with that in the control group
without cholesterol stimulation (P<0.05; Fig. 3A and B). Following stimulation with
cholesterol for 12 h, the ratios of p-AKT/AKT was significantly
decreased in the SnPP treatment group and HO-1 overexpression
group, compared with that in the control group (P<0.05).
Following stimulation with cholesterol for 24 h, the ratio of
p-AKT/AKT was significantly decreased in the SnPP treatment group,
compared with that in the control group (P<0.05), and was
significantly increased in the HO-1 overexpression group, compared
with that in the SnPP treatment group (P<0.05; Fig. 3B).
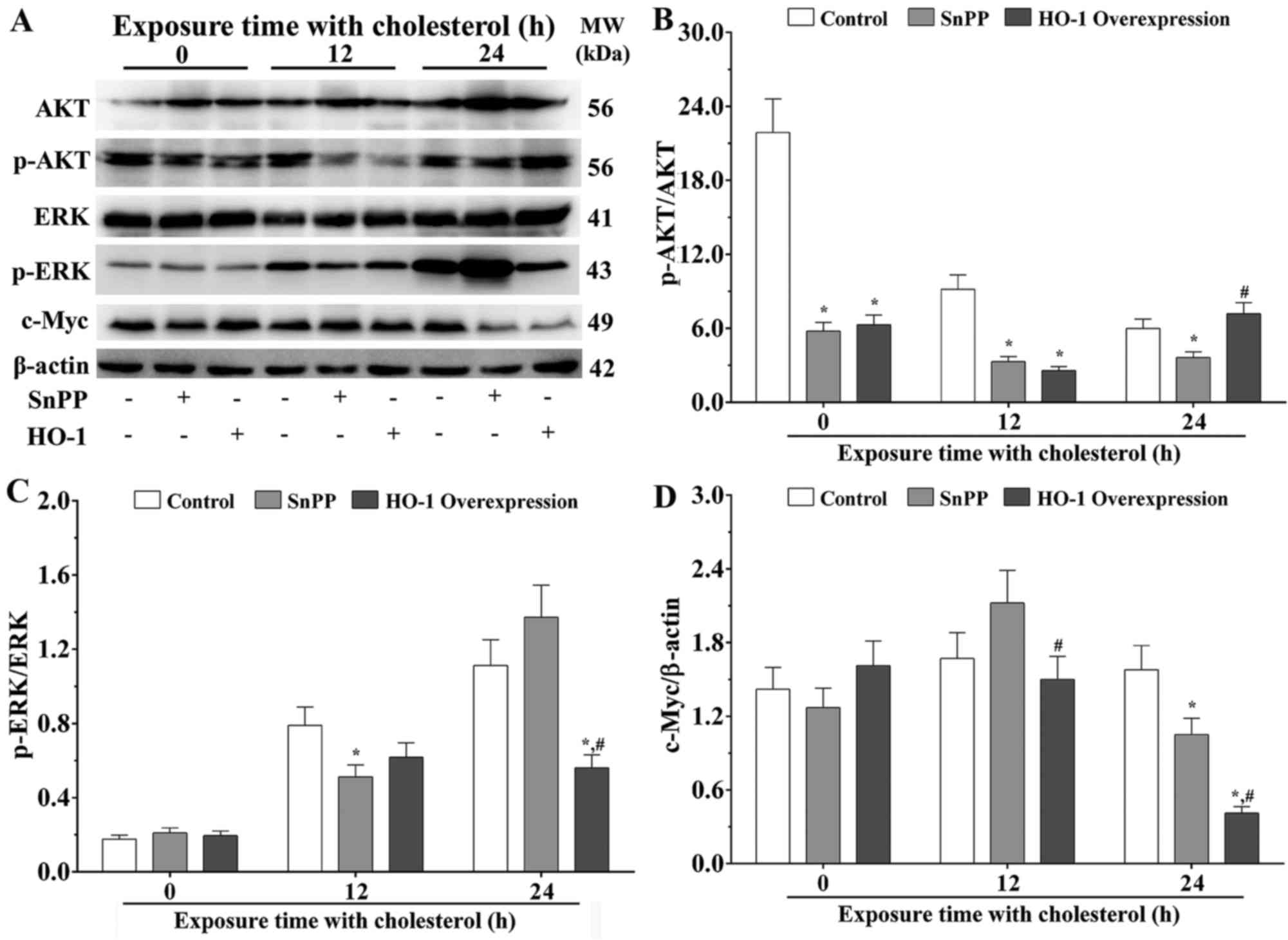 | Figure 3.Effects of cholesterol on the
expression of HO-1-associated molecules in EA.hy926 cells
stimulated with cholesterol. (A) Western blot analysis to evaluate
the expression levels of AKT, p-AKT, ERK, p-ERK and c-Myc. The
expression levels of (B) AKT, p-AKT, (C) ERK, p-ERK and (D) c-Myc
were quantified by densitometry. β-actin was used as an internal
control. Data are presented as the mean ± standard error of the
mean for each group (n=3). *P<0.05, vs. control group;
#P<0.05, vs. SnPP group. HO-1, heme oxygenase 1;
SnPP, tin protoporphyrin; ERK, extracellular signal-regulated
kinase; p-, phosphorylated. |
The present study confirmed that there were no
significant differences between the ratios of p-ERK/ERK in the
three treatment groups without exposure with cholesterol. Following
