Introduction
Alzheimer's disease (AD) is currently one of the
most common neurodegenerative disorders, and a leading cause of
memory loss and cognitive decline in patients (1). The mechanism of onset of
neurodegenerative diseases and the various components responsible
remain to be fully elucidated. It is well known that β-amyloid
treatment causes neuron death and subsequently results in dementia
(1,2). Studies have revealed that β-amyloid
is involved in regulating the activity of neurons and synapses, and
its aggregation in nervous system tissues results in the
development of neurological disorders (3). In the neuronal cells, β-amyloid
induces degeneration through a cascade of cellular processes
involving the generation of reactive oxygen species (ROS) and the
induction of cell death (4). The
use of ROS-quenching agents, including polyphenolic compounds and
tocopherol, has been shown to inhibit the harmful effects induced
by β-amyloid (5,6). The consumption of food items rich in
anti-oxidant compounds has also been shown to prevent the
development of various neurological disorders (7). The anti-oxidative compounds exhibit
their effect through inhibiting the activation of various factors
associated with several pathways (7,8).
Aminoguanidine is a low molecular weight compound,
soluble in polar solvents (e.g. water) and exhibits a broad
spectrum of activities (9). It is
important in the inhibition of tissue damage in patients with
diabetes mellitus (9). Treatment
of animals with aminoguanidine efficiently prevents injury to the
brain and stroke (10–12). Aminoguanidine treatment is
promising in the improvement of spinal cord motor function
following injury (13). It also
prevents the initiation of reactions leading to the production of
ROS and formation of inflammation following spinal cord injury
(13). The present study aimed to
investigate the role of aminoguanidine in the prevention of harmful
effects in astroglioma F98 cells induced by β-amyloid treatment. It
was observed that aminoguanidine prevented F98 cells from the
β-amyloid-induced increase in the production of ROS, and the
enhanced expression of prostaglandin E2 (PGE2) and cyclooxygenase
(COX)-2, and inhibited the activation of nuclear factor
(NF)-κB.
Materials and methods
Cell line and culture
The F98 rat glioma cell line was purchased from the
American Type Culture Collection (Rockville, MD). The cells were
grown as monolayer cultures in DMEM (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) in 5% CO2 at 37°C.
The medium was supplemented with 10% heat-inactivated fetal bovine
serum (Invitrogen; Thermo Fisher Scientific, Inc.) with
penicillin/streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.).
Chemicals and reagents
Aminoguanidine and β-amyloid were obtained from
Sigma-Aldrich; Merck Millipore (Darmstadt, Germany). Aminoguanidine
was dissolved in distilled water to prepare 1 µM stock solution and
stored at −10°C.
Cell viability assay
The F98 cells were cultured at a density of
2×105 cells per 100 µl of medium for 24 h in 96-well
cell culture microplates (Corning Life Sciences, Lowell, MA, USA).
Following incubation, the medium was replaced with a medium
containing various concentrations of aminoguanidine (10, 20, 30 and
40 µM) and incubated at 37°C for 12 h. β-amyloid (15 µM) was then
added to each of the wells, and incubation was continued for 12 h.
The control cells, after 24 h culture, were incubated for 12 h in a
medium containing β-amyloid (15 µM). Subsequently,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
5 mg/ml; Sigma-Aldrich; Merck Millipore) was added to the wells (10
µl) and incubated for 4 h at 37°C, followed by the addition of 100
µl dimethyl sulfoxide to dissolve any formazan crystals formed. The
enzyme-linked immunosorbent detector (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) was used to measure optical density at 570
nm.
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
Total RNA was isolated from the F98 glioma cells
following treatment with aminoguanidine and/or β-amylase using
TRIzol (Thermo Fisher Scientific, Inc.). The RNA samples (1 µg)
were then subjected to RT and PCR sequentially using a One Step
RT-PCR kit (Qiagen Inc., Valencia, CA, USA). The primers used were
as follows: COX-2, forward 5′-TTCAAATGAGATTGTGGGAAAAT-3′ and
reverse 5′-AGATCATCTCTGCCTGAGTATCTT-3′; iNOS, forward
5′-AGAGAGATCCGGTTCACA-3′ and reverse 5′-CACAGAGCTGAGGGTACA-3′;
COX-1, forward 5′-TGCCCAGCTCCTGGCCCGCCGCTT-3′ and reverse
5′-GTGCATCAACACAGGCGCCTCTTC-3′; GAPDH, forward
5′CGGAGTCAACGGATTTGGTCGTAT-3′ and reverse
5′-AGCCTTCTCCATGGTGGTGAAGAC-3′. A Mastercycler (Eppendorf, Hamburg,
Germany) was used to perform amplification of the cDNA strands
using the following thermocycling sequence: Initial step at 50°C
for 2 min and 95°C for 10 min, followed by 30 cycles of 95°C for 15
sec and 60°C for 1 min. Following amplification, the products were
run on 1.5% agarose gels, followed by ethidium bromide staining
(Sigma-Aldrich; Merck Millipore).
