Introduction
Lipotoxic cardiomyopathy often occurs in patients
with diabetes and obesity, and is attributed to excessive
accumulation of lipids and their intermediate products in
cardiomyocytes (1). Diabetes and
obesity usually result in the disordered lipid metabolism and
elevation of free fatty acids (FFAs) (2). Previous studies have demonstrated
that increased FFAs, particularly saturated FFAs, induce myocardial
injury, and ultimately result in heart dysfunction to some extent
(3,4). However, the underlying mechanisms
responsible for lipotoxic cardiomyopathy remain unknown.
The endoplasmic reticulum (ER) is a fundamental
organelle that has a key role in the modification, folding and
oligomerization of the majority synthesized structural and secreted
proteins (5). It has been
established that multiple physiological and pathological
conditions, including hypoxia, hyperglycemia and acidosis, can
result in ER homeostasis breakdown and an elevated accumulation of
unfolded/misfolded proteins within the ER lumen, which is commonly
termed ‘ER stress’ (6). In order
to cope with ER stress, cells initially induce the unfolded protein
response (UPR) signaling to adapt to stress conditions and maintain
the balance of ER homeostasis again. However, prolonged or
excessive ER stress will trigger ER stress-mediated apoptosis
pathway, which is an apoptosis signal pathway independent of the
death-receptor and mitochondria mediated-apoptosis pathways
(7).
Palmitate (PA), a type of saturated FFA, induces
cardiomyocyte apoptosis (8), and
thus, is used to mimic cardiomyocytes lipotoxicity in vitro
(9). Several previous studies have
reported that PA induces ER stress-mediated apoptosis in
hepatocytes (10), pancreatic β
cells (11) and mature adipocytes
(12), but whether PA induces ER
stress-mediated apoptosis in cardiomyocytes remains unknown.
Therefore, the aim of the present study was to explore the role of
ER stress-mediated apoptosis pathway in PA-induced cardiomyocyte
lipotoxicity.
Materials and methods
Cell culture and PA treatment
The H9c2 rat cardiomyocyte cell line, obtained from
the Shanghai Institutes for Biological Sciences (Shanghai, China),
was routinely cultured in Dulbecco's modified Eagle's medium
(Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented
with 10% fetal bovine serum (Hangzhou Sijiqing Biological
Engineering Materials Co., Ltd., Hangzhou, China), 100 U/ml
penicillin and 100 µg/ml streptomycin in a humidified atmosphere at
37°C with 5% CO2. When the confluence of H9c2 cells was
~80%, PA (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at doses
of 100, 200 and 400 µM was added to the medium and incubated for 12
h. An ER stress inhibitor, 4-phenyl butyric acid (4-PBA; 5 mM), was
administrated 90 min before 400 µM PA treatment.
Oil Red O staining
Oil Red O staining was used to measure the
accumulation of intracellular lipids. Briefly, 150 mg Oil Red O
powder (Sigma-Aldrich; Merck KGaA) was dissolved in 50 ml 60%
isopropanol to prepare the Oil Red O reserve solution (3 mg/ml) and
further diluted by distilled water (3:2) to prepare a working
solution. H9c2 cells were fixed in 4% paraformaldehyde for 30 min
at room temperature, washed with PBS three times, then followed by
incubation with the Oil Red O working solution for 30 min at room
temperature. After washing with PBS three times, nuclei were
counterstained with 50% hematoxylin for 2 min at room temperature.
Finally, the result of Oil Red O staining was observed under an
optical microscope (Olympus Corporation, Tokyo, Japan).
Measurement of cell viability
MTT assay was used to determine the cell viability
following PA treatment. Briefly, H9c2 cells were seeded in a
96-well plate and treated with various concentrations of PA as
described above. After 12 h, the medium was removed and replaced
with 0.5 mg/ml MTT solution (Sigma-Aldrich; Merck KGaA), 200 µl per
well. After incubation at 37°C for 4 h, the MTT solution was
removed and each well was washed with PBS three times. The
precipitated formazan in each well was solubilized by dimethyl
sulfoxide (Sigma-Aldrich; Merck KGaA) and the optical density value
was detected by an automated microplate reader (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 490 nm.
Measurement of cell injury
Lactate dehydrogenase (LDH) activity in the culture
medium was detected to determine the injury of H9c2 cells using a
commercial LDH assay kit (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China) according to the manufacturer's
protocols.
Measurement of apoptosis
Apoptosis was detected by flow cytometry analysis
using the PE-labeled Annexin V/7-aminoactinomycin D apoptosis
detection kit (BD Biosciences, Franklin Lakes, NJ, USA) according
to the manufacturer's protocols.
Western blotting analysis
Cells were harvested after PA treatment for 12 h and
mixed with radioimmunoprecipitation assay lysis buffer (Beyotime
Institute of Biotechnology, Nanjing, China) and protease inhibitor
phenylmethanesulfonyl fluoride on ice for 20 min. Proteins were
extracted from cells following 5 min of centrifugation of 13,000 ×
g at 4°C. The concentration of protein was measured using Enhanced
BCA Protein Assay kit (Beyotime Institute of Biotechnology). For
western blot analysis, 50 µg protein denatured by heating was
subjected to 10% SDS-polyacrylamide gel electrophoresis for
separation and then transferred to polyvinylidene difluoride
membranes. The membrane was blocked using 2% bovine serum albumin
(BSA) for 1 h at room temperature and then incubated overnight at
4°C with the specific primary antibodies including monoclonal
anti-78 kDa glucose-regulated protein (GRP78; cat no. 3183;
1:1,000), anti-protein kinase R-like endoplasmic reticulum kinase
(PERK; cat no. 3192; 1:1,000), anti-phospho (p)-PERK (cat no. 3179;
1:1,000), anti-eukaryotic initiation factor 2 α (eIF2α; cat no.
