Introduction
Chronic obstructive pulmonary disease (COPD) is a
devastating lung disease with a basic feature of incomplete
reversibility of airflow obstruction, which may be resulted from
lung's abnormal inflammatory response to harmful particles or gases
(1). Pulmonary tissues of COPD
have obvious vascular inflammatory changes and pathological
reconstruction in the early days even before hypoxia appears, and
pulmonary vascular remodeling of COPD is associated with the role
of chronic inflammatory cells and inflammatory mediators.
Therefore, chronic inflammation is a central part of the early
pulmonary vascular remodeling (2).
Pulmonary artery smooth muscle cells (PASMCs) not only are the main
effectors involved in pulmonary vascular remodeling, but also can
act like immune cells to synthesize and secrete inflammatory
mediators to promote the development of pulmonary vascular
inflammation, and thus serve key roles during pulmonary vascular
remodeling. However, the underlying mechanisms remain largely
unclear.
Toll-like receptors (TLRs) are receptors that have
the prominent biological function to promote synthesis and release
of cytokines to trigger inflammatory response (3). Moreover, it also can promote the
maturation of antigen presenting cells to induce the human body's
acquired immunity. TLR4 was the first identified mammalian TLR,
belonging to cytomembrane type I transmembrane glycoprotein
receptor, and can be activated by the lipopolysaccharides (LPS) of
pathogenic microorganisms, lipoproteins and peptidoglycans, to
promote cells to produce cytokines, chemokines, adhesion molecules
and acute phase protein to regulate the inflammatory responses
(4). TLR4 is widely expressed in
pulmonary macrophages, airway epithelial cells, airway smooth
muscle cells and vascular endothelial cells. Previous studies of
the authors reported that TLR4 receptor mediates PI3K/NF-κB
signaling during airway remodeling in asthma, which is an important
signal pathway to regulate the bronchial smooth muscle cells to
synthesize and secrete inflammatory factor (5). Some reports have demonstrated an
increased expression of TLR4 in lung tissue of COPD patients
(6). Yet, the role of TLR4 in
synthesis and secretion function of PASMCs is unclear. The present
study investigated the TLR4 expression level in PASMCs of COPD rats
and its role in PASMCs synthesis and secretion function.
Materials and methods
Materials
LPS was purchased from Sigma-Aldrich (Darmstadt,
Germany). The cigarettes were from Liuzhou Cigarette Factory
(Liuzhou, China). Gentamicin (cat. no. 15710-064) was from Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). Rat monoclonal antibody
against TLR4 was from Abcam (Cambridge, MA, USA). Horseradish
peroxidase-conjugated anti-rat IgG was from Beijing Zhongshan
Golden Bridge Biotechnology Co., Ltd. (Beijing, China). The DAB kit
was from Sangon Biotech Co., Ltd. (Shanghai, China). TAK-242 was
from MedChem Express (Monmouth Junction, NJ, USA). The ELISA kit
(cat. no. E-EL-R0009c) was from R&D Systems, Inc. (Minneapolis,
MN, USA).
Animal grouping and modeling
The animal protocol was reviewed and approved by the
Institutional Animal Care and Use Committee of the Affiliated
Hospital of Guilin Medical University (Guilin, China). A total of
24 specific pathogen-free Wistar rats (12 males and 12 females)
weighing 180–220 g were randomly divided into two groups: Control
group and COPD group. Rats in the control group were bred normally
for 8 weeks and then examined. Rats in the COPD group were
administered with 200 µg LPS to the airways on days 1 and 14, and
treatment of passive inhaling of cigarette smoke was administered 1
h/day for 8 weeks. Rats in control group were administered with
same volume of saline to the airways on day 1 and 14, and were
treated in the same apparatus as the COPD group with normal air 1
h/day for 8 weeks.
Lung tissue specimen preparation
Lung tissues were prepared as following: i) Middle
lobes of the right lungs of the rats were removed; ii) 10% neutral
formalin was injected into the lungs until the middle lobe of the
right lung had swelled completely; iii) the hilum of the lung was
ligated; iv) the middle lobes were soaked in 10% neutral formalin
and fixed for 24 h; v) the specimen was sliced continuously 2–3 mm
to the right of the middle lobe; vi) gradient alcohol dehydration,
paraffin embedding and slicing were performed according to standard
protocol (7); and vii) alterations
in the airway and pulmonary alveoli were observed following
conventional hematoxylin and eosin staining.
Rat PASMC culture
Rat PASMCs were cultured in medium supplemented with
25% inactivated fetal bovine serum and 100 µg/ml gentamicin, and
cells at passages 3–6 were used for experiments. PASMCs were
isolated from the 3rd level or lower artery branch of pulmonary
lobe segments from Wistar rats.