Western blot analysis
Following treatment of the F98 cells with
aminoguanidine and/or β-amyloid, F98 cells were subjected to on-ice
lysis using lysis buffer [100 mM NaCl, 20 mM Tris-HCl, (pH 7.8),
0.1% NP-40], containing protease inhibitor cocktail (Roche
Diagnostics GmbH, Mannheim, Germany) and dithiothreitol,
Na3VO4, NaF and phenylmethane sulfonyl
fluoride (1 mM each). The homogenates were centrifuged at 4°C at
14,000 × g for 15 min, and the supernatant was collected and stored
at −80°C until further analysis. Total protein (50 µg) was loaded
per lane and separated on 10% SDS-PAGE gels by electrophoresis at
100 V. The proteins were then electroblotted onto nitrocellulose
membranes (Hybond ECL; GE Healthcare Life Sciences, Chalfont, UK).
The non-specific sites on the membrane were blocked by incubation
with 5% skim milk powder and 3% bovine serum albumin (BSA; Cayman
Chemical Compnay, Ann Arbor, MI, USA) over a period of 2 h at room
temperature. The blots were then probed by incubation with mouse
monoclonal primary antibodies, diluted 1:500, overnight at 4°C. The
primary antibodies used were anti-iNOS (610600), PGE2 (610205),
COX-2 (610203) and NF-κB (558393) (all from BD Biosciences, San
Jose, CA, USA). The membranes were washed with PBS three times,
followed by incubation with a peroxidase-labeled anti-rabbit
secondary antibody (1:10,000; catalog number-611-103-122; R&D
Systems, Inc., Minneapolis, MN USA) for 1 h at room temperature.
The ECL-western blot detection kit (NEN, MA) was used for the
visualization of immunoreactivity.
Measurement of ROS generation
The production of ROS in F98 cells treated with
aminoguanidine and/or β-amyloid was determined with
dichlorofluorescein-diacetate (DCFH-DA) using flow cytometric
analysis. The cells, following aminoguanidine and β-amyloid
treatment or β-amyloid treatment alone (vehicle control), were
collected, rinsed twice with ice-cold PBS, and suspended in PBS
(2×106 cells/ml). Subsequently, 500 µl of this
suspension was incubated for 45 min at 25°C in tubes containing
DCFH-DA at a concentration of 5 µM. The generation of ROS in the
F98 cells was determined via DCF fluorescence intensity using flow
cytometry.
Analysis of the aggregation of PGE2 in
cells
The F98 cells were incubated for 12 h in a medium
containing β-amyloid and aminoguanidine (treated cells) or
β-amyloid alone (vehicle control). Following incubation, the medium
was removed and the cells were subjected to an enzyme immunoassay
using a commercial kit (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The ELISA reader system
was then used for the determination of PGE2 accumulation in the
cells.
Immunochemistry for analysis of NFκB
translocation
Immunohistochemistry was performed using the Ventana
EX system and a DAB universal kit (Ventana Medical Systems, Inc.,
Tucson, AZ, USA). Following treatment with aminoguanidine and/or
β-amyloid, the F98 cells were fixed using paraformaldehyde at 4°C
for 45 min. The cells were then rinsed in PBS twice, followed by
permeabilization in 0.2% Triton X-100. Following washing with PBS,
the cells were blocked in BSA for 45 min and finally incubated with
the anti-NF-κB antibody (dilution 1:1,000) for 3 h at 25°C.
Following rinsing with PBS, the F98 cells were incubated for 45 min
at 25°C with FITC-conjugated anti-rabbit IgG (Sigma-Aldrich; Merck
Millipore), and a fluorescence microscope (Carl Zeiss AG,
Oberkochen, Germany) was used for cell analysis.