5324; 1:1,000), anti-p-eIF2α (cat no. 3398; 1:1,000) (all from Cell
Signaling Technology, Inc., Danvers, MA, USA), anti-cleaved
caspase-12 (cat no. ab18766; 1:1,000), anti-C/EBP homologous
protein (CHOP; 1:1,000, cat no. ab11419) (both from Abcam,
Cambridge, UK), and anti-β-actin (TA-09; 1:2,000; Beijing Zhongshan
Jinqiao Biotechnology Co., Ltd., Beijing, China). Subsequently, the
membranes were incubated with horseradish peroxidase-conjugated
goat anti-rabbit (cat no. sc-2004; 1:2,000) or rabbit anti-mouse
immunoglobulin G (cat no. sc-358914; 1:2,000) (both from Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) at 37°C for 2 h. Detection of
protein band was performed using an enhanced chemiluminescence kit
(Santa Cruz Biotechnology, Inc.) according to the manufacturer's
protocols. The levels of phosphorylated proteins were normalized to
their corresponding total protein levels. Relative densitometry was
calculated using ImageJ software version 2× (National Institutes of
Health, Bethesda, MD, USA).
Statistical analysis
The data were expressed as the mean ± standard
deviation. Statistical analysis was performed by software SPSS 17.0
version (SPSS, Inc., Chicago, IL, USA). Differences between groups
were initially evaluated using one-way analysis of variance, and if
the differences were significant, multiple comparison analysis was
further performed using Fisher's least significant difference test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effect of PA on cell lipid
accumulation
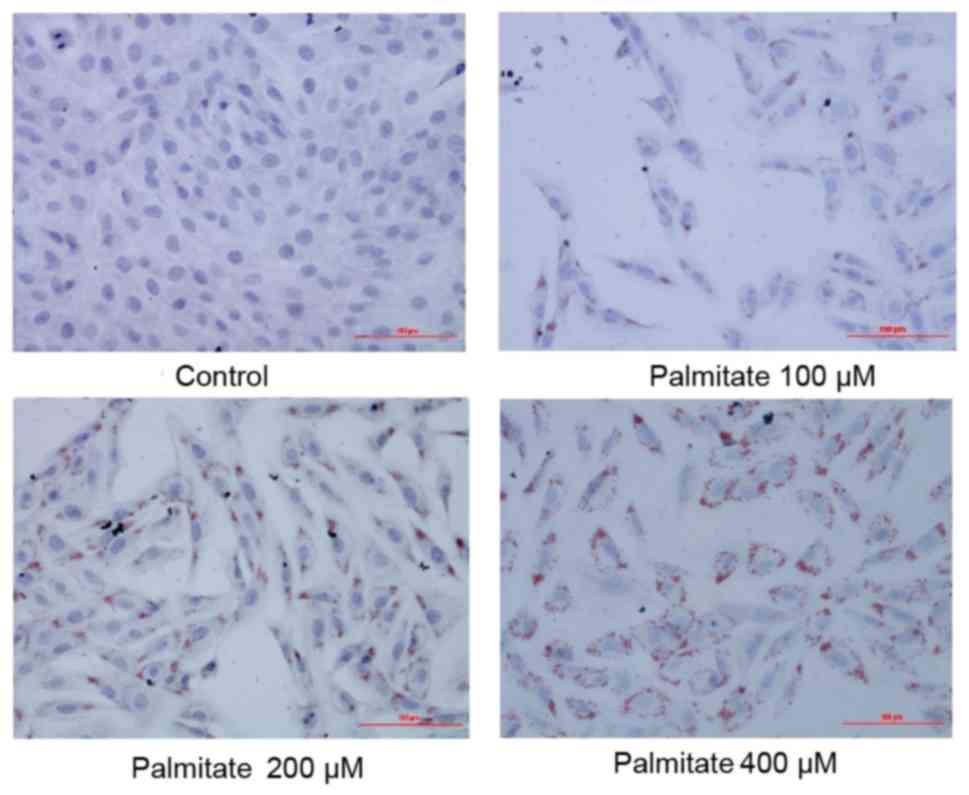
The result of Oil Red O staining demonstrated a
gradual increase in the extent of lipid accumulation in H9c2 cells
following treatment with increasing concentrations of PA (0, 100,
200 and 400 µM; Fig. 1),
suggesting PA induced excessive lipid accumulation in H9c2
cells.
