Introduction
A neuropeptide substance P (SP) is an important
mediator of neurogenic inflammation in the central and peripheral
nervous systems. SP is implicated in pain, and also plays an
important role in tumor cell proliferation, anti-apoptotic effects
on tumor cells, angiogenesis, tumor cell invasion and metastasis
(1–3). SP is released from primary afferent
nociceptors and sympathetic postganglionic neurons, and activates
neighboring receptors, thereby triggering spreading activation
(4,5). Moreover, SP has been shown to induce
the expression of proinflammatory cytokines and chemokines, such as
interleukin (IL)-6 and IL-8 (6,7),
which are involved in the pathogenesis of several human brain
disorders (such as multiple sclerosis, dementia complex, and
Alzheimer's disease) (8), although
it is currently a matter of debate whether SP really plays a
pathogenic role in these disorder. Previous report has shown that
the activation of a SP receptor [neurokinin-1 receptor (NK1R)]
elicits signals affecting inflammatory cytokine gene expression
(7). In addition, it has been
reported that SP potently triggers the activation of nuclear factor
(NF)-κB, an important transcriptional activator, which regulates
the production of various cytokines and other proinflammatory
mediators (7).
Mitogen-activated protein kinases (MAPK), a family
of protein Ser/Thr kinases, consist of at least three major
subfamilies: i) the p42/44 MAPKs, which are also called
extracellular signal regulated kinases (ERK-1 and ERK-2); ii) the
c-Jun NH2-terminal kinase/stress-activated protein kinases
(JNK/SAPK) including p46 JNK1 and p54 JNK2; and iii) the p38 MAPK
subfamily. MAPKs are activated under stress conditions in response
to a variety of extracellular stimuli, including oxidative stress.
Among these MAPKs, phosphorylation of p38 MAPK is induced in dorsal
horn of the spinal cord and dorsal root ganglia following
peripheral nerve injury or inflammation (9–11).
Gabapentin (GBP) and pregabalin (PGB) are structural
analogues of γ-amino butyric acid (GABA) with lipophilic
characteristics, and were developed as potential anticonvulsants
(12). However, many studies have
shown that these substances do not act as GABA agonists and indeed
have little demonstrable effect on any aspect of GABA transmission
(13,14). Furthermore, a recent study has
clarified the current understanding of gabapentinoid pharmacology
that GBP and PGB do not inhibit any conventional subtype of
voltage-gated calcium channel (VGCC), but rather selectively block
calcium channels that contain the α2δ-1 subunit, with
pharmacodynamics and cellular-specificities, depending on the
structural and biochemical states of the α2δ-1 protein (15). It is believed that blockade of VGCC
containing the α2δ-1 subunit is the predominant pharmacological
mechanism of both GBP and PGB (16). PGB binds potently to the α2δ-1
subunit and modulates calcium influx at nerve terminals, thereby,
reducing the release of several neurotransmitters, including
glutamate, noradrenaline, serotonin and SP (17–19).
Interestingly, GBP could be a therapeutic agent, when given
systemically, for treatment of neuropathic or postsurgical pain
(20–24), and also reduces the experimental
pain in humans after sensitization of the skin with capsaicin and
heat (25). Furthermore, GBP and
its derivative PGB reduce nociceptive behaviors of animal models
with neuropathic pain or inflammation such as nerve ligation,
injection of immune antigens, herpes infection, arthritis,
diabetes, postoperative pain and thermal injury (26–30).
In contrast, neither GBP nor PGB alters acute nociceptive responses
(31,32). Thus, it is believed that the
antinociceptive action of these substances depends on the chronic
nociceptive responses with neuropathic or inflammatory
conditions.
It has been demonstrated that the release of
peptidergic neurotransmitters (including SP) from sensory neurons
is increased during inflammation or in neuropathic pain models
(33). In addition, SP is axonally
transported to peripheral nerve endings, where it is released in
response to traumatic stimuli and induces various biological
effects (33), Importantly, GBP
and PGB modulate the release of SP under the conditions
corresponding to significant inflammation-induced sensitization of
the spinal cord, which involves the action of SP on NK1 (SP)
receptor (NK1R) (34). Based on
the findings, we hypothesized that GBP and PGB may exert
antiinflammatory action by modulating the SP-mediated NK1R
response. In this study, therefore, we investigated the
antiinflammatory effects of GBP and PGB on SP-induced activation of
a human glioblastoma astrocytoma cell line U373 MG cell, which
expresses the high levels of NK1R.
Materials and methods
Materials
U373 MG cell line (Uppsala; ECACC08061901) was
purchased from European Collection of Cell Cultures (ECACC;
Salisbury, UK). SP was obtained from Sigma-Aldrich Co., LLC., (St.
Louis, MO, USA); GBP from Tokyo Chemical Industry Co., Ltd. (Tokyo,
Japan); PGB from Toronto Research Chemicals (Toronto, ON, Canada);
Minimum essential medium (MEM), non-essential amino acids (NEAA),
sodium pyruvate and fetal bovine serum (FBS) from Gibco BRL Life
Technologies (Grand Island, NY, USA). Phosphate-buffered saline
(PBS), RIPA buffer containing protease inhibitor cocktail, sample
buffer solution containing reducing reagent (6X) for SDS-PAGE,
running buffer solution (10X) for SDS-PAGE, Blocking One, WB
Stripping Solution Strong, and Protein Ladder One Multi-color
(Broad Range) for SDS-PAGE from Nacalai Tesque, Inc., (Kyoto,
Japan). BCA protein assay reagent kit and enhanced
chemiluminescence reagent, SuperSignal West Dura and NE-PER nuclear
and cytoplasmic extraction reagents from Thermo Fisher Scientific,
Inc. (Waltham, MA, USA). Mini-PROTEAN® TGX™ Precast Gel
and Trans-Blot® Turbo™ Mini PVDF Transfer Packs from
Bio-Rad Laboratories, Inc., (Hercules, CA, USA).