Statistical analysis
The data are presented as the mean ± standard error
of the mean of three independent experiments performed in
triplicate. Statistical comparison of the mean values between data
was performed using unpaired Student's t-tests. For statistical
analysis of the data, Prism Software (GraphPad Software, Inc., La
Jolla, CA, USA) was used. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of aminoguanidine on the
β-amyloid-induced decrease inF98 glioma cell viability
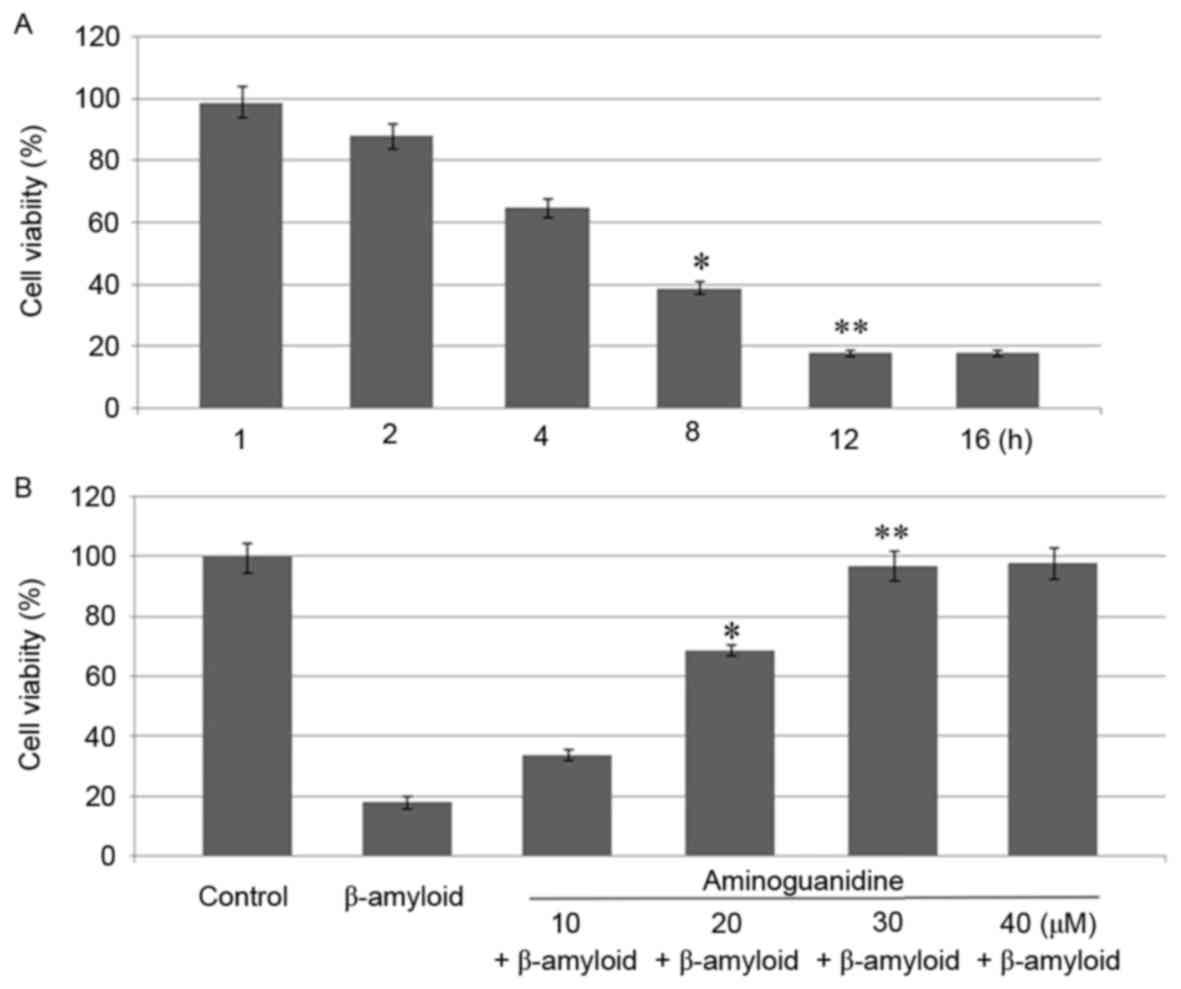
The analysis of the effect of β-amyloid revealed
inhibition in the rate of viability of the F98 glioma cells 12 h
following treatment. Incubation of the F98 cells with β-amyloid for
12 h at a concentration of 15 µM reduced the viability to 18%