Immunochemical staining
Paraffin embedded lung tissues were cut into 4 µm
thick sections. Floating sections on 0.01 M PBS (Thermo Fisher
Scientific, Inc.) were exposed to 0.25% Triton X-100 (J&K
Scientific Ltd., Beijing, China) and 3% H2O2,
and then blocked in 1% FBS (cat. no. 10099-141; Thermo Fisher
Scientific, Inc.). They were then incubated sequentially with rat
monoclonal antibody against TLR4 (1:800), horseradish
peroxidase-conjugated anti-rat IgG (1:200). Sections were developed
with a DAB kit. Mouse PASMCs were seeded in 6-well plates with or
without LPS (concentration 1 µg/ml), the cultured cells were washed
with PBS and fixed with 4% paraformaldehyde for 30 min. They were
then rinsed in PBS and incubated at room temperature with 0.5%
Triton X-100 for 15 min, followed by blocking in PBS containing 1%
BSA at room temperature for 30 min. The cells were then incubated
with mouse monoclonal antibody against TLR4 (cat. no. ab22048;
dilution 1:800) at 4°C overnight. The cultured cells were rinsed in
PBS and incubated for 1 h with biotinylated anti-mouse IgG (cat.
no. zb2305; dilution 1:200; Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd.) at room temperature, then developed with a
DAB kit.
Western blot analysis
Proteins (15 µg) were separated with SDS-PAGE and