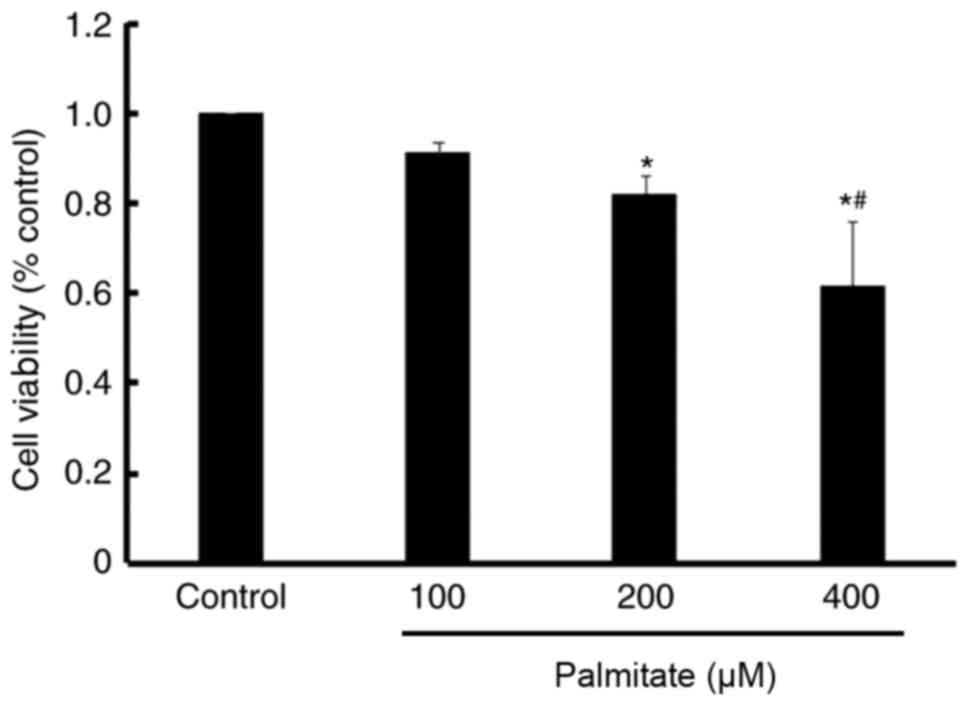
Effect of PA on cell viability
An MTT assay demonstrated a significant decrease in
cell viability following treatment with 200 and 400 µM PA compared
with the control. Furthermore, PA reduced cell viability in a
dose-dependent manner (Fig.
2).
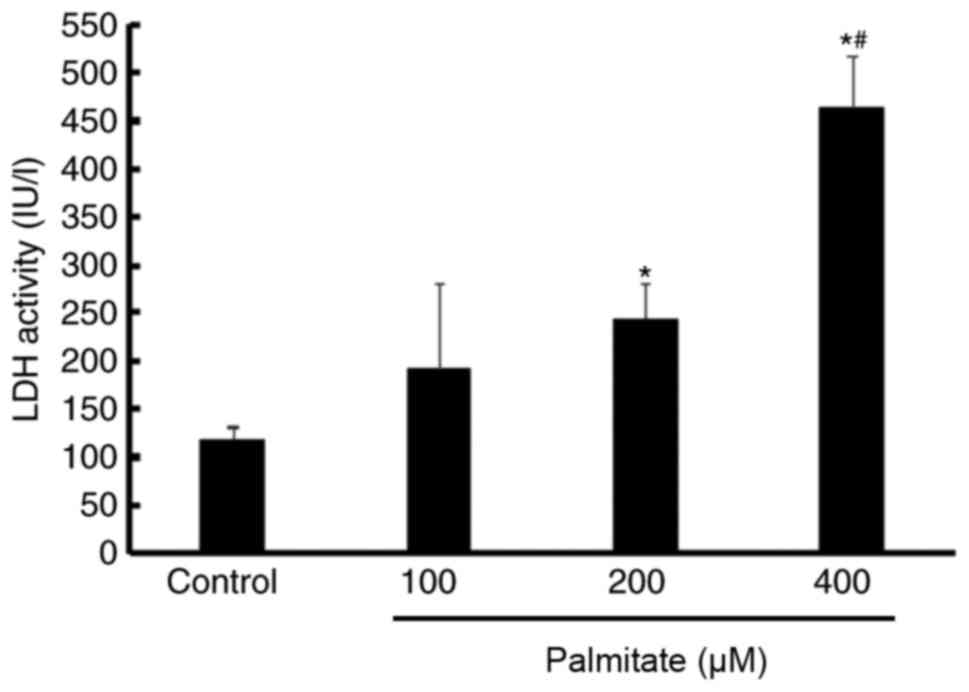
Effect of PA on cell injury
An LDH assay indicated that LDH activity was
significantly increased by 200 and 400 µM PA compared with the
control. Furthermore, LDH activity in the 400 µM PA treatment group
was higher than in the 200 µM PA group, which suggested that PA
induced H9c2 cell injury in a dose-dependent manner (Fig. 3).