(Fig. 1A). β-amyloid reduced the
viability of F98 cells in a concentration- and time-dependent
manner. The effect of aminoguanidine on the β-amyloid-induced
inhibition in F98 cell viability was analyzed following treatment
with concentrations of 10, 20, 30 and 40 µM. The results showed
that pretreatment with 30 µM aminoguanidine for 12 h completely
prevented the β-amyloid-induced decrease in the viability of the
F98 cells (Fig. 1B).
Aminoguanidine inhibits
β-amyloid-induced increases in the expression of NO and iNOS
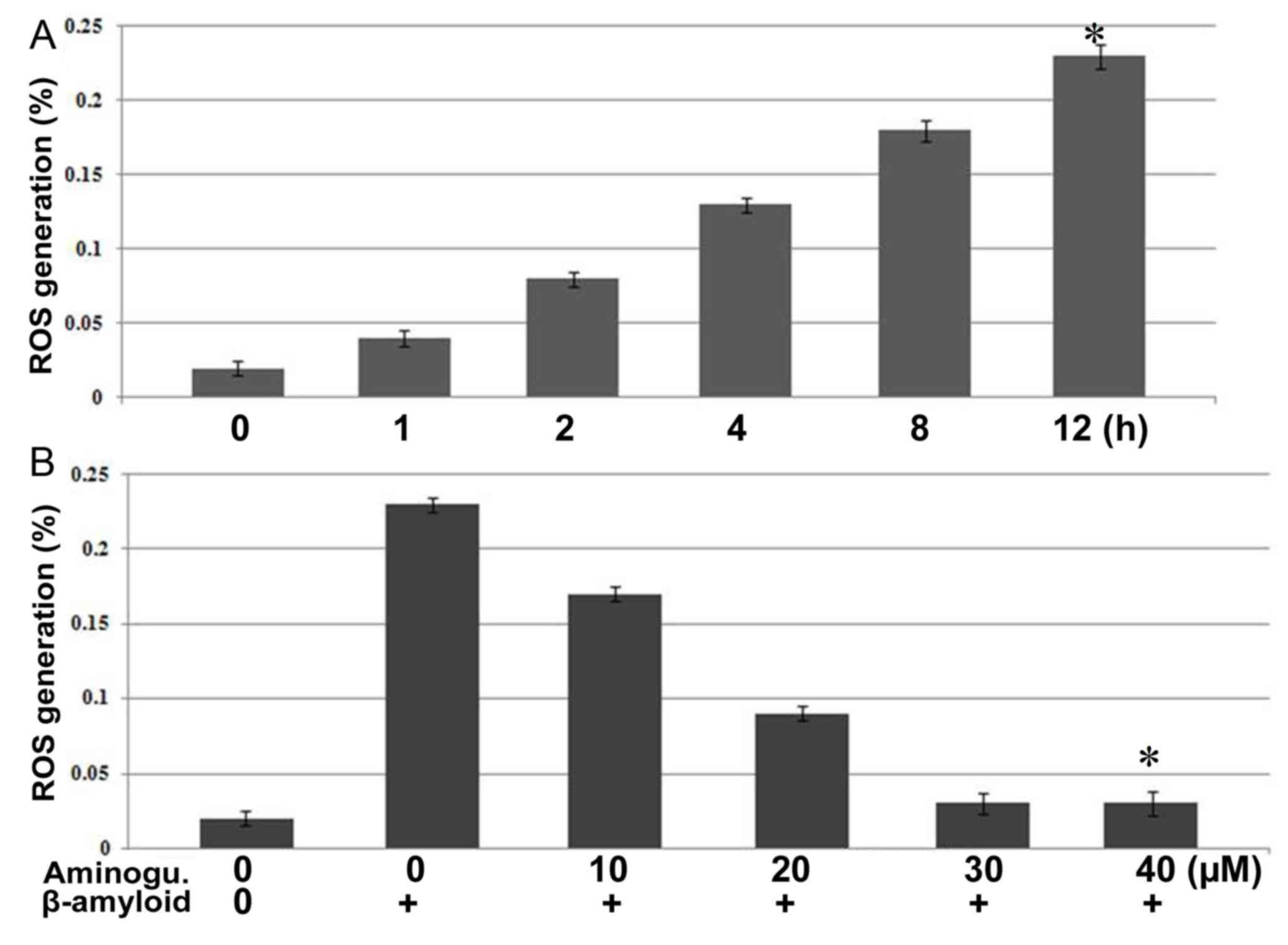
The analysis of the expression levels of ROS and
iNOS revealed significantly (P<0.005) higher expression
following β-amyloid treatment in the F98 cells. Incubation of the
F98 cells with a 15 µM concentration of β-amyloid for 12 h led to a