Antibodies
Anti-phospho-ERK1/2 MAPK (Thr202/Tyr204) rabbit
antibody (no. 9101), anti-ERK1/2 MAPK rabbit antibody (no. 9102),
anti-phospho-NF-κB p65 (Ser536) rabbit monoclonal antibody (mAb;
no. 3033), anti-NF-κB p65 rabbit mAb (no. 8242), and histone H3
(D1H2) XP rabbit mAb (no. 4499) from Cell Signaling Technology,
Inc. (Danvers, MA, USA), anti-phospho p38 MAPK rabbit antibody
(Thr180/Tyr182; V121A) from Promega Corporation (Madison, WI, USA),
anti-p38 MAPK (p38/SAPK2α) mouse mAb (no. 612168) from BD
Biosciences (San Jose, CA, USA). Horseradish peroxidase
(HRP)-conjugated goat anti-rabbit IgG (AP132P), HRP-conjugated goat
anti-mouse IgG/IgM (AP308P) from Chemicon International (Temecula,
CA, USA).
Cell culture
U373 MG cells were cultured in MEM supplemented with
1% (v/v) penicillin/streptomycin, and NEAA, 1 mM sodium pyruvate
and 10% heat-inactivated FBS. The cells were maintained at 37°C in
a 5% CO2 humidified atmosphere.
Preparation of whole cell lysate and
western blot analysis
U373 MG cells were plated into 12-well tissue
culture plates at a density of 1×105 cells/well and
incubated in MEM with 10% FBS for 12 h, followed by incubation in
MEM with 0.5% FBS for 12 h at 37°C. Subsequently, the cells were
incubated with GBP or PGB (1 mM) for 60 min, and then stimulated
with SP (100 nM) for 10 or 15 min. Thereafter, the cells were
washed three times with ice-cold PBS and lysed in 0.1 ml of RIPA
buffer (50 mmol/l Tris-HCl pH 7.6, 150 mmol/l NaCl, 1% Nonidet P40,
0.5% sodium deoxycholate, 0.1% SDS and Protease Inhibitor
Cocktail). Protein concentrations of cell lysates were measured
with BCA protein assay reagent (Thermo Scientific). The lysates
were mixed with SDS-polyacrylamide gel electrophoresis (PAGE)
sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol,
0.05% bromphenol blue, and 5% 2-mercaptoethanol), and applied to
SDS-PAGE in 10% gels (Mini-PROTEAN® TGX™ Precast Gel;
10–20 µg protein/lane). Thereafter, separated proteins were
electroblotted onto polyvinylidine fluoride membranes
(Trans-Blot® Turbo™ Mini PVDF Transfer Packs). After
incubation with Blocking One (Nacalai Tesque, Inc.), blots were
proved with a 1,000-fold dilution of rabbit anti-phospho p38 MAPK
antibody, anti-phospho ERK1/2 antibody or anti-phospho NF-κB
antibody, and further proved with a 10,000-fold dilution of
HRP-conjugated goat anti-rabbit IgG. Signals were detected with
SuperSignal West Dura Chemiluminescent Substrate (Thermo Fisher
Scientific, Inc.), and quantified using LAS-3000 luminescent image
analyzer (Fujifilm, Tokyo, Japan) and MultiGauge software
(Fujifilm). Thereafter, the antibody was stripped using WB
Stripping Solution Strong (Nacalai Tesque, Inc.) at room
temperature for 15 min. Blots were proved with a 1,000-fold
dilution of mouse anti-p38 MAPK antibody, rabbit anti-ERK1/2
antibody or anti-NF-κB antibody, and further proved with a
10,000-fold dilution of HRP-conjugated goat anti-rabbit IgG or
HRP-conjugated goat anti-mouse IgG/IgM. Signals were detected and
analyzed, as described above.
Preparation of nuclear extract and
western blot analysis
U373 MG cells were plated into 12-well tissue
culture plates at a density of 2×105 cells/well and
incubated in MEM with 10% FBS for 12 h, followed by incubation in
MEM with 0.5% FBS for 12 h at 37°C. Subsequently, the cells were
incubated with GBP or PGB (1.0 mM) for 60 min, and then stimulated
with SP (100 nM) for 15 min. Thereafter, the cells were washed
three times with ice-cold PBS. Cells were detached by trypsin
treatment, and washed in ice-cold PBS containing phosphatase
inhibitors, and then centrifuged at 300 g for 5 min. Nuclear
fractions were prepared using NE-PER nuclear and cytoplasmic
extraction reagents by suspending the cell pellet in a hypotonic
buffer, and centrifugation at 14,000 × g for 30 min. After
collection of the supernatant (cytoplasmic fraction), the pellet
(nuclear fraction) was lysed and solubilized in lysis buffer
containing proteasome inhibitors. Protein concentrations were
determined using with BCA protein assay reagent. The nuclear
extract was mixed with SDS-PAGE sample buffer), and applied to
SDS-PAGE in 10% gels, followed by western blot analysis using a
1,000-fold dilution of rabbit anti-NF-κB antibody and a 10,000-fold
dilution of HRP-conjugated goat anti-rabbit IgG, and a 1,000-fold
dilution of rabbit histone H3 and a 10,000-fold dilution of
HRP-conjugated goat anti-rabbit IgG, as described in the above
section.
Quantification of IL-6 and IL-8
U373 MG cells were plated into 12-well tissue
culture plates at a density of 1×105 cells/well and
incubated in MEM with 10% FBS for 12 h, followed by incubation in
MEM with 0.5% FBS for 12 h at 37°C. Then, the cells were incubated
with GBP or PGB (1 mM) for 60 min, and stimulated with SP (100 nM)
for 24 h. Culture media were recovered, centrifuged for 10 min at
12,000 × g, and the levels of IL-6 in the supernatants were
measured by a sandwich enzyme-linked immunosorbent assay (ELISA)
kit, according to the manufacturer's instructions (eBioscience, San
Diego, CA, USA). Moreover, the levels of IL-8 in the media were
measured by ELISA kit, according to the manufacturer's instructions
(R&D systems, Inc., Minneapolis, MN, USA).