then transferred onto polyvinylidene difluoride membrane, which was
then blocked in 5% non-fat milk at room temperature for 2 h. The
membrane was washed with TBS with 0.1% Tween-20 then incubated with
mouse monoclonal antibody against TLR4 (dilution 1:1,000) at 4°C
overnight, followed with 3×10 min TBS-T washes. The membrane was
then incubated at room temperature with horseradish
peroxidase-conjugated anti-rat IgG (cat. no. ZB-2305; dilution
1:5,000) for 2 h. After 3×10 min TBS-T washes, the membrane was
developed with ECL Plus western blotting detection reagents
(Beyotime Institute of Biotechnology, Haimen, China). The ChemiDoc™
XRS+System (using Image Lab 3.0 software; Bio-Rad Laboratories,
Inc., Hercules, CA, USA) was used to capture and analyze images of
the membrane.
ELISA
PASMCs were plated in 6-well plates
(5×104 cells/well), and incubated for 24 h in serum-free
media, There were four groups analyzed: The control group, LPS (the
final concentration is 1 µg/ml) treatment group, TAK-242 (the final
concentration is 1 µM/l) treatment group, LPS+TAK-242 treatment
group (LPS final concentration was 1 µg/ml, TAK-242 final
concentration was 1 µM/l). Each group had four duplicates, and
three independent experiments were performed. Cellular supernatants
were analyzed by an ELISA kit for mouse IFN-γ.
Statistical analysis