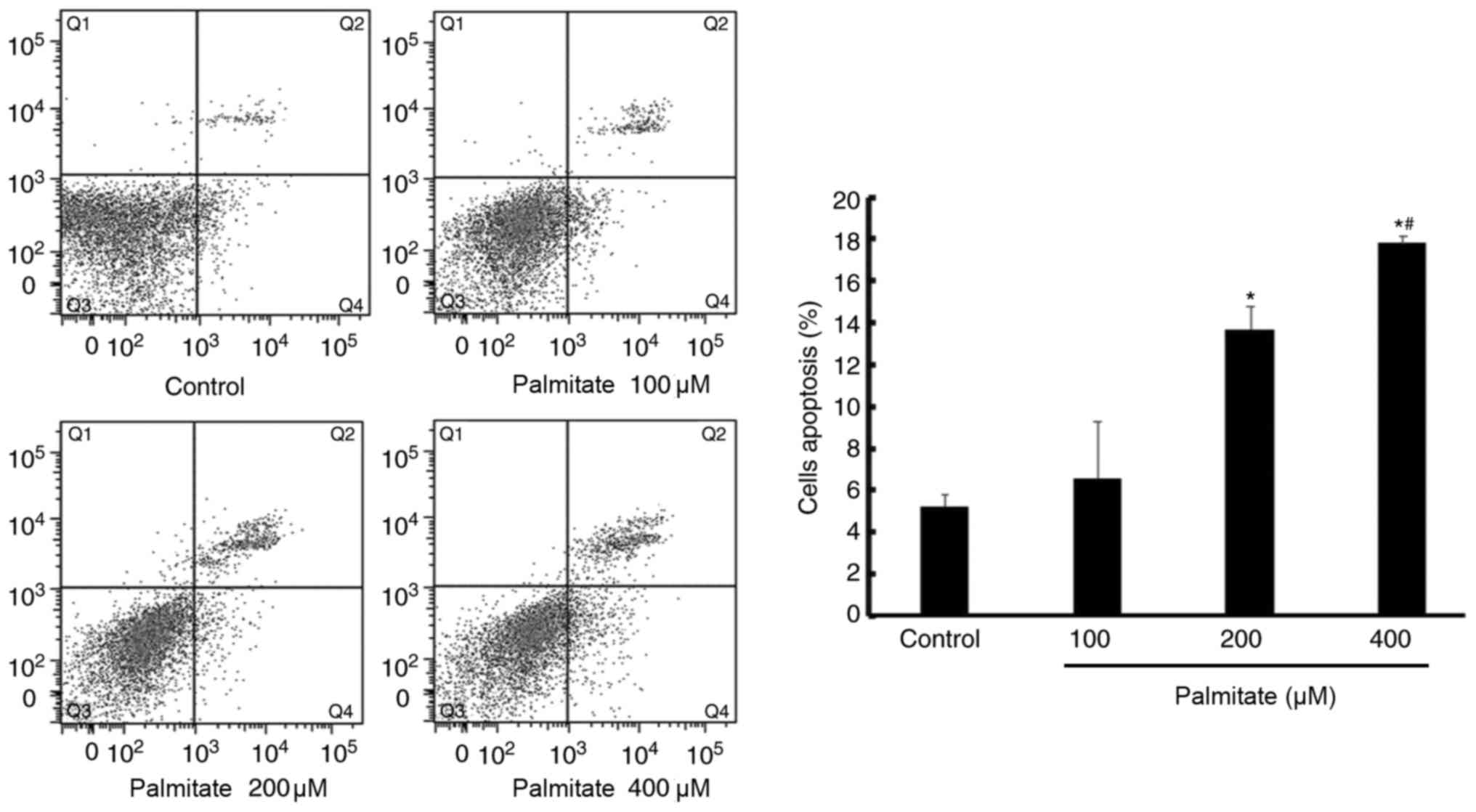
Effect of PA on cell apoptosis
PA (200 and 400 µM) significantly increased H9c2
cell apoptosis compared with the control, demonstrated by increased
apoptosis rate. In addition, apoptosis induced by PA was also
displayed in a dose-dependent manner (Fig. 4).
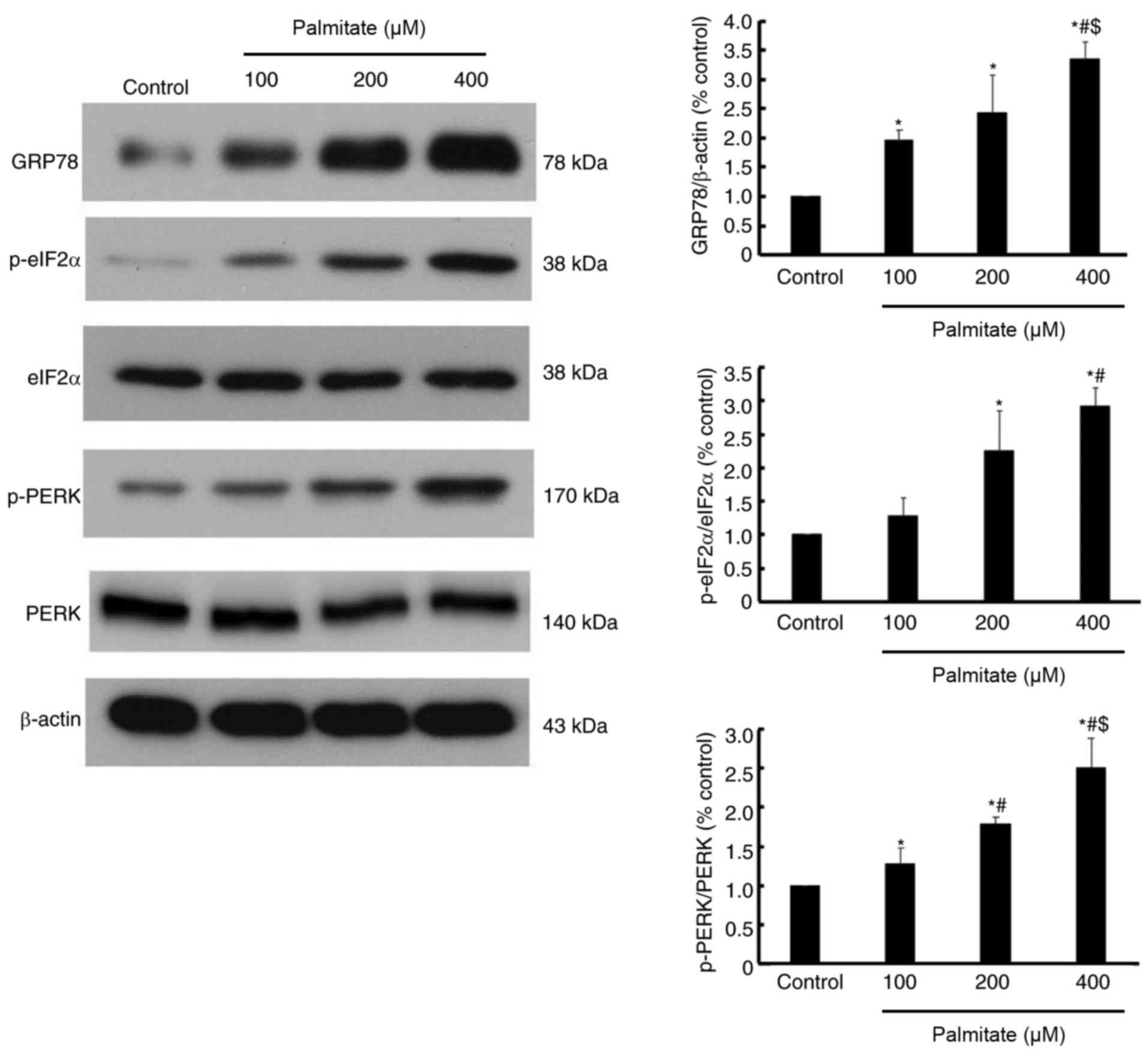
Effect of PA on the expression of ER
stress markers
As presented in Fig.