Statistical analysis
Data are expressed as mean ± standard deviation, and
analyzed for significant difference by a one-way ANOVA with
multiple comparison test using GraphPad Prism version 6.0 for
Windows (GraphPad Software, San Diego, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Suppression of SP-induced activation
of p38 MAPK and ERK1/2 by GBP and PGB
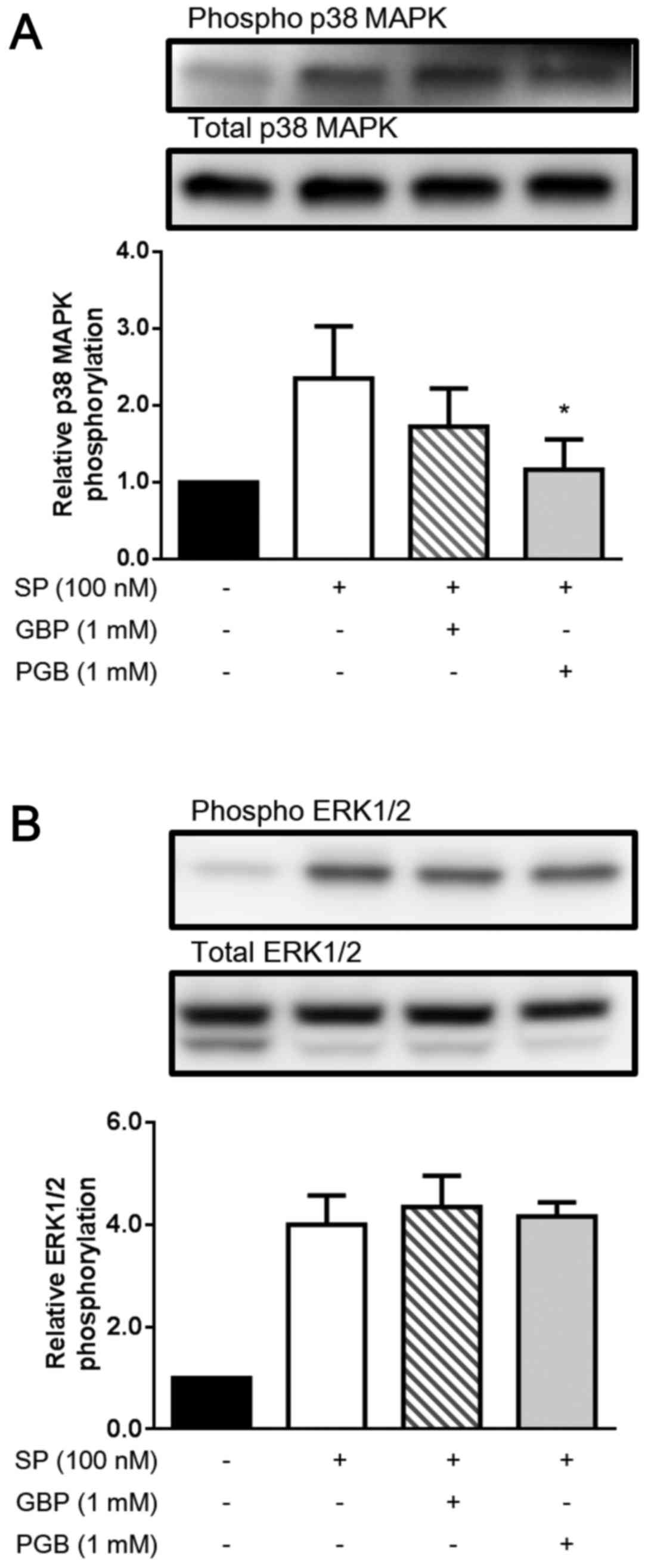
First, we evaluated the effect of GBP and PGB on the
phosphorylation of p38 MAPK. As shown in Fig. 1A, SP stimulation (100 nM) clearly
induced the phosphorylation of p38 MAPK in U373 MG cells.
Interestingly, GBP (1 mM) and PGB (1 mM) substantially suppressed
the SP-induced phosphorylation of p38 MAPK, although only the
suppression by PGB was statistically significant (P<0.05).
Next, we evaluated the effect of GBP and PGB on the
phosphorylation of ERK1/2. As shown in Fig. 1B, SP stimulation (100 nM) markedly
induced the phosphorylation of ERK1/2 in U373 MG cells; however,
GBP (1 mM) and PGB (1 mM) did not essentially suppress the
SP-induced phosphorylation of ERK1/2.
Suppression of SP-induced activation
of NF-κB and nuclear translocation of p65 by GBP and PGB
Furthermore, we evaluated the effect of GBP and PGB
on the phosphorylation of NF-κB. SP stimulation (100 nM)
substantially induced the phosphorylation of NF-κB in U373 MG
cells. Importantly, GBP and PGB significantly abolished the
SP-induced phosphorylation of NF-κB (Fig. 2A; P<0.05).