SPSS statistical software (version, 17.0; SPSS,
Inc., Chicago, IL, USA) was used to analyze data. A one-way
analysis of variance was used for statistical analysis between
groups. The SNK (homogeneous variance) or Games-Howell method
(inhomogeneous variance) was used to compare two groups with normal
distributions. P<0.05 was considered to indicate a statistically
significant difference.
Results
Lung tissue pathological changes and
enhanced TLR4 expression in COPD rats
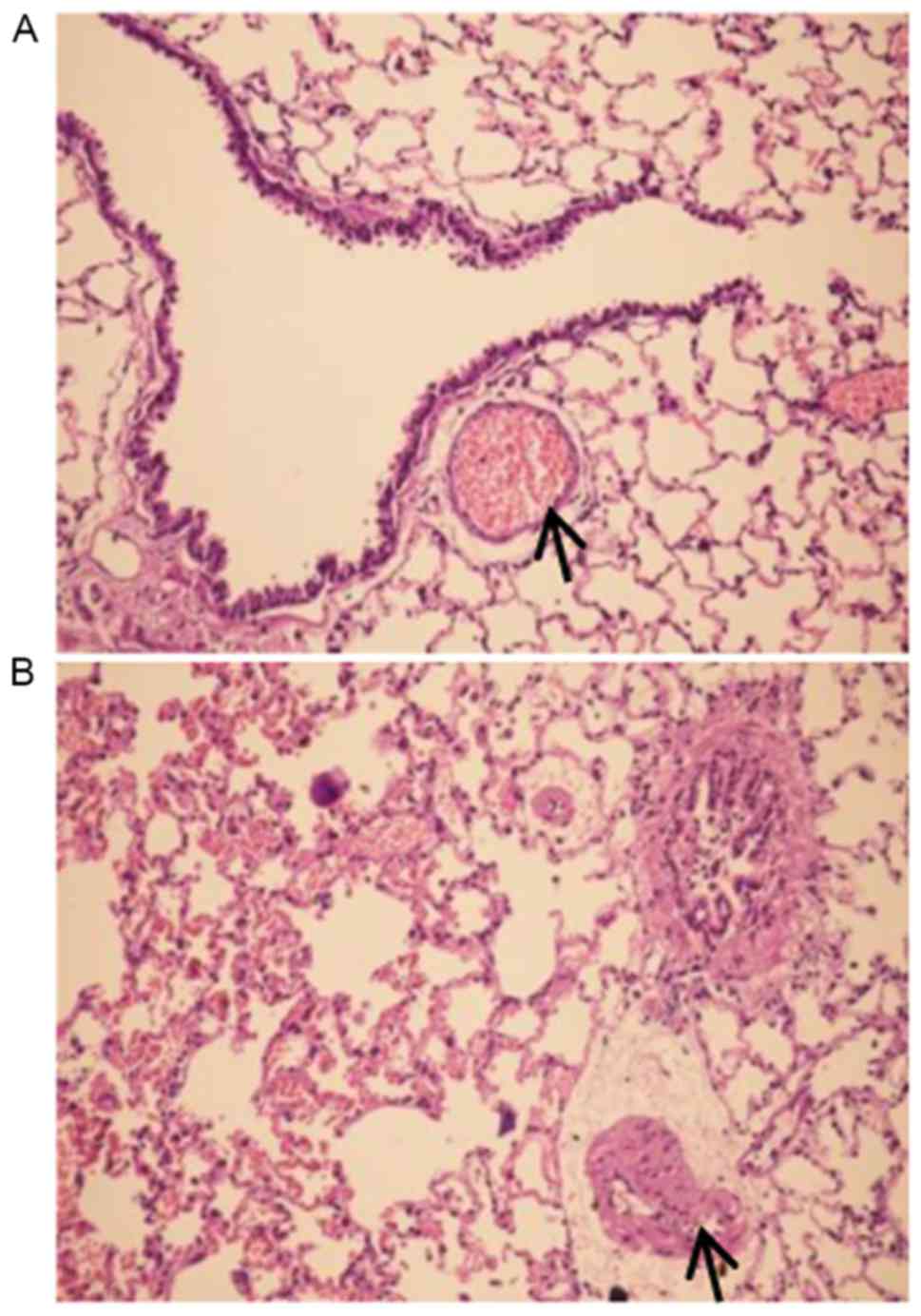
Pathological exam revealed that the control group
exhibited normal airway structures, intact walls and no
inflammatory cell infiltration (Fig.
1A). In contrast, the COPD group had inflammatory cell
infiltration in airway walls, the airway epithelium cell
proliferation, dramatically thickened PASMCs layer in artery and
apparent emphysema lung bullae (Fig.
1B). These pathological changes were consistent with COPD
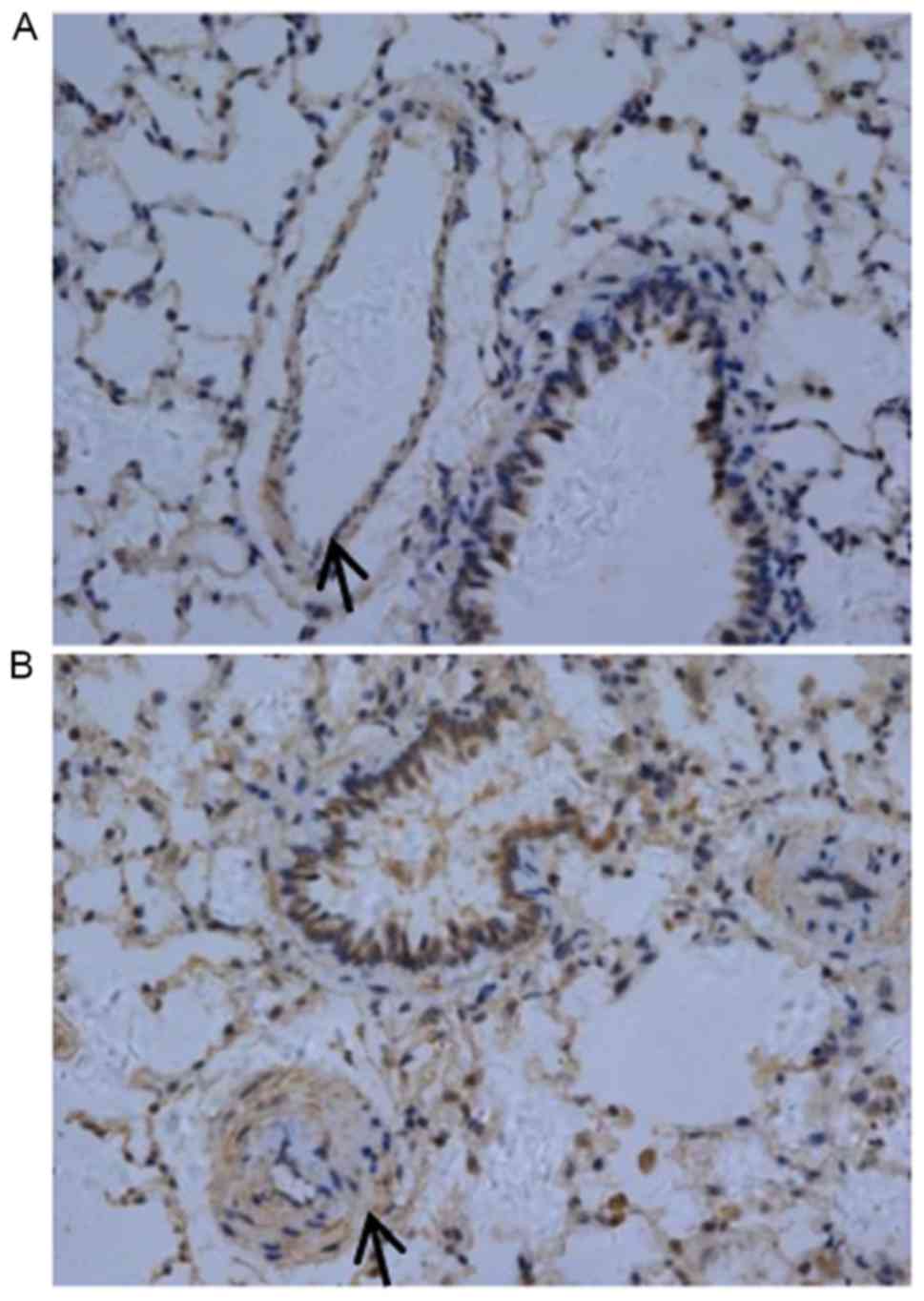
typical features. In particular, the authors observed enhanced TLR4
expression in pulmonary artery smooth muscle layer of COPD rats
(Fig. 2).
TLR4 expression is enhanced by LPS and
inhibited by TAK-242 in PASMCs
As previous reports demonstrated that human aorta
smooth muscle cells express TLR4, which is upregulated by LPS