5, the expression of GRP78, a well-established ER stress
marker, was increased by various dose of PA in a dose-dependent
manner. Furthermore, the phosphorylation of eIF2α and PERK were
also increased in a dose-dependent manner (Fig. 5), which indicated that PERK/eIF2α
signaling may be activated by PA.
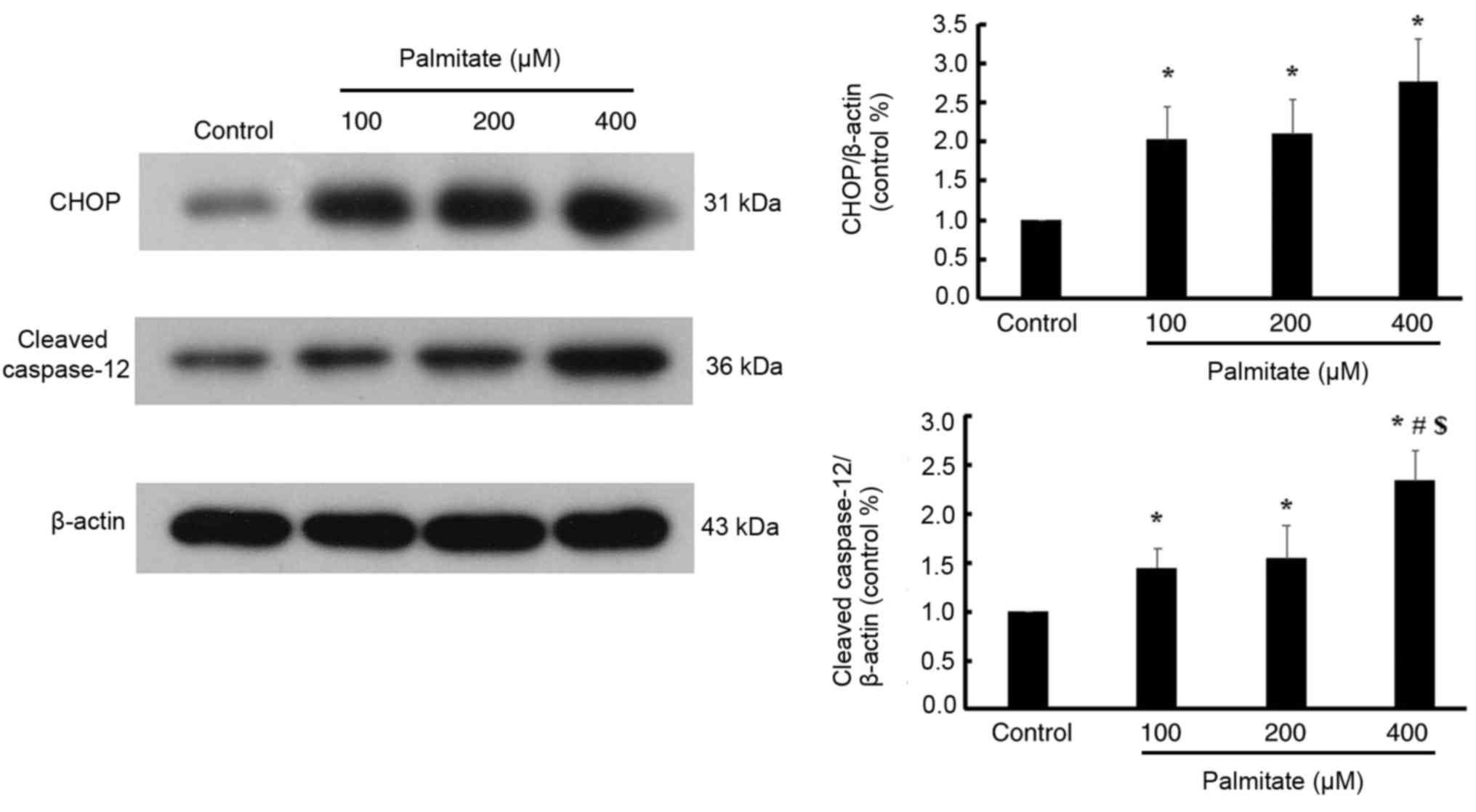
Effect of PA on the ER stress-mediated
apoptosis pathway
Various doses of PA significantly increased the
expression of CHOP compared with the control; however, no
significant differences in the expression of CHOP were detected
among the different PA-treated groups. Furthermore, various dose of
PA significantly increased the expression of cleaved caspase-12 in
a dose-dependent manner (Fig.