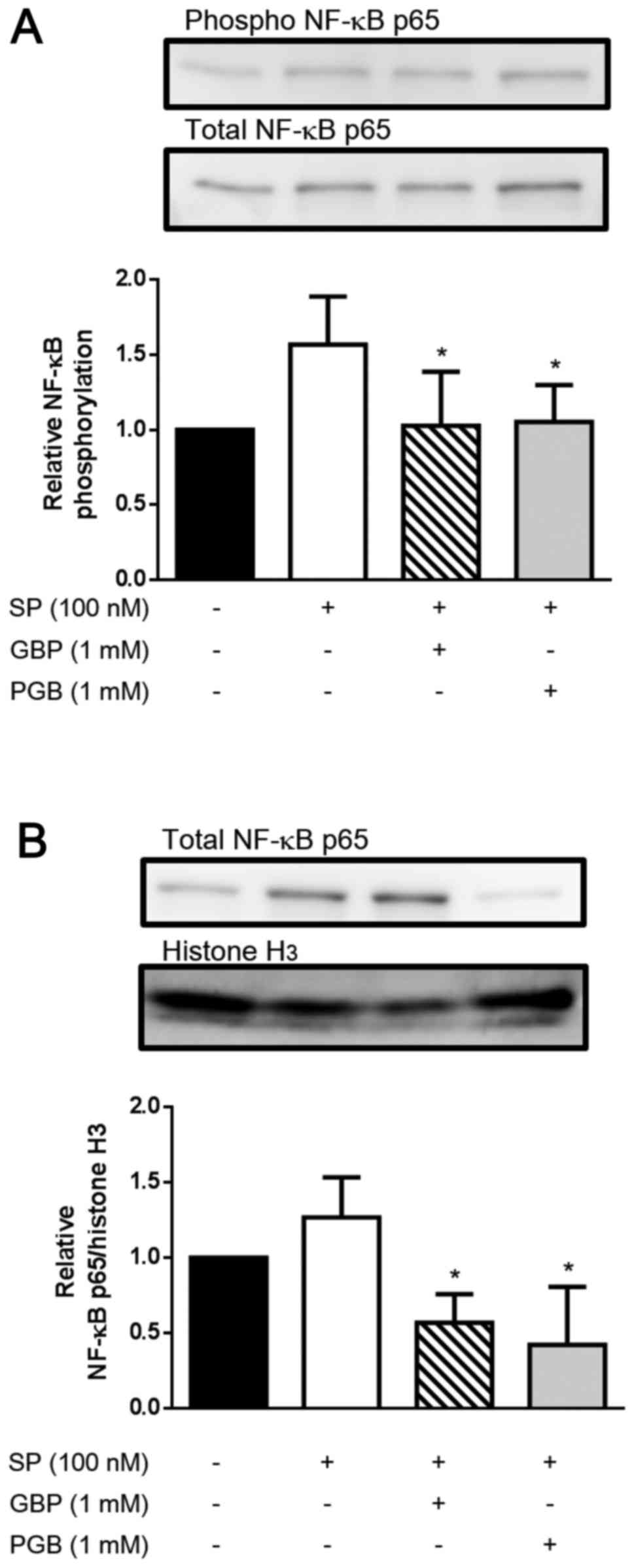 | Figure 2.Evaluation of the effects of
gabapentin (GBP) and pregabalin (PGB) on substance P (SP)-induced
phosporylation of nuclear factor (NF)-κB and SP-induced nuclear
translocation of NF-κB. U373 MG cells were plated into 12-well
tissue culture plates at a density of 2×105 cells/well
and incubated in minimum essential medium (MEM) with 10% FBS for 12
h, followed by incubation in MEM with 0.5% fetal bovine serum (FBS)
for 12 h at 37°C. Subsequently, the cells were incubated with or
without GBP (1 mM) or PGB (1 mM) for 60 min, and then incubated
with or without SP (100 nM) for 15 min. (A) The expression of
phosphorylation of NF-κB were detected by probing with anti-phospho
NF-κB p65 (Ser536) rabbit mAb and HRP-conjugated goat anti-rabbit
immunoglobulin G (IgG). In order to confirm that equal amount of
proteins were analyzed in each samples, the blots were stripped,
and total NF-κB were detected by reprobing with anti-NF-κB p65
rabbit mAb and horseradish peroxidase (HRP)-conjugated goat
anti-rabbit IgG. (B) After incubation of U373 MG cells with SP in
the absence or presence of GBP or PGB, nuclear fractions were
prepared, and the nuclear levels of NF-κB p65 were analyzed by
probing with anti-NF-κB p65 rabbit mAb and HRP-conjugated goat
anti-rabbit IgG. In order to confirm that equal amount of proteins
were analyzed in each samples, the blots were stripped, and histone
H3 were detected by reprobing with anti-histone H3 and
HRP-conjugated goat anti-rabbit IgG. A representative image is
shown. Data are the means ± standard deviation of 3 separate
experiments, and expressed as relative to the cells incubated
without SP, GBP, and PGB. Data are compared between the
SP-stimulated cells incubated without and with GBP or PGB.
*P<0.05. |
To further characterize the effect of GBP and PGB on
the SP-induced activation of NF-κB, we evaluated the nuclear
translocation of NF-κB. Thus, U373 cells were pretreated with GBP
(1 mM) and PGB (1 mM) followed by stimulation with SP (100 nM), and
the nuclear levels of p65 were analyzed. Nuclear p65 level was
increased after SP stimulation compared with unstimulated cells;
however, SP-induced p65 level was significantly reduced by GBP and
PGB (Fig. 2B; P<0.05).