(8), the authors hypothesized that
rat pulmonary arterial smooth muscle cells may have similar
features. To test the hypothesis, rat PASMCs were isolated and
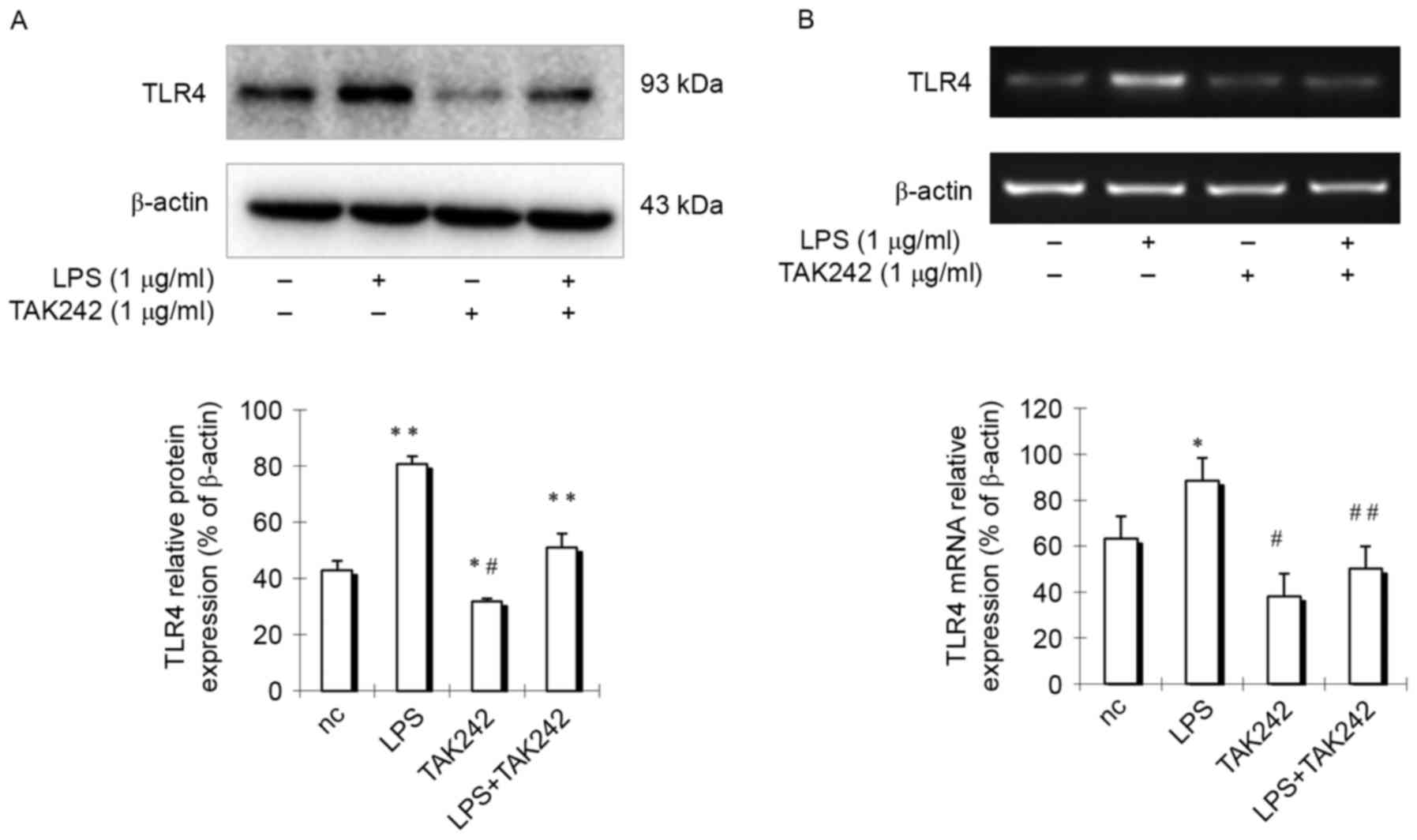
cells were stimulated with LPS for 24 h, then TLR4 expression was
detected in PASMCs. Results indicated that TLR4 expression in cells
treated with LPS was increased obviously compared with control
group cells (Fig. 3), indicating
that TLR4 expression is enhanced by LPS treatment. However, if
cells were treated with LPS at the presence of TLR4 inhibitor
TAK-242, which selectively inhibits TLR4 (9), TLR4 expression was remarkably
suppressed (Fig. 3). These
findings indicated that there is an active TLR4 signaling pathway
in PASMCs.
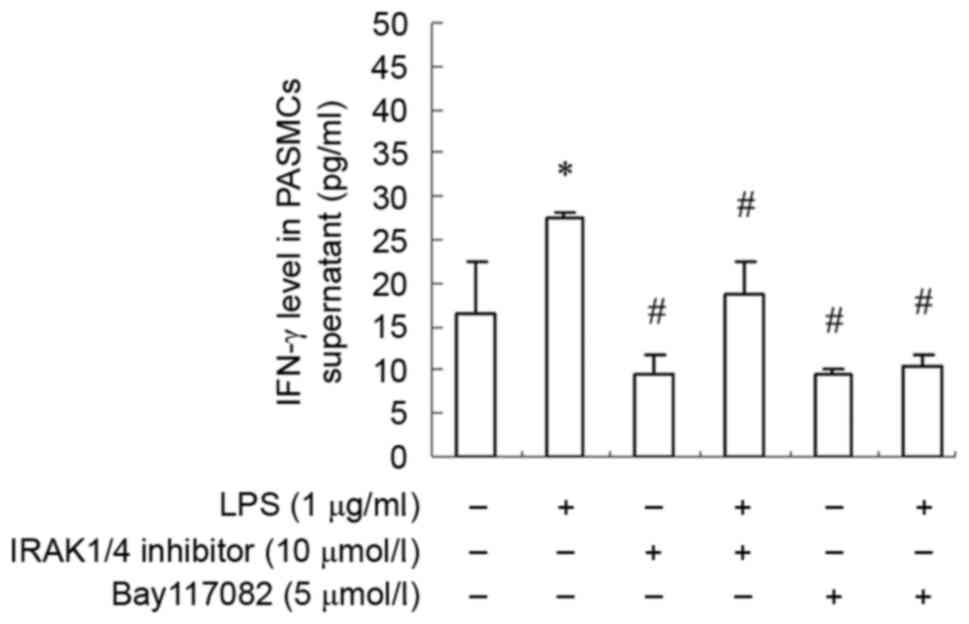
IFN-γ production is enhanced by LPS
and inhibited by TAK-242 in PASMCs
As TLR4 pathway serve a key role in inflammatory
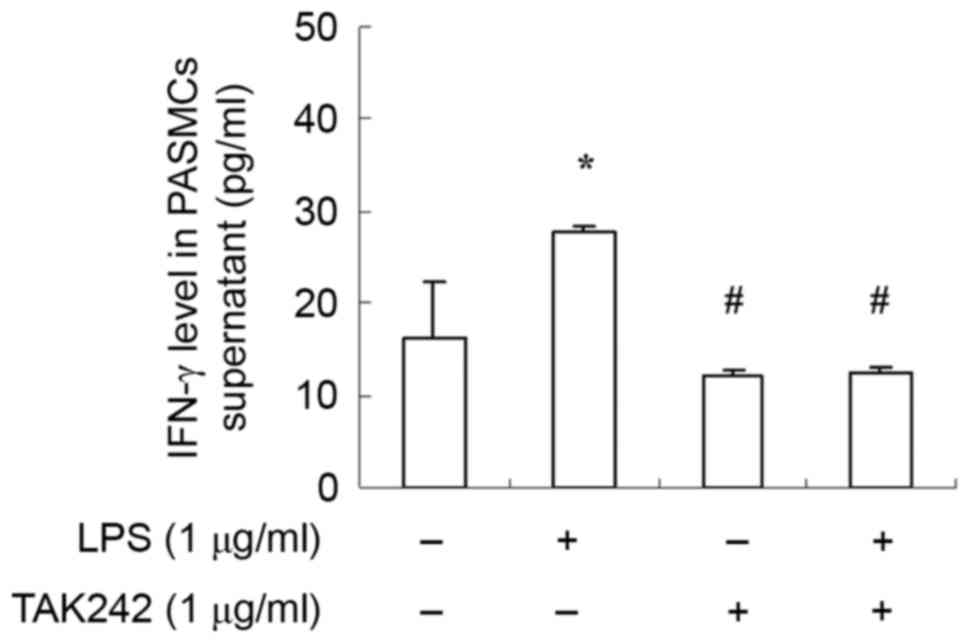
response in immune cells. To further examine the function of the
TLR4 signaling pathway in PASMCs, cells were treated with LPS at
the presence or absence of TAK-242, and the amount of IFN-γ was
measured with ELISA. Results demonstrated that IFN-γ expression was
increased significantly by LPS stimulation (Fig. 4). Moreover, TLR4 inhibitor TAK-242
antagonized the effect of LPS (Fig.