stimulation with cholesterol for 12 h, the ratio of p-ERK/ERK was
significantly decreased in the SnPP treatment group, compared with
that in the control group (P<0.05). Following stimulation with
cholesterol for 24 h, the ratio of p-ERK/ERK was significantly
decreased in the HO-1 overexpression group (P<0.05), compared
with the ratios in the control group or SnPP treatment group
(Fig. 3C).
The present study also measured the expression level
of c-Myc, a downstream molecule of the MARK/ERK pathway. No
significant differences were found between the expression levels of
c-Myc in the three groups without cholesterol stimulation.
Following stimulation with cholesterol for 12 h, the expression of
c-Myc in the HO-1 overexpression group was significantly decreased,
compared with than in SnPP group (P<0.05). Following stimulation
with cholesterol for 24 h, the expression level of c-Myc in the
SnPP treatment group was significantly decreased, compared with
that in the control group (P<0.05), and the expression level of
c-Myc in the HO-1 overexpression group was significantly decreased,
compared with the ratios in the control group and SnPP treatment
group (P<0.05; Fig. 3D).
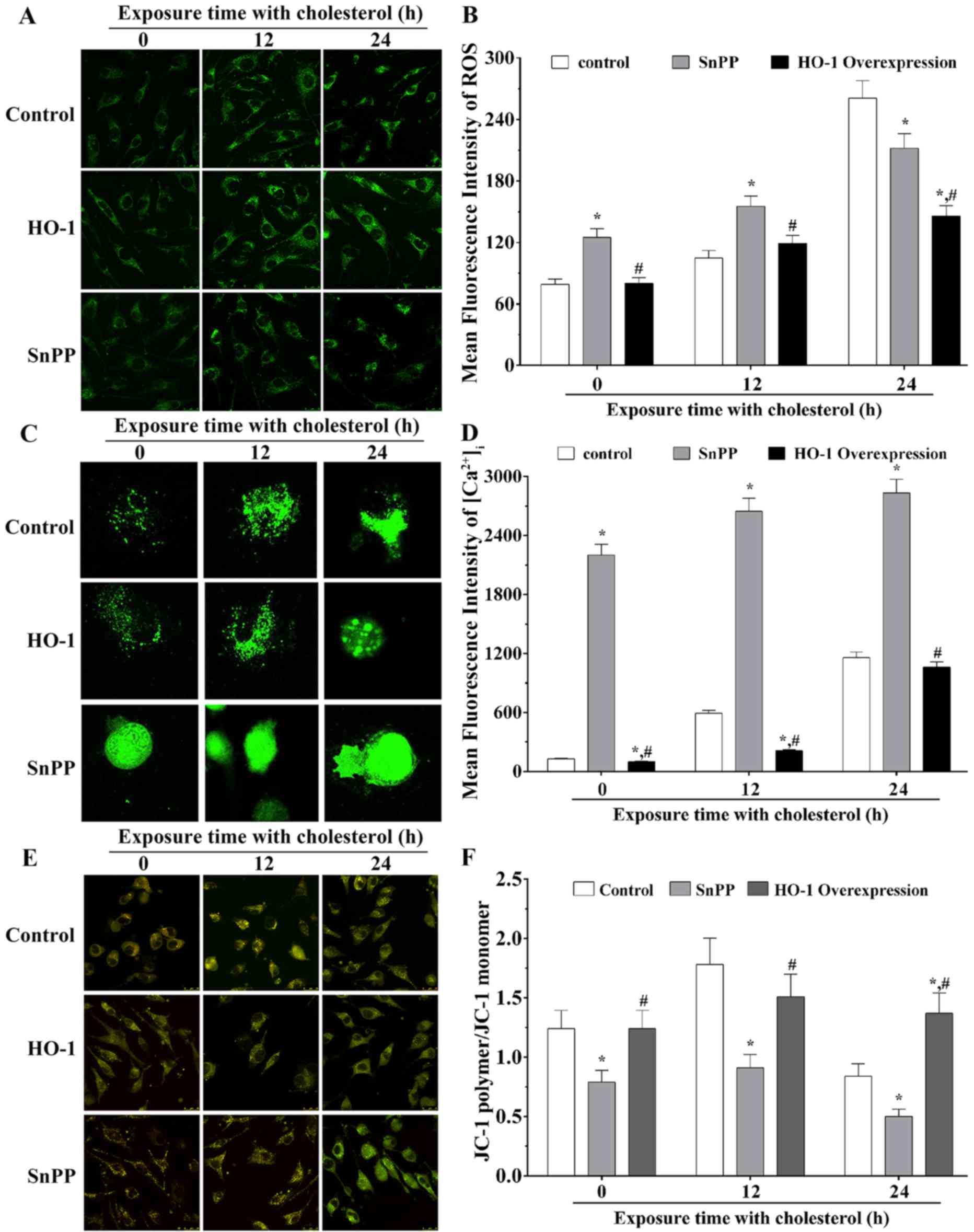
Measurements of concentrations of
intracellular ROS ([ROS]i)
Using confocal imaging, the present study measured
the concentrations of [ROS]i (Fig. 4A). The mean optical intensity (MOD)