Suppression of SP-induced IL-6 and
IL-8 production by GBP and PGB
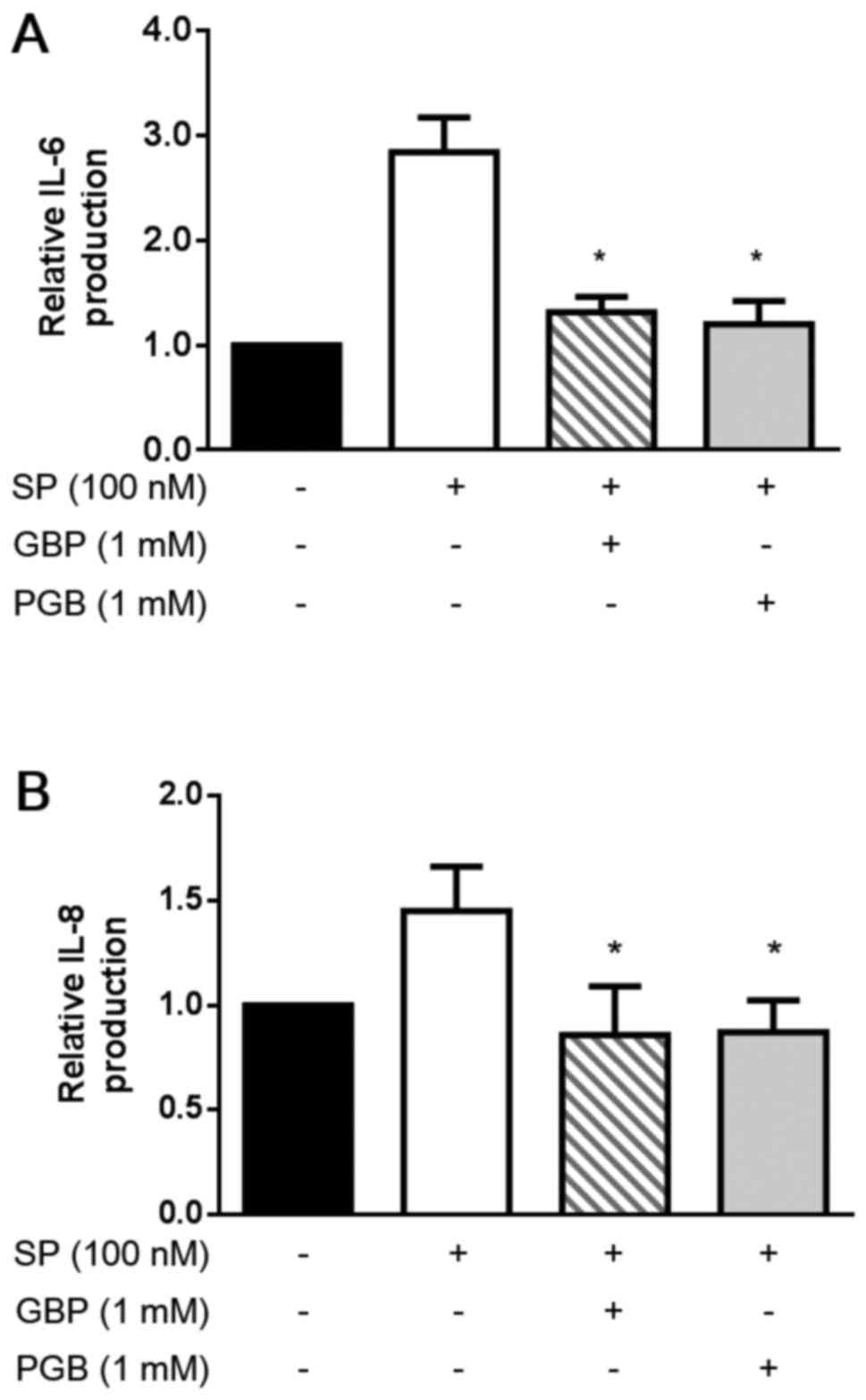
Finally, we evaluated the effect of GBP and PGB on
the IL-6 and IL-8 production by U373 MG cells. As shown in Fig. 3, both IL-6 and IL-8 were produced
by resting U373 MG cells without SP. Importantly, the production of
IL-6 and IL-8 was substantially increased by SP stimulation (100
nM). Noticeably, GBP and PGB (1 mM) significantly suppressed both
the IL-6 and IL-8 production (P<0.05).
Discussion
To our knowledge, this is the first study to
demonstrate the effects of GBP and PGB on the SP-induced
inflammatory responses in glioblastoma astrocytoma U373 MG cells.
U373 MG glioblastoma astrocytoma cells express a functional
high-affinity NK1R (a SP receptor) (35) and are able to produce IL-6 and IL-8
in response to SP (6). In this
study, we revealed that GBP and PGB suppressed the SP-induced
production of IL-6 and IL-8 in U373 MG cells. Furthermore, GBP and
PGB inhibited the SP-induced phosphorylation of p38 MAPK and NF-κB,
and nuclear translocation of NF-κB in U373 MG cells. Thus, GBP and
PGB likely prevent the SP-induced IL-6 and IL-8 production in U373
MG cells by inhibiting signaling molecules including p38 MAPK and
NF-κB, thereby exhibiting anti-inflammatory action.
According to pharmacokinetic study (36,37),
the maximum plasma concentration of GBP after administration of a
single 400 mg GBP tablet to 12 volunteers was 3.33±1.19 µg/ml
(approximately 20 µM). Moreover, it has been shown that 25 µM GBP
and PGB reduced the SP-induced NF-κB activation in glioblastoma
cell line in vitro (38).
In our preliminary experiments, we observed that 100 µM GBP weakly
but 1 mM GBP clearly suppressed the SP-induced phosphorylation of
p38 MAPK and NF-κB, and production of IL-6 and IL-8. Thus, we
examined the effect of GBP and PGB at a concentration of 1 mM on
the SP-induced phosphorylation of signaling molecules, and
production of cytokines.
The concentration of GBP used in this study (1 mM)
was 50-fold higher than that of the maximum plasma concentration;
however, we observed that GBP at 100 µM weakly suppressed the
SP-induced phosphorylation of p38 MAPK and NF-κB, and production of
IL-6 and IL-8 (data not shown). Thus, we speculate that GBP could
exert the anti-inflammatory action in vivo, based on our
findings.
GBP and PGB are anticonvulsants originally developed
as spasmolytic agents for the management of generalized or partial
epileptic seizures resistant to conventional therapies (12). However, subsequent single center
and multicenter, randomized double-blind trials demonstrated that
GBP is also effective for the management of pain of inflammatory
and neuropathic origin, such as post herpetic neuralgia and painful
diabetic neuropathy (39,40). Although PGB has not been
extensively investigated as GBP, recent double-blind trials showed
that PGB is also effective in the management of postoperative pain
and diabetic neuropathy (39). In
animal models of nociception, GBP reduces the mechanical or thermal
hypersensitivity associated with nerve injury (31,41),
incisional injury (26),
inflammatory injury (27,41,42),
and formalin-induced injury (42–44).
PGB similarly reduces the mechanical or thermal hypersensitivity
associated with injuries, described above (27,42,45).
In contrast, both compounds do not exhibit substantive effect on
acute pain in uninjured animal models.
An interesting action of SP is the induction and
modulation of proinflammatory cytokine secretion by glial cells
(7). The mechanism of SP-induced
signal transduction is incompletely understood. However, it has
been suggested that the activation of NF-κB is involved in
SP-induced cytokine expression (7). Importantly, it is demonstrated that
GBP and PGB decrease SP-induced NF-κB activation in human
neuroblastoma and rat glioma cells, and that these drugs also
inhibit NF-κB activation in rat spinal dorsal root ganglia cells
pre-treated with SP (38).