6).
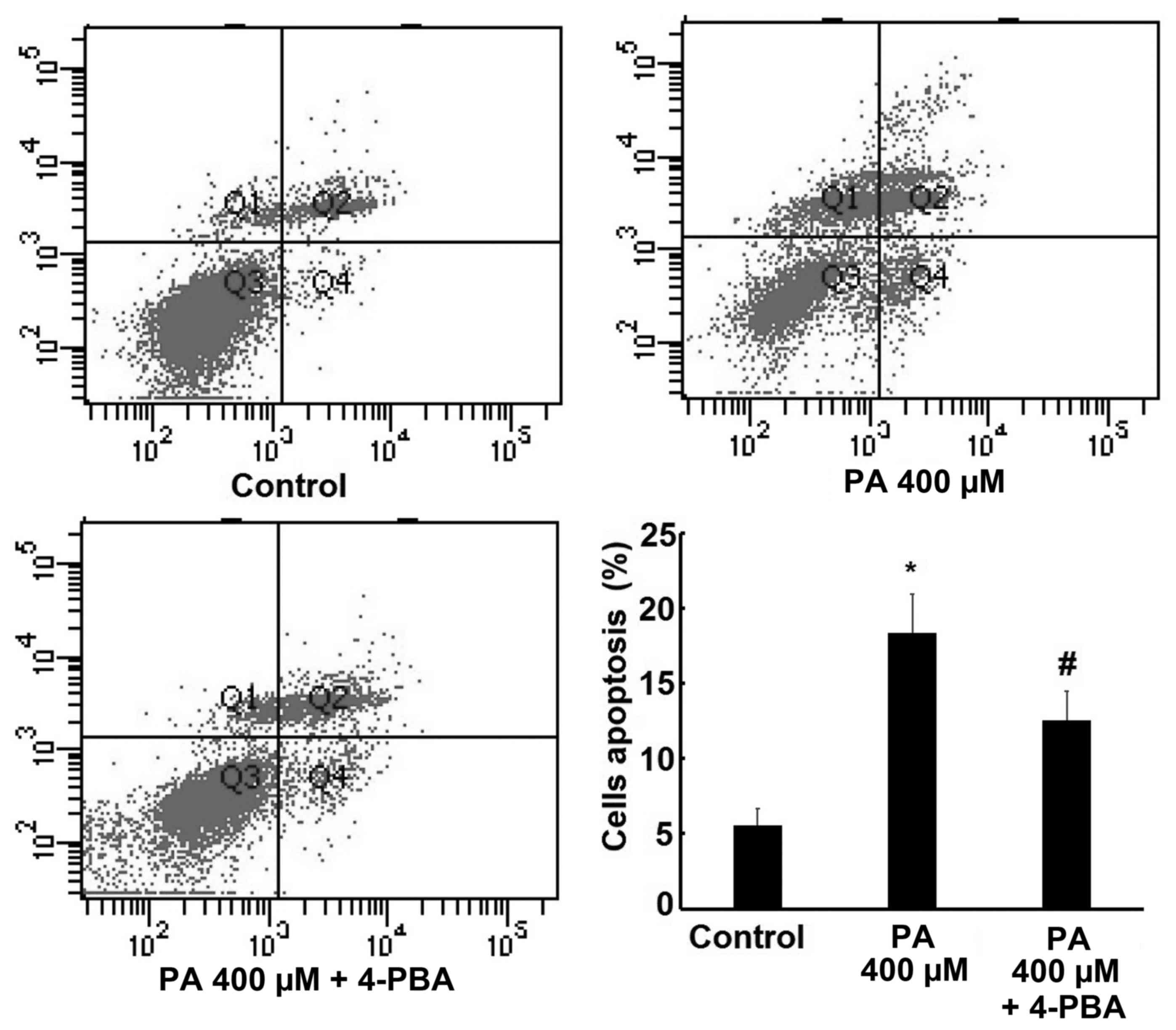
Effect of 4-PBA on PA-induced
apoptosis
In order to reconfirm the role of ER stress in PA
induced apoptosis, 4-PBA, a specific ER stress inhibitor, was
administered to examine the changes in cell apoptosis rate and the
expression of CHOP and cleaved caspase-12 following ER stress
inhibition. The results revealed that 400 µM PA significantly
increased H9c2 cells apoptosis rate, but this effect was reversed
by treatment with 4-PBA (Fig. 7).