marked increase in expression levels of ROS and iNOS (Fig. 2A). In addition, F98 cells were
pre-treated with aminoguanidine for 12 h prior to incubation with
β-amyloid and the expression of ROS was analyzed. It was observed
that aminoguanidine pre-treatment at a concentration of 30 µM for
12 h inhibited the β-amyloid-induced increase in the expression of
ROS completely (Fig. 2B).
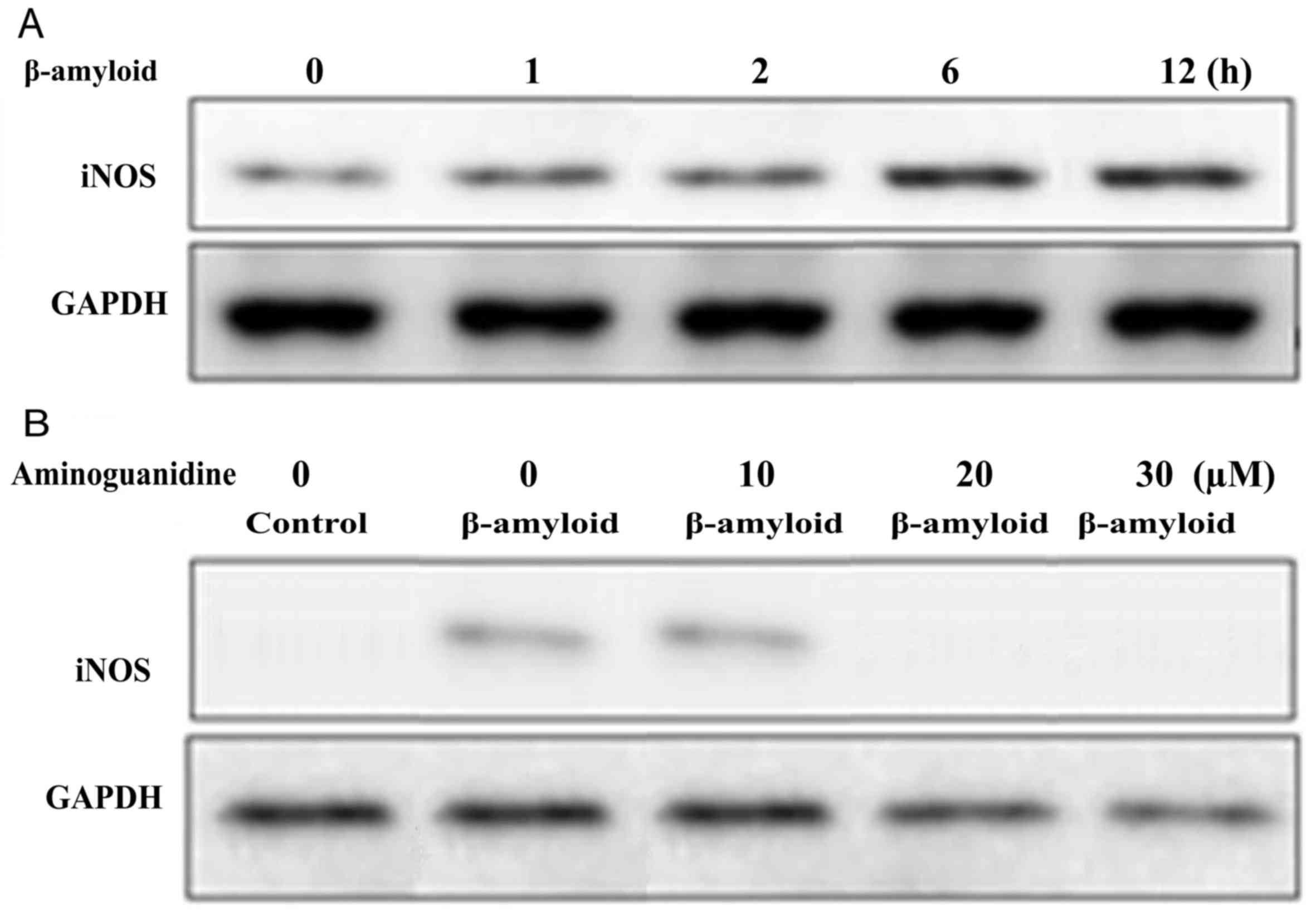
Aminoguanidine pre-treatment for 12 h at a 30 µM concentration
inhibited the iNOS protein expression in the F98 cells (Fig. 3).
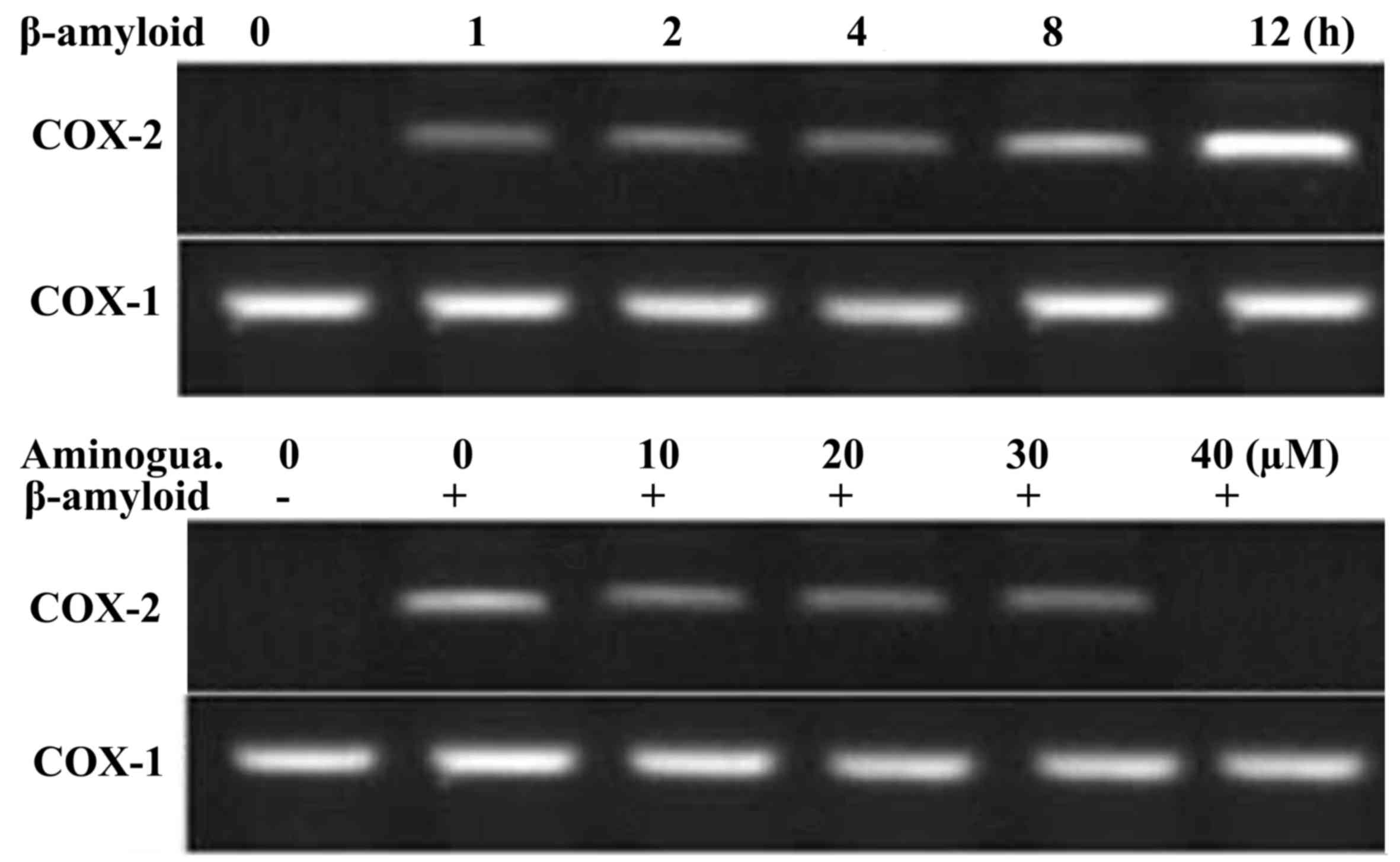
Aminoguanidine inhibits
β-amyloid-induced increase in the expression of PGE2 and COX-2 in
F98 cells
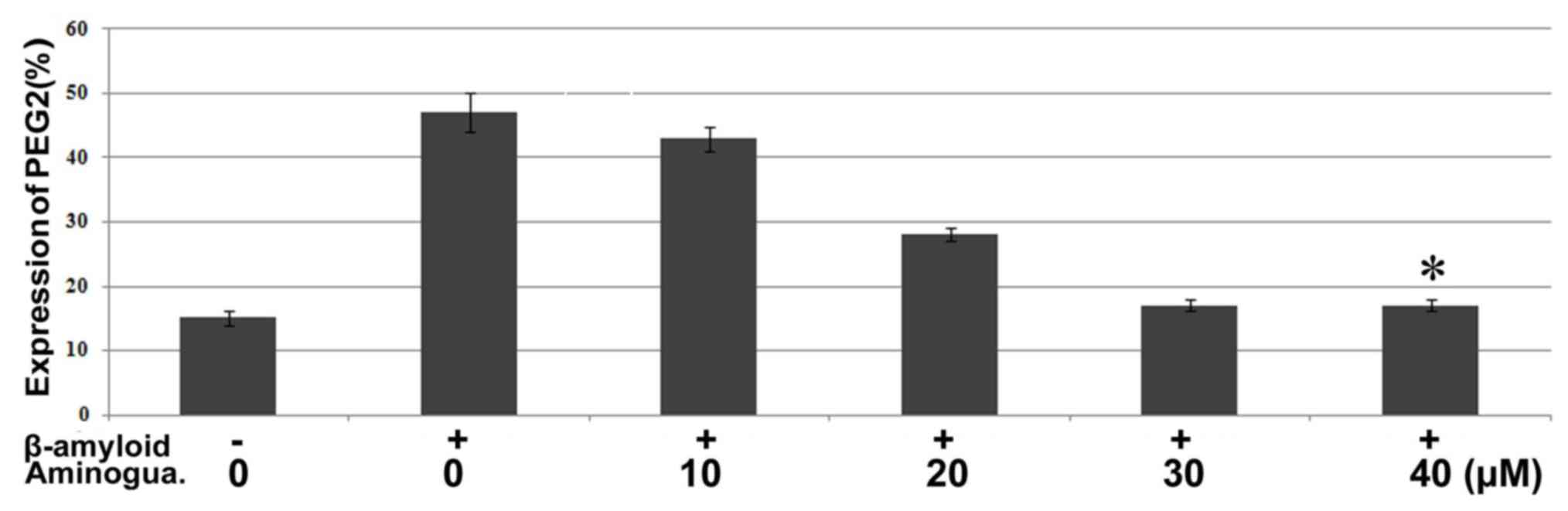
Treatment of the F98 cells with β-amyloid for 12 h
at a concentration of 15 µM led to a marked increase in the
expression of COX-2 (Fig. 4).
However, pre-treatment of the F98 cells with various concentrations
of aminoguanidine exhibited a concentration-dependent reduction in
the expression of PGE2. Pre-treatment with a 30 µM concentration of
aminoguanidine for 12 h resulted in reductions in the mRNA levels
corresponding to the expression of COX-2 to the same level as in
the control cells (Fig. 5). The
expression of PGE2 in the β-amyloid-treated cells was also markedly
increased, compared with that in the control cells. However,
pre-treatment of the F98 cells with aminoguanidine at a 30 µM
concentration for 12 h prior to incubation with β-amyloid
significantly (P<0.002) reduced the expression of PGE2.