values of the control group, SnPP treatment group and HO-1
overexpression group were 79.22±5.28, 125.18±8.35 and 80.33±5.36,
respectively, without stimulation with cholesterol. Following
stimulation with cholesterol for 12 h, the MOD values of three
groups were 105.10±7.00, 155.25±10.35 and 119.03±7.94. Following
stimulation with cholesterol for 24 h, the MOD values were
260.80±17.39, 212.02±14.13 and 146.07±9.74. These results indicated
that, without cholesterol stimulation, [ROS]i in the
SnPP treatment group was significantly increased, compared with
that in the control group (P<0.05), whereas that of the HO-1
overexpression group was significantly decreased, compared with
that of the SnPP treatment group (P<0.05). Following stimulation
with cholesterol for 12 h, [ROS]i in the SnPP treatment
group was significantly increased, compared with that in the
control group (P<0.05), and [ROS]i in the HO-1 overexpression
group was significantly decreased compared with that in the SnPP
treatment group (P<0.05). Following stimulation with cholesterol
for 24 h, [ROS]i in the SnPP treatment group was
significantly decreased, compared with that in the control group
(P<0.05), and [ROS]i in the HO-1 overexpression group was
significantly decreased (P<0.05), compared with that in the SnPP
treatment group. The quantification results of mean fluorescence
intensity of [ROS]i are presented in Fig. 4B.