Interestingly, it has been reported that GBP and PGB attenuate the
release of neuropeptides (such as SP) from inflamed spinal cord
(34). Importantly, the present
study demonstrated that GBP and PGB prevent the SP-induced IL-6 and
IL-8 production in U373 MG cells by inhibiting p38 MAPK and NF-κB
activation. Based on these findings, it could be speculated that
gabapentinoids may exhibit anti-inflammatory action by suppressing
not only the SP-induced cytokine production via the inhibition of
p38 MAPK and NF-κB activation, but also the secretion of
inflammatory neuropeptides (such as SP) at the inflamed neural
tissue. In fact, it has been demonstrated that PGB binds potently
to the α2δ-1 subunit of calcium channels and modulates calcium
influx at nerve terminals, thereby reducing the release of several
neurotransmitters, including SP (46). However, it has been also reported
that PGB and GBP suppress the degradation of IκB, thereby
inhibiting nuclear localization of NF-κB (p65) in SH-SY5Y
glioblastoma cells (45). Thus,
gabapentinoid-mediated modulation of nuclear localization of NF-κB
is also considered as a molecular mechanism for the actions of GBP
and PGB.
The α2δ-1 subunit of VGCC is involved in propagation
of excitatory signals mediated by glutamate, calcitonin
gene-related protein (CGRP), and SP (34). Its upregulation under pathological
conditions is associated with hyperexcitatory states such as
seizures and neuropathic pain. The causal link between
α2δ-1-subunit expression and hyperalgesia has been demonstrated in
several experimental animal models of spinal nerve injury (47,48).
Notably, PGB binds potently to the α2δ-1 subunit and suppresses
calcium influx at nerve terminals, thereby modulating
hyperexcitatory states of neuronal synapse.
SP stimulates a number of intracellular signaling
molecules including the members of MAPK family (ERK1/2 and p38
MAPK) via the action of NK1R. Moreover, SP induce the IL-6 and IL-8
production via the activation of p38 MAPK, ERK1/2 and NF-κB by U373
MG (49). In this study, GBP and
PGB did not reduce the SP-induced ERK1/2 phosphorylation (Fig. 1B) but reduced p38 MAPK
phosphorylation in U373 MG cells (Fig.
1A). Thus, our results suggest that GBP and PGB reduce the
SP-induced production of IL-6 and IL-8 by the NK1R (a principal
receptor for SP)-expressing U373 MG cells via the inhibition of
signaling molecules p38 MAPK but not ERK1/2. It is possible that
GBP and PGB may reduce the expression of NK1R, thereby suppressing
the SP-induced production of IL-6 and IL-8. However, GBP and PGB
did not reduce the SP-induced ERK1/2 phosphorylation, as mentioned
above. Thus, GBP and PGB unlikely modulate the expression of
NK1R.
In a neuropathic pain model, it is well documented
that microglial activation, accompanying with p38 MAPK
phosphorylation in the spinal cord, plays an important role in the
development of neuropathic pain (50–52).
Both peripheral inflammation and nerve injury induce p38 MAPK
activation in spinal microglia, and p38 MAPK inhibitor SB203580
suppresses the inflammation-induced thermal hyperalgesia and spinal
nerve ligation-induced mechanical allodynia (53). Moreover, intrathecal injection of
minocycline, which inhibits spinal microglia activation by blocking
p38 MAPK, exerts antinociceptive effect in both inflammation and
neuropathic pain models (52).
These observations indicate that p38 MAPK activation plays
important role in development of neuroinflammation. In addition,
IL-6 is speculated to be involved in the development of neuropathic
pain, because IL-6 mRNA was significantly elevated in both the
dorsal and ventral horns in a neuropathic pain model of spinal
nerve cryoneurolysis and spinal nerve tight ligation (54,55).
IL-8 also contributes to the pathophysiology of inflammatory pain,
because the expression of IL-8 was critically upregulated in the
ipsilateral spinal cord dorsal horn after chronic constriction
injury (56,57). In this study, GBP and PGB reduced
IL-6 and IL-8 production possibly by suppressing p38 MAPK in U373
MG cell, and the results explain the pharmacological action of GBP
and PGB on neuroinflammation (neuropathic pain).
In conclusion, we demonstrated that GBP and PGB
suppressed the production of IL-6 and IL-8 in U373 MG cells.
Furthermore, GBP and PGB inhibited the SP-induced activation of p38
MAPK, and NF-κB in U373 MG cells. Thus, GBP and PGB likely prevent
the SP-induced IL-6 and IL-8 production by U373 MG cells via the
inhibition of signaling molecules including p38 MAPK and NF-κB,
thereby exhibiting anti-neuroinflammatory action.
Acknowledgements
This study was supported by Grant-in-Aid for
Scientific Research (C) (15K10564).
References
|
1
|
Samsam M, Coveñas R, Ahangari R, Yajeya J,
Narváez JA and Tramu G: Simultaneous depletion of neurokinin A
substance P and calcitonin gene-related peptide from the caudal
trigeminal nucleus of the rat during electrical stimulation of the
trigeminal ganglion. Pain. 84:389–395. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnson MB, Young AD and Marriott I: The
therapeutic potential of targeting substance P/NK-1R interactions
in inflammatory CNS disorders. Front Cell Neurosci. 10:2962017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Muñoz M and Coveñas R: Involvement of
substance P and the NK-1 receptor in cancer progression. Peptides.
48:1–9. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ebner K and Singewald N: The role of
substance P in stress and anxiety responses. Amino Acids.
31:251–272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garcia-Recio S and Gascón P: Biological
and pharmacological aspects of the NK1-receptor. Biomed Res Int.
2015:4957042015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lieb K, Schaller H, Bauer J, Berger M,
Schulze-Osthoff K and Fiebich BL: Substance P and histamine induce
interleukin-6 expression in human astrocytoma cells by a mechanism
involving protein kinase C and nuclear factor-IL-6. J Neurochem.
70:1577–1583. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lieb K, Fiebich BL, Berger M, Bauer J and
Schulze-Osthoff K: The neuropeptide substance P activates
transcription factor NF-kappa B and kappa B-dependent gene
expression in human astrocytoma cells. J Immunol. 159:4952–4958.