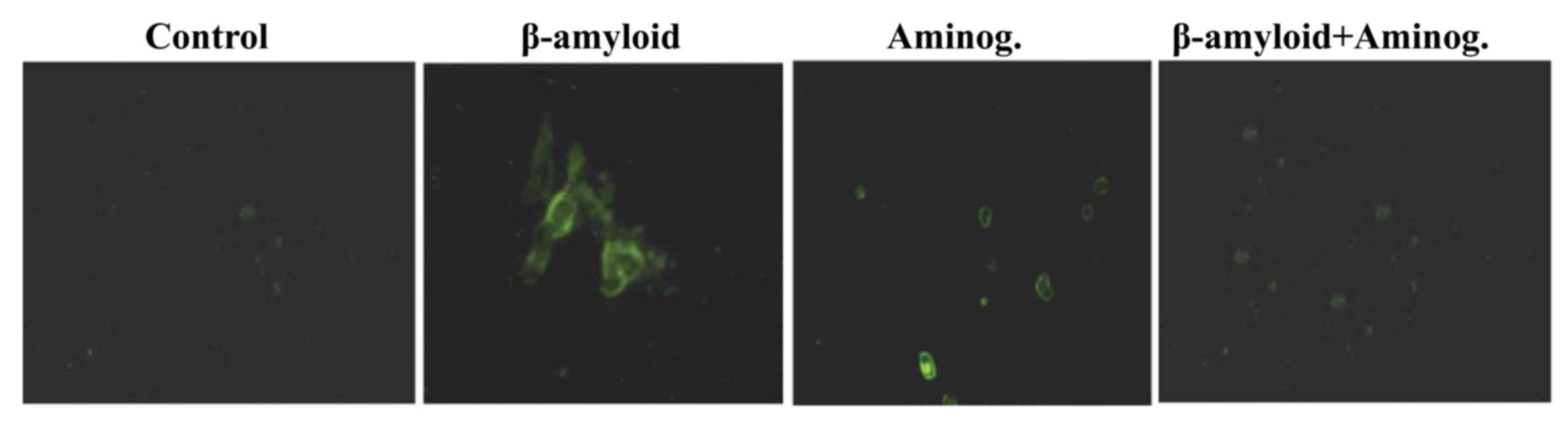
Repression of β-amyloid-induced
nuclear translocation of NF-κB by aminoguanidine
Analysis of the localization of NF-κB p65 in
β-amyloid-treated F98 cells revealed its presence in the cell
cytosol. By contrast, in the control cells, NF-κB p65 was confined
to the cell nucleus alone (Fig.
6). In the F98 cells, pre-treatment with aminoguanidine
resulted in the inhibition of NF-κB p65 translocation into the
cytosol in a concentration-dependent manner. Following treatment
with a 30 µM concentration of aminoguanidine, the translocation of
NF-κB p65 from the nucleus to the cytosol was completely inhibited
(Fig. 6).
Discussion
Alzheimer's disease, a frequently observed
neurological disorder, is caused by the aggregation of β-amyloid in
the central nervous system tissues (14). Alterations in the structure and
function of neurons by the accumulation of β-amyloid results in the
development of neuronal inflammation and the induction of
apoptosis, which is characteristic of Alzheimer's disease (15). The expression of cytokines is
higher in the nervous system tissues of patients with Alzheimer's
disease (16). This suggests that
the compounds, which inhibit the expression of cytokines can be
important for the treatment of neurological disorders (16). COX-2 has been found to exhibit an
important effect on the induction of inflammation in cells. It has
been reported that, under normal circumstances, the expression of
COX-2 in cells is negligible, whereas its expression is increased
during various neurological disorders (17). The examination of nervous system
tissues obtained from patients with neurological disorders has
revealed markedly higher expression levels of COX-2 (17). It has been suggested that the
expression of COX-2 leads to an increase in the production of ROS,
which leads to neuron death (17).
Studies have also demonstrated higher expression levels of PGE2 in
the neuronal tissues of those suffering from neurological disorders
(18). Thus, reducing the
expression of PGE2 through the use of chemotherapeutic agents is
considered to be an important strategy for the treatment of
Alzheimer disease. The results from the present study demonstrated
a significant decrease in the β-amyloid-induced expression of COX-2
and PGE2 when F98 cells were pre-treated with aminoguanidine.
The generation of ROS, including NO, in cells leads
to alterations in several cellular processes and the development of
disorders, including Alzheimer disease (19). The production of NO takes place
through the involvement of the enzyme, NOS (20). iNOS generates an increased quantity
of NO, resulting in the development of inflammatory reactions. The
results of the present study demonstrated that aminoguanidine
pre-treatment of the F98 cells inhibited the β-amyloid-induced
generation of NO and secretion of iNOS. The inhibition of iNOS
secretion was evident at the mRNA and protein levels.
The activation of NF-κB has been found in the
neurological tissues of patients with Alzheimer disease during
postmortem examination (21). The
treatment of F98 cells with β-amyloid has also been found to induce
the activation of NF-κB (22). In
the present study, treatment of F98 cells with aminoguanidine
resulted in the inactivation of NF-κB and the translocation of
NF-κB into the nucleus.
In conclusion, aminoguanidine prevented
β-amyloid-induced Alzheimer disease through reductions in the
expression levels of NO, iNOS, PGE2 and COX-2, and the inactivation
of NF-κB. Therefore, aminoguanidine offers potential for use in the
treatment of neurological disorders, including Alzheimer's
disease.
Acknowledgements
We would like to acknowledge the financial help
offered by Ankang City Central Hospital (Shaanxi, China).