Measurements of
[Ca2+]i
The [Ca2+]i was measured using
confocal images, the results are shown in Fig. 4C. The MOD of the control, SnPP
treatment and HO-1 overexpression groups were 127.62±6.38,
2201.20±110.06 and 98.50±4.93, respectively, in the absence of
cholesterol stimulation. Following stimulation with cholesterol for
12 h, the MOD values of these three groups were 592.91±29.65,
2,645.78±132.29 and 212.36±10.62. Following stimulated with
cholesterol for 24 h, the MOD values were 1,157.45±57.87,
2,831.36±141.57 and 1.062.75±53.14. Without stimulation with
cholesterol, [Ca2+]i in the SnPP treatment
group was significantly increased, compared with that in the
control group (P<0.05), and [Ca2+]i in the
HO-1 overexpression group was significantly decreased, compared
with that in the control and SnPP treatment groups (P<0.05).
Following stimulation with cholesterol for 12 h, the
[Ca2+]i in the SnPP treatment group was
significantly increased, compared with that in the control group
(P<0.05), and [Ca2+]i in the HO-1
overexpression group was significantly decreased, compared with
that in the control or SnPP treatment group (P<0.05). Following
stimulation with cholesterol for 24 h,
[Ca2+]i in the SnPP treatment group was
significantly increased, compared with that in control group
(P<0.05), whereas the level of [Ca2+]i in
the HO-1 overexpression group was significantly decreased, compared
with that in the SnPP treatment group (P<0.05). The
quantification results of mean fluorescence intensity of
[Ca2+]i are presented in in Fig. 4D.
Measurements of ΔΨm
The present measured the ΔΨm in three groups using
confocal imaging (Fig. 4E). The
ratios of red fluorescence (JC-1 polymer)/green fluorescence (JC-1
monomer) in the three groups are shown in Fig. 4F. Without cholesterol stimulation
and with cholesterol stimulation for 12 h, the ratio of JC-1
polymer/JC-1 monomer in the SnPP treatment group was significantly
decreased (P<0.05), compared with the ratios in the control or
HO-1 overexpression group. Following stimulation with cholesterol
for 24 h, the ratio in the SnPP treatment group was significantly
decreased, compared with that in the control group (P<0.05), and
that in the HO-1 overexpression group was significantly increased,
compared with the ratios in the control and SnPP treatment groups
(P<0.05).
Discussion
HO-1, an essential enzyme in heme catabolism, is
activated in conditions of high concentrations of heme and other
pathophysiological statuses, including oxidative stress, high
glucose and viral infection, in which it exhibits cytoprotective
effects (18,19). The results of the present study
indicated that the expression of HO-1 in EA.hy926 cells following
stimulation with cholesterol for 12 h was increased. However, this
expression decreased following stimulation with cholesterol for 24
h. These results suggested that EA.hy926 cells gradually restored
to a normal physiological status. The overexpression HO-1 at the
transcriptional level is essential to adapt to cholesterol-mediated
oxidative stress. Previous studies have confirmed that HO-1 can be
activated by hypoxia, and the present study showed that the
expression of HO-1 was increased with alleviation of the cell
injury caused by a combination of cholesterol stimulation and SnPP
inhibition in the EA.hy926 cells (18).
Several signal transduction pathways can induce HO-1
expression via the activation of different transcription factors,
including BTB domain and CNC homolog 2, P53, cAMP response element
binding protein and Nrf2 (20).
EA.hy926 cells have different defense systems in response to
oxidative stress, including phase I enzymes and phase II enzymes
(21). The results of the present
study showed that the expression levels of Nrf2 in EA.hy926 cells
stimulated with cholesterol were increased, and the activation of
Nrf2 promoted the upregulation in the expression of HO-1. Under
oxidative stress, Nrf2 is released from Keap1 and translocated to
the nucleus, where it binds to AREs in the promoter/enhancer region
of antioxidant enzyme genes of the phase II anti-oxidative system,
including HO-1. Nrf2 can upregulate the expression of these
antioxidant enzymes, and has a cytoprotective role (22,23).