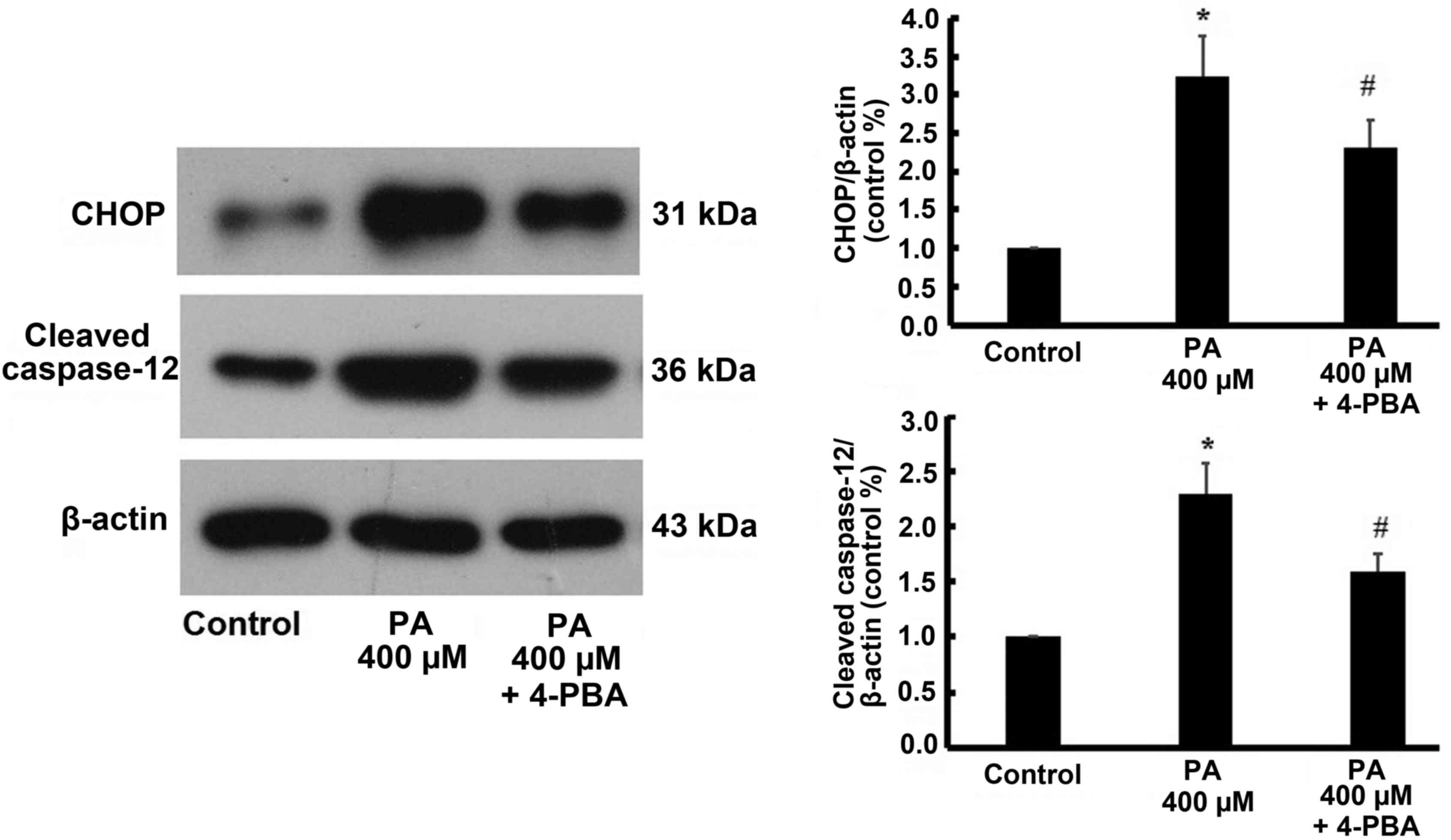
Similarly, the expression of CHOP and cleaved caspase-12 were
increased by 400 µM treatment, but were also reversed by 4-PBA
treatment (Fig. 8).
Discussion
Myocardial damage caused by hyperlipemia
predominantly occurs by two mechanisms: In most cases,
hyperlipemia, a well-known independent risk factor of coronary
heart disease, accelerates the development of coronary
atherosclerosis and results in myocardial ischemia, and even
myocardial necrosis, eventually (13); additionally, sustained and
excessive hyperlipemia can also directly result in myocardial
damage, termed ‘myocardial lipotoxic injury’ during severe
metabolic disorders, including diabetes and severe obesity
(14,15). The present study demonstrated that
PA promoted excessive lipid deposition in cardiomyocytes and
resulted in decreased cell viability, increased LDH activity and
apoptosis rate in a dose-dependent manner, which is consistent with
the findings of Wei et al (4,16).
Therefore, the present study reconfirmed that PA could induce
myocardial lipotoxic injury in vitro.
ER stress, an important adaptive response in
eukaryotic cells, is often activated under the conditions of
various pathophysiological procedures, including anoxia (17), poisoning (18) and infection (19). In particular, previous studies have
demonstrated that ER stress is involved in different cardiovascular
diseases including atherosclerosis (20), hypertension (21) and heart failure (22). The current study demonstrated that
GRP78, a marker of ER stress, was elevated in cardiomyocytes
following PA treatment. Furthermore, PERK/eIF2α, part of a
well-established ER stress-associated pathway, were activated by PA
treatment, as demonstrated by increased phosphorylation of PERK and
eIF2α. The current results were similar to previous reports
indicating that ER stress is activated in response to chronically
elevated free fatty acids in hepatocytes (10) and pancreatic β cells (11).
However, ER stress is a double-edged sword, in that
prolonged or excessive ER stress will trigger an ER stress-mediated
apoptosis pathway (7). Unlike the
death receptor- and mitochondria-mediated apoptosis pathways,
specific ER stress-induced apoptosis proteins, including CHOP and
caspase-12, were activated (23).
One of the notable novel findings of the current study is that
myocardial lipotoxic injury induced by various doses of PA were
involved in the activation of ER stress-mediated apoptosis pathway.
Although, Park et al (24)
demonstrated that ER stress-mediated autophagy had an important
role in regulating myocardial lipotoxic injury induced by PA,
whether ER stress-mediated apoptosis is implicated in the onset of
myocardial lipotoxic injury remains unknown. The present study
demonstrated that CHOP and cleaved caspase-12 were significantly
up-regulated in cardiomyocytes when treated with different dose of
PA, but the effect reversed following ER inhibition using 4-PBA.