References
|
1
|
Lewczuk P, Kamrowski-Kruck H, Peters O,
Heuser I, Jessen F, Popp J, Bürger K, Hampel H, Frölich L, Wolf S,
et al: Soluble amyloid precursor proteins in the cerebrospinal
fluid as novel potential biomarkers of Alzheimer's disease: A
multicenter study. Mol Psychiatry. 15:138–145. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weiner MW: Dementia in 2012: Further
insights into Alzheimer disease pathogenesis. Nat Rev Neurol.
2:65–66. 2013. View Article : Google Scholar
|
|
3
|
Tan L, Yu JT, Hu N and Tan L: Non-coding
RNAs in Alzheimer's disease. Mol Neurobiol. 47:382–393. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eckert A, Keil U, Marques CA, Bonert A,
Frey C, Schüssel K and Müller WE: Mitochondrial dysfunction,
apoptotic cell death, and Alzheimer's disease. Biochem Pharmacol.
66:1627–1634. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Canevari L, Abramov AY and Duchen MR:
Toxicity of amyloid beta peptide: Tales of calcium, mitochondria,
and oxidative stress. Neurochem Res. 29:637–650. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grundman M, Grundman M and Delaney P:
Antioxidant strategies for Alzheimer's disease. Proc Nutr Soc.
61:191–202. 2002; View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Komatsu M and Hiramatsu M: The efficacy of
an antioxidant cocktail on lipid peroxide level and superoxide
dismutase activity in aged rat brain and DNA damage in iron-induced
epileptogenic foci. Toxicology. 148:143–148. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bastianetto S, Zheng WH and Quirion R:
Neuroprotective abilities of resveratrol and other red wine
constituents against nitric oxide-related toxicity in cultured
hippocampal neurons. Br J Pharmacol. 131:711–720. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abdel-Rahman E and Bolto WK: Pimagedine: A
novel therapy for diabetic nephropathy. Expert Opin Investig Drugs.
11:565–574. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Di F, Yan-Ting G, Hui L, Tao T, Zai-Hua X,
Xue-Ying S, Hong-Li X and Yun-Jie W: Role of aminoguanidine in
brain protection in surgical brain injury in rat. Neurosci Let.
448:204–207. 2008. View Article : Google Scholar
|
|
11
|
Sugimoto K and Iadecola C: Effects of
aminoguanidine on cerebral ischemia in mice: Comparison between
mice with and without inducible nitric oxide synthase gene.
Neurosci Lett. 331:25–28. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Louin G, Marchand-Verrecchia C, Palmier B,
Plotkine M and Jafarian-Tehrani M: Selective inhibition of
inducible nitric oxide synthase reduces neurological deficit but
not cerebral edema following traumatic brain injury.
Neuropharmacology. 50:182–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pearse DD, Chatzipanteli K, Marcillo AE,
Bunge MB and Dietrich WD: Comparison of iNOS inhibition by
antisense and pharmacological inhibitors after spinal cord injury.
J Neuropathol Exp Neurol. 62:1096–1107. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hardy JA and Higgins GA: Alzheimer's
disease: The amyloid cascade hypothesis. Science. 256:184–185.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yankner BA, Dawes LR, Fisher S,
Villa-Komaroff L, Oster-Granite ML and Neve RL: Neurotoxicity of a
fragment of the amyloid precursor associated with Alzheimer's
disease. Science. 245:417–420. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Raivich G, Jones LL, Werner A, Bluthmann
H, Doetschmann T and Kreutzberg GW: Molecular signals for glial
activation: Pro- and anti-inflammatory cytokines in the injured
brain. Acta Neurochir Suppl. 73:S21–S30. 1999.
|
|
17
|
Pasinetti GM: From epidemiology to
therapeutic trials with anti-inflammatory drugs in Alzheimer's
disease: The role of NSAIDs and cyclooxygenase in beta-amyloidosis
and clinical dementia. J Alzheimers Dis. 4:435–445. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Montine TJ, Sidell KR, Crews BC,
Markesbery WR, Marnett LJ, Roberts LJ and Morrow JD: Elevated CSF
prostaglandin E2 levels in patients with probable AD. Neurology.
53:1495–1498. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Duncan AJ and Heales SJ: Nitric oxide and
neurological disorders. Mol Aspects Med. 26:67–96. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
MacMicking J, Xie QW and Nathan C: Nitric
oxide and macrophage function. Annu Rev Immunol. 15:323–350. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Terai K, Matsuo A and McGeer PL:
Enhancement of immunoreactivity for NF-kappa B in the hippocampal
formation and cerebral cortex of Alzheimer's disease. Brain Res.
735:159–168. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Akama KT and van Eldik LJ: Beta-amyloid
stimulation of inducible nitric-oxide synthase in astrocytes is
interleukin-1beta- and tumor necrosis factor-alpha
(TNFalpha)-dependent, and involves a TNFalpha receptor-associated
factor- and NFkappaB-inducing kinase-dependent signaling mechanism.
J Biol Chem. 275:7918–7924. 2000. View Article : Google Scholar : PubMed/NCBI
|