4). These findings suggested that LPS stimulate PASMCs to
produce IFN-γ via the TLR4 signaling pathway.
The cellular response to LPS depends
on IRAK and NF-κB in PASMCs
To further elucidate the roles of TLR4 signaling
pathway in the cellular response to LPS in PASMCs, the authors
analyzed the phosphorylation status or expression of some key
components of this pathway, including interleukin-1
receptor-associated kinase (IRAK), IκB kinase (IKK), I-κB and
nuclear factor (NF)-κB p65, upon LPS treatment. The authors
reported increased amount of phosphorylated IRAK, IKK and I-κB, as
well as increased p65 total protein following LPS treatment
(Fig. 5). However, this effect of
LPS could be inhibited by TAK-242 (Fig. 5). To further verify this
observation, cells were firstly treated with IRAK1/4 inhibitor or
NF-κB inhibitor Bay117082, and then treated with LPS.
Phosphorylation of IRAK, IKK and I-κB was suppressed remarkably,
and p65 total protein was decreased dramatically as well (Fig. 6). Moreover, IFN-γ production was
also decreased significantly (Fig.
7). These finding suggested that PASMCs' response to LPS
stimulation depends on IRAK and NF-κB.
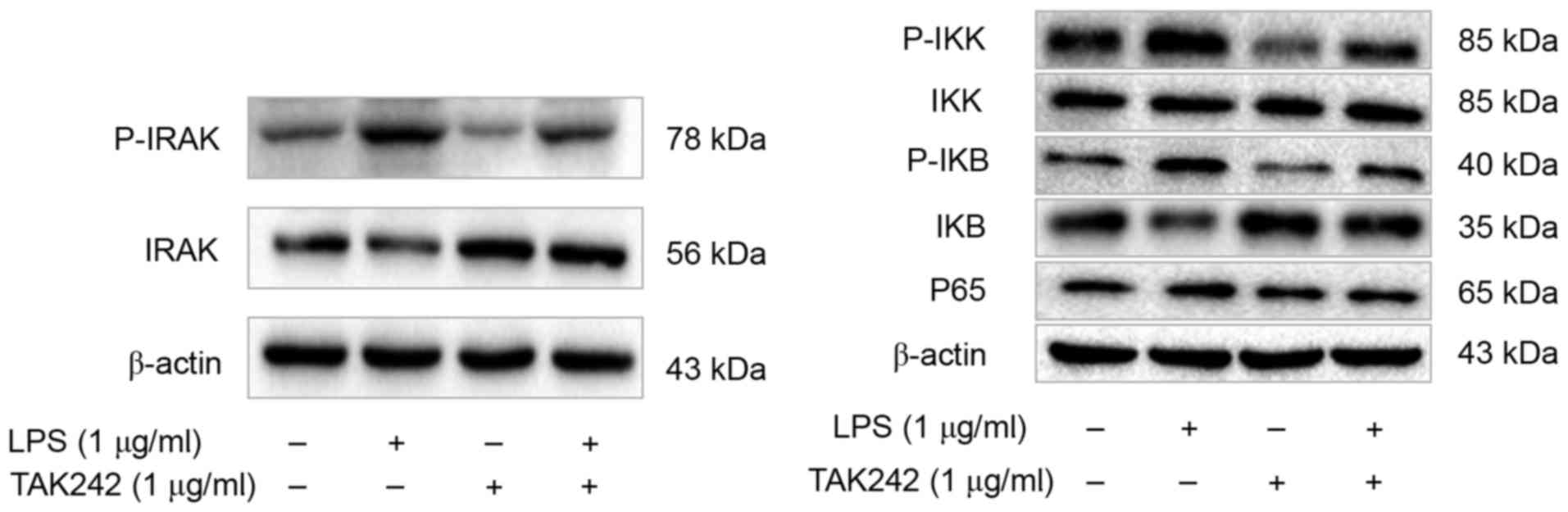 | Figure 5.TLR4 inhibitor TAK-242 suppresses the
TLR4/IRAK/NF-ΚB signaling pathway in PASMCs. Western blot analysis
was performed to detect the levels of p-IRAK, IRAK, p-IKK, IKK,
p-IκB, IκB and NF-κB p65, and the representative blot results are
shown. TLR4, Toll-like receptor 4; IRAK, interleukin-1
receptor-associated kinase; NF-κB, nuclear factor-κB; PASMCs,
pulmonary artery smooth muscle cells; IKK, IκB kinase; LPS,
lipopolysaccharide. |
 | Figure 6.IRAK1/4 inhibitor and Bay117082
suppress the TLR4/IRAK/NF-ΚB signaling pathway in PASMCs. Western
blot analysis was performed to detect the levels of p-IRAK, IRAK,
p-IKK, IKK, p-IκB, IκB and NF-κB p65, and the representative blots
are shown. IRAK, interleukin-1 receptor-associated kinase; TLR4,
Toll-like receptor 4; NF-κB, nuclear factor-κB; PASMCs, pulmonary
artery smooth muscle cells; IKK, IκB kinase; LPS,
lipopolysaccharide. |
Discussion
COPD is a chronic inflammatory disease, which has
high incidence and mortality rate, and has become a significant
public health issue. Pulmonary hypertension is closely related to
the prognosis of COPD (10).
However, the underlying mechanisms remain unclear. In the present
study, the authors found PASMCs has a functional TLR4 signaling
pathway, which enables cells to respond to LPS and produce cytokine
IFN-γ. These findings may provide experimental evidences to
understand how pulmonary hypertension occurs in COPD patients.
Vascular inflammation and vascular remodeling are
important pathological features of COPD, and PASMCs is the main
effector of pulmonary vascular remodeling. Reports have