This indicated that myocardial lipotoxic injury induced by PA may
be involved in the activation of the ER stress-mediated apoptosis
pathway.
In conclusion, the current study demonstrated that
PA induces myocardial lipotoxic injury, potentially by triggering
ER stress and the ER stress-mediated apoptosis pathway.
Acknowledgements
This study was supported by Fund for National
Natural Science Foundation of China (grant no. 81670320), the
Scientific Research of The First Hospital of China Medical
University (grant no. fsfh1501) and the Natural Science Foundation
of Liaoning Province (grant no. 201602826).
References
|
1
|
Ussher JR: The role of cardiac
lipotoxicity in the pathogenesis of diabetic cardiomyopathy. Expert
Rev Cardiovasc Ther. 12:345–358. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carley AN and Severson DL: Fatty acid
metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys
Acta. 1734:112–126. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gambert S, Vergely C, Filomenko R, Moreau
D, Bettaieb A, Opie LH and Rochette L: Adverse effects of free
fatty acid associated with increased oxidative stress in
postischemic isolated rat hearts. Mol Cell Biochem. 283:147–152.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wei CD, Li Y, Zheng HY, Tong YQ and Dai W:
Palmitate induces H9c2 cell apoptosis by increasing reactive oxygen
species generation and activation of the ERK1/2 signaling pathway.
Mol Med Rep. 7:855–861. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Phillips MJ and Voeltz GK: Structure and
function of ER membrane contact sites with other organelles. Nat
Rev Mol Cell Biol. 17:69–82. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Groenendyk J, Sreenivasaiah PK, Kim DH,
Agellon LB and Michalak M: Biology of endoplasmic reticulum stress
in the heart. Circ Res. 107:1185–1197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sano R and Reed JC: ER stress-induced cell
death mechanisms. Biochim Biophys Acta. 1833:3460–3470. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Listenberger LL, Ory DS and Schaffer JE:
Palmitate-induced apoptosis can occur through a
ceramide-independent pathway. J Biol Chem. 276:14890–14895. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haffar T, Bérubé-Simard FA and Bousette N:
Cardiomyocyte lipotoxicity is mediated by Il-6 and causes
down-regulation of PPARs. Biochem Biophys Res Commun. 459:54–59.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cao J, Dai DL, Yao L, Yu HH, Ning B, Zhang
Q, Chen J, Cheng WH, Shen W and Yang ZX: Saturated fatty acid
induction of endoplasmic reticulum stress and apoptosis in human
liver cells via the PERK/ATF4/CHOP signaling pathway. Mol Cell
Biochem. 364:115–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lai E, Bikopoulos G, Wheeler MB,
Rozakis-Adcock M and Volchuk A: Differential activation of ER
stress and apoptosis in response to chronically elevated free fatty
acids in pancreatic beta-cells. Am J Physiol Endocrinol Metab.
294:E540–E550. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yin J, Wang Y, Gu L, Fan N, Ma Y and Peng
Y: Palmitate induces endoplasmic reticulum stress and autophagy in
mature adipocytes: Implications for apoptosis and inflammation. Int
J Mol Med. 35:932–940. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
van Rooy MJ and Pretorius E: Obesity,
hypertension and hypercholesterolemia as risk factors for
atherosclerosis leading to ischemic events. Curr Med Chem.
21:2121–2129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wende AR and Abel ED: Lipotoxicity in the
heart. Biochim Biophys Acta. 1801:311–319. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Goldberg IJ, Trent CM and Schulze PC:
Lipid metabolism and toxicity in the heart. Cell Metab. 15:805–812.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wei CD, Li Y, Zheng HY, Sun KS, Tong YQ,
Dai W, Wu W and Bao AY: Globular adiponectin protects H9c2 cells
from palmitate-induced apoptosis via Akt and ERK1/2 signaling
pathways. Lipids Health Dis. 11:1352012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
López-Hernández B, Ceña V and Posadas I:
The endoplasmic reticulum stress and the HIF-1 signalling pathways
are involved in the neuronal damage caused by chemical hypoxia. Br
J Pharmacol. 172:2838–2851. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen S, Melchior WB Jr and Guo L:
Endoplasmic reticulum stress in drug- and environmental
toxicant-induced liver toxicity. J Environ Sci Health C Environ
Carcinog Ecotoxicol Rev. 32:83–104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cui Y, Zhao D, Barrow PA and Zhou X: The
endoplasmic reticulum stress response: A link with tuberculosis?
Tuberculosis (Edinb). 97:52–56. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chistiakov DA, Sobenin IA, Orekhov AN and
Bobryshev YV: Role of endoplasmic reticulum stress in
atherosclerosis and diabetic macrovascular complications. Biomed
Res Int. 2014:6101402014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo XF and Yang XJ: Endoplasmic reticulum
stress response in spontaneously hypertensive rats is affected by
myocardial ischemia reperfusion injury. Exp Ther Med. 9:319–326.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang J, Hu X and Jiang H: ER
stress-induced apoptosis: A novel therapeutic target in heart
failure. Int J Cardiol. 177:564–565. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jing G, Wang JJ and Zhang SX: ER stress
and apoptosis: A new mechanism for retinal cell death. Exp Diabetes
Res. 2012:5895892012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park M, Sabetski A, Chan Y Kwan, Turdi S
and Sweeney G: Palmitate induces ER stress and autophagy in H9c2
cells: Implications for apoptosis and adiponectin resistance. J
Cell Physiol. 230:630–639. 2015. View Article : Google Scholar : PubMed/NCBI
|