The results of the present study also showed that the
[Ca2+]i was increased following stimulation with
cholesterol, and the concentration of [Ca2+]i
in the HO-1-overexpressing group was significantly decreased,
indicating that the expression of HO-1 may be a negative regulator
of [Ca2+]i. [Ca2+]i, as
a second messenger in several signaling pathways, promotes the
translocation of Nrf2 from the cytoplasm to the nucleus, and
functions as a transcriptional factor to regulate downstream
molecules (24). The activation of
Nrf2 signaling increases the expression of HO-1, activates the
phase II antioxidant response, and exerts its anti-oxidative
function to protect endothelial cells.
NF-κB consists of a family of transcription factors,
including p52, p50, c-rel, RelA (p65) and RelB, and is a key
transcription factor, which mediates immune responses to
inflammation and cell proliferation, and protects against UV
radiation (25). Several studies
have shown that the generation of ROS can subsequently activate
NF-κB (26,27). The results of the present study
showed that expression levels of the NF-κB p65 subunit were
increased following exposure to cholesterol, indicating that
cholesterol induced the accumulation of ROS in EA.hy926 cells
(28) and then caused the
upregulation of NF-κB. Studies have also shown that the p65 subunit
of NF-κB assists in increasing the level of nuclear Keap1. Keap1
can bind with Nrf2, decreasing the binding of Nrf2 with AREs in the
promoter/enhancer region, and then inhibits the activation of
antioxidant enzyme genes in the phase II anti-oxidative system,
including the expression of HO-1 (29,30).
In addition, p65 can promote the interaction of histone deacetylase
3 with MAF bZIP transcription factor K (MafK), preventing the
heterodimer formation of MafK with Nrf2 and decreasing the
expression of ARE-associated genes (31).
The MAPK signaling pathways, including ERK1/2, p38
and c-Jun N-terminal kinase, are the regulators of the expression
of HO-1 under extracellular stimulation. MAPKs act as positive and
negative regulators between the extracellular stimulation and
expression of HO-1, depending on different cell types and various
extracellular stimulations (32).
The results of the present study showed that the ratios of
p-ERK/ERK were increased in the three groups following stimulation
with cholesterol. MAPKs, as critical signaling pathways, are
involved in the activation and translocation of Nrf2 for the
synthesis of essential proteins, particularly HO-1. ERK signaling
can promote the Nrf2 phosphorylation process, which can induce the
release of Nrf2 from the Keap1-Nrf2 complex and cause the
translocation of Nrf2 from the cytoplasm into the nucleus, inducing
the upregulation of HO-1. ERK is involved in cellular responses
under the stimulation of a number of growth and differentiation
factors; however, several studies have confirmed that HO-1 is also
induced by the ERK signaling pathway under physiological conditions
(23,33). According to the results of the
present study, the ERK signaling pathway was activated under
stimulation with cholesterol, which resulted in the upregulated
expression of HO-1 and a cytoprotective effect in EA.hy926
cells.
The present study showed that the expression levels
of NF-κB in the three groups were upregulated following stimulation
with cholesterol for 12 h, and were decreased following stimulation
for 24 h. The ratio of p-ERK/ERK was increased following
stimulation with cholesterol for 12 and 24 h in the control and
SnPP treatment groups. The ratio in the HO-1 overexpression group
was increased following stimulation with cholesterol for 12 h.
Therefore, it was hypothesized that endothelial cells can alleviate
cholesterol-induced oxidative stress via the activation and
translocation of Nrf2, activation of the MAPK/ERK signaling pathway
and increased concentration of [Ca2+]i to
promote the expression of HO-1 in EA.hy926 cells.
Several studies have shown that oxidative stress can
decrease the expression of several survival signaling molecules,
including PI3K, p-AKT and Bcl-2. PI3K pathway activation is
involved in cell proliferation and cell survival in response to
cytokines and growth factors (34–36).
In the present study, it was found that the ratio of p-AKT/AKT was
decreased following stimulation with cholesterol, indicating that
the PI3K/AKT signaling pathway was inhibited by stimulation of
cholesterol. p-AKT can inhibit several cytosolic pro-apoptotic
substrates. The results of the present study suggested that
cholesterol increased ROS concentrations, which may significantly
increase the expression of HO-1. The ratio of p-AKT/AKT deceased as
the expression of HO-1 increased, therefore, the overexpression of
HO-1 decreased the ratio of p-AKT/AKT in endothelial cells under
cholesterol stimulation. These results suggested that the
upregulation of HO-1 significantly inhibited the
cholesterol-activated PI3K/AKT signaling pathway.