1997.PubMed/NCBI
|
|
8
|
Horuk R, Martin AW, Wang Z, Schweitzer L,
Gerassimides A, Guo H, Lu Z, Hesselgesser J, Perez HD, Kim J, et
al: Expression of chemokine receptors by subsets of neurons in the
central nervous system. J Immunol. 158:2882–2890. 1997.PubMed/NCBI
|
|
9
|
Kim SY, Bae JC, Kim JY, Lee HL, Lee KM,
Kim DS and Cho HJ: Activation of p38 MAP kinase in the rat dorsal
root ganglia and spinal cord following peripheral inflammation and
nerve injury. Neuroreport. 13:2483–2486. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ji RR, Befort K, Brenner GJ and Woolf CJ:
ERK MAP kinase activation in superficial spinal cord neurons
induces prodynorphin and NK-1 upregulation and contributes to
persistent inflammatory pain hypersensitivity. J Neurosci.
22:478–585. 2002.PubMed/NCBI
|
|
11
|
Jin SX, Zhuang ZY, Woolf CJ and Ji RR: p38
mitogen-activated protein kinase is activated after a spinal nerve
ligation in spinal cord microglia and dorsal root ganglion neurons
and contributes to the generation of neuropathic pain. J Neurosci.
23:4017–4022. 2003.PubMed/NCBI
|
|
12
|
Bryans JS and Wustrow DJ: 3-substituted
GABA analogs with central nervous system activity: a review. Med
Res Rev. 19:149–177. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Taylor CP, Gee NS, Su TZ, Kocsis JD, Welty
DF, Brown JP, Dooley DJ, Boden P and Singh L: A summary of
mechanistic hypotheses of gabapentin pharmacology. Epilepsy Res.
29:233–249. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Maneuf YP, Gonzalez MI, Sutton KS, Chung
FZ, Pinnock RD and Lee K: Cellular and molecular action of the
putative GABA-mimetic, gabapentin. Cell Mol Life Sci. 60:742–750.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brown JT and Randall A: Gabapentin fails
to alter P/Q-type Ca2+ channel-mediated synaptic
transmission in the hippocampus in vitro. Synapse. 55:262–269.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
SILLS G: The mechanisms of action of
gabapentin and pregabalin. Curr Opin Pharmacol. 6:108–113. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maneuf YP, Hughes J and McKnight AT:
Gabapentin inhibits the substance P-facilitated K (+)-evoked
release of [(3)H]glutamate from rat caudial trigeminal nucleus
slices. Pain. 93:191–196. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cunningham MO, Woodhall GL, Thompson SE,
Dooley DJ and Jones RSG: Dual effects of gabapentin and pregabalin
on glutamate release at rat entorhinal synapses in vitro. Eur J
Neurosci. 20:1566–1576. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dooley DJ, Mieske CA and Borosky SA:
Inhibition of K(+)-evoked glutamate release from rat neocortical
and hippocampal slices by gabapentin. Neurosci Lett. 280:107–110.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rosenberg JM, Harrell C, Ristic H, Werner
RA and de Rosayro AM: The effect of gabapentin on neuropathic pain.
Clin J Pain. 13:251–255. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Backonja M, Beydoun A, Edwards KR,
Schwartz SL, Fonseca V, Hes M, LaMoreaux L and Garofalo E:
Gabapentin for the symptomatic treatment of painful neuropathy in
patients with diabetes mellitus: A randomized controlled trial.
JAMA. 280:1831–1836. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rowbotham M, Harden N, Stacey B, Bernstein
P and Magnus-Miller L: Gabapentin for the treatment of postherpetic
neuralgia: A randomized controlled trial. JAMA. 280:1837–1842.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dirks J, Fredensborg BB, Christensen D,
Fomsgaard JS, Flyger H and Dahl JB: A randomized study of the
effects of single-dose gabapentin versus placebo on postoperative
pain and morphine consumption after mastectomy. Anesthesiology.
97:560–564. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Serpell MG; Neuropathic pain study group,
: Gabapentin in neuropathic pain syndromes: A randomised,
double-blind, placebo-controlled trial. Pain. 99:557–566. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Werner MU, Perkins FM, Holte K, Pedersen
JL and Kehlet H: Effects of gabapentin in acute inflammatory pain
in humans. Reg Anesth Pain Med. 26:322–328. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Field MJ, Holloman EF, McCleary S, Hughes
J and Singh L: Evaluation of gabapentin and S-(+)-3-isobutylgaba in
a rat model of postoperative pain. J Pharmacol Exp Ther.
282:1242–1246. 1997.PubMed/NCBI
|
|
27
|
Houghton AK, Lu Y and Westlund KN:
S-(+)-3-isobutylgaba and its stereoisomer reduces the amount of
inflammation and hyperalgesia in an acute arthritis model in the
rat. J Pharmacol Exp Ther. 285:533–538. 1998.PubMed/NCBI
|
|
28
|
Partridge BJ, Chaplan SR, Sakamoto E and
Yaksh TL: Characterization of the effects of gabapentin and
3-isobutyl-gamma-aminobutyric acid on substance P-induced thermal
hyperalgesia. Anesthesiology. 88:196–205. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen SR, Xu Z and Pan HL: Stereospecific
effect of pregabalin on ectopic afferent discharges and neuropathic
pain induced by sciatic nerve ligation in rats. Anesthesiology.
95:1473–1479. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takasaki I, Andoh T, Nojima H, Shiraki K
and Kuraishi Y: Gabapentin antinociception in mice with acute
herpetic pain induced by herpes simplex virus infection. J
Pharmacol Exp Ther. 296:270–275. 2001.PubMed/NCBI
|
|
31
|
Hunter JC, Gogas KR, Hedley LR, Jacobson
LO, Kassotakis L, Thompson J and Fontana DJ: The effect of novel
anti-epileptic drugs in rat experimental models of acute and
chronic pain. Eur J Pharmacol. 324:153–160. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stanfa LC, Singh L, Williams RG and
Dickenson AH: Gabapentin, ineffective in normal rats, markedly
reduces C-fibre evoked responses after inflammation. Neuroreport.