demonstrated that the cellular phenotype transitions of PASMCs
caused by the stimulation factors, such as inflammation and trauma,
raise inflammatory cell aggregation and inflammatory cascades
amplification, and promote the proliferation and migration of
PASMCs, differentiation of fibroblasts, and ultimately promote the
pulmonary vascular remodeling. This process develops in the early
stages of disease, which are the key starting steps prior to cell
proliferation and migration (5,11–13).
The results indicated that PASMCs themselves are able of producing
IFN-γ, indicating PASMCs serve dual roles as both effectors and
regulator during pathogenesis of COPD.
TLR4 is the first identified mammalian TLR, which is
often expressed in inflammatory cells and can be activated by
ligand binding, which eventually activates NF-κB via cellular
signaling cascades. NF-κB then translocates to the nucleus to
regulate target gene expression. TLR4 in lung tissue may be
activated by risk factors such as cigarette smoking, inhaling
polluted air, infection of bacteria and virus, which ultimately
activates NF-κB and upregulates expression of inflammatory
mediators, especially the primary mediator such as tumor necrosis
factor (TNF)-α and interleukin (IL)-1, which induces alveolar
macrophages to produce a large number of secondary inflammatory
cytokines such as IL-6, IL-8, These further induce the neutrophils
and CD8 + T lymphocytes to participate in the inflammatory reaction
of COPD (14,15). Passive smoking and intratracheal
instillation of LPS may cause lung injury similar to chronic
obstructive pulmonary disease via the NF-κB signaling pathway, and
TLR4 serves an important role in this process (16). A systemic defect in TLR4 signaling
increases lipopolysaccharide-induced suppression of IL-2-dependent
T-cell proliferation in COPD (3).
The TLR4/MyD88 pathway is activated and its downstream inflammatory
cytokines such as TNF-α and IL-6 were increased in macrophages from
COPD patients (17). The present
study indicated that TLR4 signaling in PASMCs depends on IRAK and
NF-κB, suggesting potential therapeutic targets for treatment of
pulmonary hypertension occurred in COPD patients. To the best of
the authors' knowledge, the current study is the first regarding
the role of TLR4 signaling in the synthesis and secretion functions
of PASMCs. Since TLR4 signaling is able to regulate expression of a
variety of inflammatory factors and cytokines in other types of
cells, further experiments are needed to investigate whether it is
also the case in PASMCs. Moreover, further elucidating the role of
TLR4 signaling in proliferation of PASMCs upon LPS stimulation will
provide more evidence to understand the mechanisms of COPD and
pulmonary hypertension.