The present study also found that expression levels
of c-Myc were decreased under stimulation with cholesterol.
Upregulation of the expression of HO-1 may indirectly suppress the
expression level of c-Myc. Studies have shown that the
overexpression of c-Myc leads to a marked reduction in the
phosphorylation of enhancer of zeste homolog 2 (EZH2) in phoenix
cells and immortalizes mammary epithelial cells (37). c-Myc can phosphorylate AKT at the
S21 site and suppress the activation of AKT in parallel with
reduced phosphorylation of EZH2. Similarly, the apoptotic effect or
mechanisms of c-Myc are primarily involved in the regulation of BCL
family members at the transcription stage, including pro-apoptotic
Bcl-2-associated X protein, Bcl-2 antagonist killer and p53
upregulated modulator of apoptosis/Bcl-2-binding component 3.
Studies have shown that the potent apoptotic potential of c-Myc can
also be enforced via the p14ARF/p53 axis (38,39).
Therefore, the results of the present study
suggested that cholesterol induced the oxidative stress status and
increased the generation of ROS in endothelial cells. This
accumulation of ROS subsequently resulted in the upregulated
expression of HO-1. The present study found that cholesterol
stimulation increased the expression of Nrf2 and the concentration
of [Ca2+]i, activating ERK signaling and
inducing the overexpression of HO-1. HO-1 exerted a cytoprotective
effect through the inhibition of PI3K/AKT signaling and by
decreasing the expression of c-Myc in EA.hy926 cells.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 81570335), the
Tianjin Natural Science Foundation (grant no. 15ZXJZSY00010 and
12JCYBJC15900) and the Opening Funding for Tianjin Key Laboratory
of Cardiovascular Remodeling and Target Organ Injury (grant no.
TJC1401).
Glossary
Abbreviations
Abbreviations:
|
HO-1
|
heme oxygenase-1
|
|
ROS
|
reactive oxygen species
|
|
ΔΨm
|
mitochondrial membrane potential
|
|
[Ca2+]i
|
intercellular Ca2+
|
References
|
1
|
Morgan AE, Mooney KM, Wilkinson SJ,
Pickles NA and McAuley MT: Cholesterol metabolism: A review of how
ageing disrupts the biological mechanisms responsible for its
regulation. Ageing Res Rev. 27:108–124. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Janoudi A, Shamoun FE, Kalavakunta JK and
Abela GS: Cholesterol crystal induced arterial inflammation and
destabilization of atherosclerotic plaque. Eur Heart J.
37:1959–1967. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Korytowski W, Wawak K, Pabisz P, Schmitt
JC, Chadwick AC, Sahoo D and Girotti AW: Impairment of macrophage
cholesterol efflux by cholesterol hydroperoxide trafficking:
Implications for atherogenesis under oxidative stress. Arterioscler
Thromb Vasc Biol. 35:2104–2113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu
Y and Dong W: ROS and ROS-mediated cellular signaling. Oxid Med
Cell Longev. 2016:43509652016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martínez-Reyes I and Cuezva JM: The
H(+)-ATP synthase: A gate to ROS-mediated cell death or cell
survival. Biochim Biophys Acta. 1837:1099–1112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fang J, Seki T and Maeda H: Therapeutic
strategies by modulating oxygen stress in cancer and inflammation.
Adv Drug Deliv Rev. 61:290–302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cao J, Inoue K, Sodhi K, Puri N, Peterson
SJ, Rezzani R and Abraham NG: High-fat diet exacerbates renal
dysfunction in SHR: Reversal by induction of HO-1-adiponectin axis.
Obesity (Silver Spring). 20:945–953. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liemburg-Apers DC, Willems PH, Koopman WJ
and Grefte S: Interactions between mitochondrial reactive oxygen
species and cellular glucose metabolism. Arch Toxicol.