8:587–590. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oku R, Satoh M and Takagi H: Release of
substance P from the spinal dorsal horn is enhanced in
polyarthritic rats. Neurosci Lett. 74:315–319. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fehrenbacher JC, Taylor CP and Vasko MR:
Pregabalin and gabapentin reduce release of substance P and CGRP
from rat spinal tissues only after inflammation or activation of
protein kinase C. Pain. 105:133–141. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Heuillet E, Ménager J, Fardin V, Flamand
O, Bock M, Garret C, Crespo A, Fallourd AM and Doble A:
Characterization of a human NK1 tachykinin receptor in the
astrocytoma cell line U 373 MG. J Neurochem. 60:868–876. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bahrami G and Mohammadi B: Sensitive
microanalysis of gabapentin by high-performance liquid
chromatography in human serum using pre-column derivatization with
4-chloro-7-nitrobenzofurazan: Application to a bioequivalence
study. J Chromatogr B Analyt Technol Biomed Life Sci. 837:24–28.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jalalizadeh H, Souri E, Tehrani MB and
Jahangiri A: Validated HPLC method for the determination of
gabapentin in human plasma using pre-column derivatization with
1-fluoro-2,4-dinitrobenzene and its application to a
pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life
Sci. 854:43–47. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Park S, Ahn ES, Han DW, Lee JH, Min KT,
Kim H and Hong YW: Pregabalin and gabapentin inhibit substance
P-induced NF-kappaB activation in neuroblastoma and glioma cells. J
Cell Biochem. 105:414–423. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tremont-Lukats IW, Megeff C and Backonja
MM: Anticonvulsants for neuropathic pain syndromes: Mechanisms of
action and place in therapy. Drugs. 60:1029–1052. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mao J and Chen LL: Gabapentin in pain
management. Anesth Analg. 91:680–687. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Patel S, Naeem S, Kesingland A, Froestl W,
Capogna M, Urban L and Fox A: The effects of GABA(B) agonists and
gabapentin on mechanical hyperalgesia in models of neuropathic and
inflammatory pain in the rat. Pain. 90:217–226. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Field MJ, Oles RJ, Lewis AS, McCleary S,
Hughes J and Singh L: Gabapentin (neurontin) and
S-(+)-3-isobutylgaba represent a novel class of selective
antihyperalgesic agents. Br J Pharmacol. 121:1513–1522. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shimoyama N, Shimoyama M, Davis AM,
Inturrisi CE and Elliott KJ: Spinal gabapentin is antinociceptive
in the rat formalin test. Neurosci Lett. 222:65–67. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kaneko M, Mestre C, Sánchez EH and Hammond
DL: Intrathecally administered gabapentin inhibits formalin-evoked
nociception and the expression of Fos-like immunoreactivity in the
spinal cord of the rat. J Pharmacol Exp Ther. 292:743–751.
2000.PubMed/NCBI
|
|
45
|
Field MJ, McCleary S, Hughes J and Singh
L: Gabapentin and pregabalin, but not morphine and amitriptyline,
block both static and dynamic components of mechanical allodynia
induced by streptozocin in the rat. Pain. 80:391–398. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gajraj NM: Pregabalin: Its pharmacology
and use in pain management. Anesth Analg. 105:1805–1815. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Davies A, Hendrich J, Van Minh AT, Wratten
J, Douglas L and Dolphin AC: Functional biology of the
alpha(2)delta subunits of voltage-gated calcium channels. Trends
Pharmacol Sci. 28:220–228. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Luo ZD, Chaplan SR, Higuera ES, Sorkin LS,
Stauderman KA, Williams ME and Yaksh TL: Upregulation of dorsal
root ganglion (alpha)2(delta) calcium channel subunit and its
correlation with allodynia in spinal nerve-injured rats. J
Neurosci. 21:1868–1875. 2001.PubMed/NCBI
|
|
49
|
Fiebich BL, Schleicher S, Butcher RD,
Craig A and Lieb K: The neuropeptide substance P activates p38
mitogen-activated protein kinase resulting in IL-6 expression
independently from NF-kappa B. J Immunol. 165:5606–5611. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hu P, Bembrick AL, Keay KA and McLachlan
EM: Immune cell involvement in dorsal root ganglia and spinal cord
after chronic constriction or transection of the rat sciatic nerve.
Brain Behav Immun. 21:599–616. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Scholz J, Abele A, Marian C, Häussler A,
Herbert TA, Woolf CJ and Tegeder I: Low-dose methotrexate reduces
peripheral nerve injury-evoked spinal microglial activation and
neuropathic pain behavior in rats. Pain. 138:130–142. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cao H and Zhang YQ: Spinal glial
activation contributes to pathological pain states. Neurosci
Biobehav Rev. 32:972–983. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Obata K and Noguchi K: MAPK activation in
nociceptive neurons and pain hypersensitivity. Life Sci.
74:2643–2653. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Arruda JL, Colburn RW, Rickman AJ,
Rutkowski MD and DeLeo JA: Increase of interleukin-6 mRNA in the
spinal cord following peripheral nerve injury in the rat: Potential
role of IL-6 in neuropathic pain. Brain Res Mol Brain Res.
62:228–235. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lee HL, Lee KM, Son SJ, Hwang SH and Cho
HJ: Temporal expression of cytokines and their receptors mRNAs in a
neuropathic pain model. Neuroreport. 15:2807–2811. 2004.PubMed/NCBI
|
|
56
|
Wang XM, Hamza M, Wu TX and Dionne RA:
Upregulation of IL-6, IL-8 and CCL2 gene expression after acute
inflammation: Correlation to clinical pain. Pain. 142:275–283.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang F, Xu S, Shen X, Guo X, Peng Y and
Yang J: Spinal macrophage migration inhibitory factor is a major
contributor to rodent neuropathic pain-like hypersensitivity.
Anesthesiology. 114:643–659. 2011. View Article : Google Scholar : PubMed/NCBI
|