In summary, this study revealed the LPS enhances
expression of TLR4 and IFN-γ via the TLR4/IRAK/NF-κB signaling
pathway in PASMCs, these findings may provide potential therapeutic
targets for COPD. However, further experiments are needed to find
more details to understand the role of the TLR4 signaling pathway
during the pathogenesis of COPD.
Acknowledgements
The current study was supported in part by the
Natural Science Foundation of Guangxi (grant nos. 2014GXNSFAA118151
and 2015GXNSFAA139178), the National Natural Science Foundation of
China (grant nos. 81360010 and 81560453) and the Medical and
Healthy Technology R&D Project from the Guangxi Health and
Family Planning Commission (grant no. S2015-34). G.H. was supported
by Hundred Talents Program of Guangxi.
References
|
1
|
Bagdonas E, Raudoniute J, Bruzauskaite I
and Aldonyte R: Novel aspects of pathogenesis and regeneration
mechanisms in COPD. Int J Chron Obstruct Pulmon Dis. 10:995–1013.
2015.PubMed/NCBI
|
|
2
|
Barbera JA and Blanco I: Pulmonary
hypertension in patients with chronic obstructive pulmonary
disease: Advances in pathophysiology and management. Drugs.
69:1153–1171. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Knobloch J, Chikosi SJ, Yanik S, Rupp J,
Jungck D and Koch A: A systemic defect in Toll-like receptor 4
signaling increases lipopolysaccharide-induced suppression of
IL-2-dependent T-cell proliferation in COPD. Am J Physiol Lung Cell
Mol Physiol. 310:L24–L39. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Molteni M, Gemma S and Rossetti C: The
role of toll-like receptor 4 in infectious and noninfectious
inflammation. Mediators Inflamm. 2016:69789362016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yi B, Cui J, Ning JN, Wang GS, Qian GS and
Lu KZ: Over-expression of PKGIα inhibits hypoxia-induced
proliferation, Akt activation, and phenotype modulation of human
PASMCs: The role of phenotype modulation of PASMCs in pulmonary
vascular remodeling. Gene. 492:354–360. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bauer EM, Shapiro R, Zheng H, Ahmad F,
Ishizawar D, Comhair SA, Erzurum SC, Billiar TR and Bauer PM: High
mobility group box 1 contributes to the pathogenesis of
experimental pulmonary hypertension via activation of Toll-like
receptor 4. Mol Med. 18:1509–1518. 2012. View Article : Google Scholar :
|
|
7
|
Donaldson JG: Immunofluorescence staining.
Curr Protoc Cell Biol Chapter. 4:Unit 4.3. 2001. View Article : Google Scholar
|
|
8
|
Li H, He Y, Zhang J, Sun S and Sun B:
Lipopolysaccharide regulates toll-like receptor 4 expression in
human aortic smooth muscle cells. Cell Biol Int. 31:831–835. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kawamoto T, Ii M, Kitazaki T, Iizawa Y and
Kimura H: TAK-242 selectively suppresses Toll-like receptor
4-signaling mediated by the intracellular domain. Eur J Pharmacol.
584:40–48. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Orr R, Smith LJ and Cuttica MJ: Pulmonary
hypertension in advanced chronic obstructive pulmonary disease.
Curr Opin Pulm Med. 18:138–143. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nazari-Jahantigh M, Wei Y and Schober A:
The role of microRNAs in arterial remodelling. Thromb Haemost.
107:611–618. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kawai T and Akira S: The role of
pattern-recognition receptors in innate immunity: Update on
Toll-like receptors. Nat Immunol. 11:373–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ospelt C and Gay S: TLRs and chronic
inflammation. Int J Biochem Cell Biol. 42:495–505. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pace E, Giarratano A, Ferraro M, Bruno A,
Siena L, Mangione S, Johnson M and Gjomarkaj M: TLR4 upregulation
underpins airway neutrophilia in smokers with chronic obstructive
pulmonary disease and acute respiratory failure. Hum Immunol.
72:54–62. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Geraghty P, Dabo AJ and D'Armiento J: TLR4
protein contributes to cigarette smoke-induced matrix
metalloproteinase-1 (MMP-1) expression in chronic obstructive
pulmonary disease. J Biol Chem. 286:30211–30218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meng Y, Yu CH, Li T, Li W, Cai SX and Li
X: Expression and significance of Toll-like receptor-4 in rats lung
established by passive smoking or associated with intratracheal
instillation of lipopolysaccharide. Zhonghua yi xue za zhi.
93:2230–2234. 2013.(In Chinese). PubMed/NCBI
|
|
17
|
Zeng X, Liu X, Bao H, Zhang Y, Wang X, Shi
K and Pang Q: Effects of sulforaphane on Toll-like receptor
4/myeloid differentiation factor 88 pathway of monocyte-derived
macrophages from patients with chronic obstructive pulmonary
disease. Zhonghua Jie He He Hu Xi Za Zhi. 37:250–254. 2014.(In
Chinese). PubMed/NCBI
|