89:1209–1226. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Andrisse S, Koehler RM, Chen JE, Patel GD,
Vallurupalli VR, Ratliff BA, Warren DE and Fisher JS: Role of GLUT1
in regulation of reactive oxygen species. Redox Biol. 2:764–771.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lv X, Song DM, Niu YH and Wang BS:
Inhibition of heme oxygenase-1 enhances the chemosensitivity of
laryngeal squamous cell cancer Hep-2 cells to cisplatin. Apoptosis.
21:489–501. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gozzelino R, Jeney V and Soares MP:
Mechanisms of cell protection by heme oxygenase-1. Annu Rev
Pharmacol Toxicol. 50:323–354. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Song G, Zong C, Zhang Z, Yu Y, Yao S, Jiao
P, Tian H, Zhai L, Zhao H, Tian S, et al: Molecular hydrogen
stabilizes atherosclerotic plaque in low-density lipoprotein
receptor-knockout mice. Free Radic Biol Med. 87:58–68. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu BJ, Chen K, Shrestha S, Ong KL, Barter
PJ and Rye KA: High-density lipoproteins inhibit vascular
endothelial inflammation by increasing 3β-hydroxysteroid-Δ24
reductase expression and inducing heme oxygenase-1. Circ Res.
112:278–288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O'Reilly S, Ciechomska M, Cant R and van
Laar JM: Interleukin-6 (IL-6) trans signaling drives a
STAT3-dependent pathway that leads to hyperactive transforming
growth factor-β (TGF-β) signaling promoting SMAD3 activation and
fibrosis via Gremlin protein. J Biol Chem. 289:9952–9960. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li X, Ren Y, Sorokin V, Poh KK, Ho HH, Lee
CN, de Kleijn D, Lim SK, Tam JP and Sze SK: Quantitative profiling
of the rat heart myoblast secretome reveals differential responses
to hypoxia and re-oxygenation stress. J Proteomics. 98:138–149.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li J, Song J, Bi S, Zhou S, Cui J, Liu J
and Wu D: Electrochemical estrogen screen method based on the
electrochemical behavior of MCF-7 cells. J Hazard Mater.
313:238–243. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu F, Tang W, Chen D, Li M, Gao Y, Zheng
H and Chen S: Expression of TGF-β1 and CTGF is associated with
fibrosis of denervated sternocleidomastoid muscles in mice. Tohoku
J Exp Med. 238:49–56. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Abraham NG and Kappas A: Pharmacological
and clinical aspects of heme oxygenase. Pharmacol Rev. 60:79–127.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li X, Ye F, Li L, Chang W, Wu X and Chen
J: The role of HO-1 in protection against lead-induced
neurotoxicity. Neurotoxicology. 52:1–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ning W, Song R, Li C, Park E, Mohsenin A,
Choi AM and Choi ME: TGF-beta1 stimulates HO-1 via the p38
mitogen-activated protein kinase in A549 pulmonary epithelial
cells. Am J Physiol Lung Cell Mol Physiol. 283:L1094–L1102. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Buendia I, Michalska P, Navarro E, Gameiro
I, Egea J and León R: Nrf2-ARE pathway: An emerging target against
oxidative stress and neuroinflammation in neurodegenerative
diseases. Pharmacol Ther. 157:84–104. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen B, Lu Y, Chen Y and Cheng J: The role
of Nrf2 in oxidative stress-induced endothelial injuries. J
Endocrinol. 225:R83–R99. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Na HK and Surh YJ: Oncogenic potential of
Nrf2 and its principal target protein heme oxygenase-1. Free Radic
Biol Med. 67:353–365. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Santofimia-Castaño P, Ruy D Clea,
Garcia-Sanchez L, Jimenez-Blasco D, Fernandez-Bermejo M, Bolaños
JP, Salido GM and Gonzalez A: Melatonin induces the expression of
Nrf2-regulated antioxidant enzymes via PKC and Ca2+
influx activation in mouse pancreatic acinar cells. Free Radic Biol
Med. 87:226–236. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Y, Zhou G, Wang Z, Guo X, Xu Q, Huang
Q and Su L: NF-κB signaling is essential for resistance to heat
stress-induced early stage apoptosis in human umbilical vein
endothelial cells. Sci Rep. 5:135472015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu B, Wang S, Li R, Chen K, He L, Deng M,
Kannappan V, Zha J, Dong H and Wang W: Disulfiram/copper
selectively eradicates AML leukemia stem cells in vitro and in vivo
by simultaneous induction of ROS-JNK and inhibition of NF-κB and
Nrf2. Cell Death Dis. 8:e27972017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chao W, Deng JS, Li PY, Liang YC and Huang
GJ: 3,4-Dihydroxybenzalactone suppresses human non-small cell lung
carcinoma cells metastasis via suppression of epithelial to
mesenchymal transition, ROS-Mediated PI3K/AKT/MAPK/MMP and NFκB
signaling pathways. Molecules. 22:pii: E537. 2017. View Article : Google Scholar
|
|
28
|
Tornatore L, Thotakura AK, Bennett J,
Moretti M and Franzoso G: The nuclear factor kappa B signaling
pathway: Integrating metabolism with inflammation. Trends Cell
Biol. 22:557–566. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hoetzel A, Vagts DA, Loop T, Humar M,
Bauer M, Pahl HL, Geiger KK and Pannen BH: Effect of nitric oxide
on shock-induced hepatic heme oxygenase-1 expression in the rat.
Hepatology. 33:925–937. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang CS, Lin AH, Yang TC, Liu KL, Chen HW
and Lii CK: Shikonin inhibits oxidized LDL-induced monocyte
adhesion by suppressing NFκB activation via up-regulation of
PI3K/Akt/Nrf2-dependent antioxidation in EA.hy926 endothelial
cells. Biochem Pharmacol. 93:352–361. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wardyn JD, Ponsford AH and Sanderson CM:
Dissecting molecular cross-talk between Nrf2 and NF-κB response
pathways. Biochem Soc Trans. 43:621–626. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu BJ, Chen K, Barter PJ and Rye KA:
Niacin inhibits vascular inflammation via the induction of heme
oxygenase-1. Circulation. 125:150–158. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cai C, Teng L, Vu D, He JQ, Guo Y, Li Q,
Tang XL, Rokosh G, Bhatnagar A and Bolli R: The heme oxygenase 1
inducer (CoPP) protects human cardiac stem cells against apoptosis
through activation of the extracellular signal-regulated kinase
(ERK)/NRF2 signaling pathway and cytokine release. J Biol Chem.
287:33720–33732. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ying C, Chen L, Wang S, Mao Y, Ling H, Li
W and Zhou X: Zeaxanthin ameliorates high glucose-induced mesangial
cell apoptosis through inhibiting oxidative stress via activating
AKT signalling-pathway. Biomed Pharmacother. 90:796–805. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peng HB, Wang RX, Deng HJ, Wang YH, Tang
JD, Cao FY and Wang JH: Protective effects of oleanolic acid on
oxidative stress and the expression of cytokines and collagen by
the AKT/NF-κB pathway in silicotic rats. Mol Med Rep. 15:3121–3128.
2017.PubMed/NCBI
|
|
36
|
Tang R, Xu X, Yang W, Yu W, Hou S, Xuan Y,
Tang Z, Zhao S, Chen Y, Xiao X, et al: MED27 promotes melanoma
growth by targeting AKT/MAPK and NF-κB/iNOS signaling pathways.
Cancer Lett. 373:77–87. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kaur M and Cole MD: MYC acts via the PTEN
tumor suppressor to elicit autoregulation and genome-wide gene
repression by activation of the Ezh2 methyltransferase. Cancer Res.
73:695–705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Calvisi DF, Ladu S, Hironaka K, Factor VM
and Thorgeirsson SS: Vitamin E down-modulates iNOS and NADPH
oxidase in c-Myc/TGF-alpha transgenic mouse model of liver cancer.
J Hepatol. 41:815–822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
McMahon SB: MYC and the control of
apoptosis. Cold Spring Harb Perspect Med. 4:a0144072014. View Article : Google Scholar : PubMed/NCBI
|