Introduction
Thalassemia is an inherited hemoglobinopathy that is
caused by an imbalance in the ratio of α-globin and β-globin
synthesis, which results in hemolytic anemia owing to the shortened
lifespan of red blood cells (1).
Clinically, α- and β-thalassemias occur as a result of genetic
defects (2). β-thalassemia is
characterized by a defect in the synthesis of β-globin chains of
the hemoglobin tetramer. Mutations in the hemoglobin subunit
β(HBB) gene lead to an abnormal formation of hemoglobin,
which results in improper oxygen transportation and the destruction
of red blood cells. These mutations either partially or completely
terminate β-globin chain synthesis, which are classified as
β+ and β0 mutations, respectively. Every
year, ~60,000 newborns are diagnosed with β-thalassemia worldwide
(3,4). The type of genetic mutation and
distribution frequency of β-thalassemia exhibits obvious regional
differences and ethnic characteristics. High incidence rates of
β-thalassemia have been reported in the southern China (3,4), and
the carrier rates of the populations of those regions were 3–24%.
Currently, >200 types of β-globin gene mutations have been
identified around the world, and >30 mutations have been
recorded in China (5). A previous
study demonstrated that compound mutations may lead to severe
thalassemia or thalassemia intermedia (6). Many of these compound mutations are
familial alterations, and spontaneous somatic mutations are quite
rare. The present study reported a case of compound heavy
β-thalassemia plus α-thalassemia caused by both familial and
somatic mutations. To treat the proband, all family members of the
patient were subjected to human leukocyte antigen (HLA) typing, and
bone marrow transplantation (BMT) was successfully performed
between the proband and his HLA-identical sister.
Case report
Written informed consent was obtained from all
participants in the present study. The proband was a boy (age, 2
years 6 months) with severe anemia (hemoglobin level, 62 g/l)
accompanied by yellowish skin, enlarged liver and spleen, among
other symptoms present since he was 6 months old. Based on these
clinical and hematological characteristics (Table I), the patient was diagnosed with
heavy β-thalassemia (2). Sanger
sequencing was performed to identify any mutations in the proband
and other family members. To conduct Sanger sequencing, three pairs
of polymerase chain reaction (PCR)/sequencing primers were designed
for hemoglobin subunit α1 (HBA1), HBA2 and hemoglobin subunit β
(HBB; PCR primer sequences are patented and withheld by the Beijing
Genomics Institute-Shenzhen, Shenzhen, Guangdong, China). Genomic
DNA was extracted from peripheral blood using the PureLink™ Genomic
DNA Mini kit (cat. no. K182000; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
instructions. The PCR mixture was prepared in a 25 µl reaction
solution containing 0.4 µM of each primer, 25 ng genomic DNA and
12.5 µl 2X GoldStarTaq Master Mix (CWBiotech, Beijing, China). PCR
was performed in a ABI 9700 PCR machine (Applied Biosystems; Thermo
Fisher Scientific, Inc.) with the following thermocycling
conditions: An initial hot-start step at 95°C for 10 min, followed
by 35 cycles of 30 sec at 95°C, 30 sec at 60°C and 90 sec at 72°C,
and a final extension of 10 min at 72°C. The PCR products were
purified using Centricon®-100columns (cat. no.
P/NN930-2119; EMD Millipore, Billerica, MA, USA) according to the
manufacturer's protocol, and then sequenced using the ABI 3730×l
DNA Analyzer (Applied BioSystems; Thermo Fisher Scientific, Inc.).
The sequencing results were analyzed using Lasergene software v7.1
(DNASTAR, Inc., Madison, WI, USA). These analyses demonstrated that
the proband was a carrier of the thalassemia gene mutation
IVS-II-654(C>T)β+. To further diagnose the proband
and their family members, additional blood tests with Sysmex
XE-5000 Automatic Hematology Analyzer and Sebia automatic
electrophoresis were performed. Peripheral blood (~2 ml) was
collected from each individual, maintained in tubes with EDTA
anticoagulant, subjected to blood cell examination using the Sysmex
XE-5000 analyzer (Sysmex Corporation, Kobe, Japan) and then
analyzed using Laboman EasyAccess v4.2 software (Sysmex
Corporation). For Sebia automatic electrophoresis, 2 ml peripheral
blood was collected and centrifuged at 3,000 × g for 5 min at room
temperature. The plasma was then removed and the samples were
subjected to Sebia automatic electrophoresis using the Sebia
MINICAP (Sebia, Lisses, France) and analyzed with Phoresis CORE
v7.4.7 software (Sebia). The results revealed that the proband had
β-thalassemia, whereas his mother and sister suffered from
microcytic hypochromic anemia (Table
I); the father was phenotypically normal.
 | Table I.Results of routine blood test and
hemoglobin electrophoresis. |
Table I.
Results of routine blood test and
hemoglobin electrophoresis.
| Variable | Normal range | Proband | Father | Mother | Sister |
|---|
| RBC
(×1012) | 4.0–5.5 | 2.85 | 3.9 | 2.74 | 2.67 |
| MCV (f1) | 80–100 | 69.4 | 82 | 75 | 74 |
| MCH (pg/cell) | 26–38 | 21.9 | 29.5 | 23.7 | 23.5 |
| MCHC (g/l) | 300–360 | 316 | 378 | 340 | 334 |
| Hb (g/l) | 120–160 | 62.0 | 112 | 105 | 90 |
| HbA (%) | >95 | 78.2 | 97.3 | 95.9 | 95.2 |
| HbA2 (%) | 1–3 | 3.8 | 2.0 | 3.5 | 3.6 |
| HbF (%) | <2 | 18.0 | 0.1 | 0.2 | 0.5 |
To determine the genetic background of this case,
peripheral blood DNA was obtained from all available family members
for full clinical genetic testing for thalassemia. Control
peripheral blood DNA was collected from two disease-free volunteers
(34-year-old male; 25-year-old female; classed as disease-free
based on normal hemoglobin levels identified via routine blood
tests and Sebia automatic electrophoresis) who were recruited from
within the Department of Pediatrics (Affiliated Hospital of Zunyi
Medical University, Zunyi, Guizhou). Sanger sequencing combined
with reverse transcription-quantitative PCR (RT-qPCR) were used to
detect the 338 known types of globin gene sequence mutations that
lead to thalassemia. RT-qPCR was performed to detect the unknown α-
and β-globin genes, and was performed as described previously
(7,8). Densitometry was performed using
StepOne™ v2.0 software (Thermo Fisher Scientific, Inc.). The
results were normalized to those of GAPDH and the 2−ΔΔCq
method was used (9). For
statistical analysis, a one-way analysis of variance was performed
on SPSS v17.0 software (SPSS, Inc., Chicago, IL, USA), and a
Dunnett's post hoc test was used for multiple comparison; P<0.05
was considered to indicate a statistically significant difference.
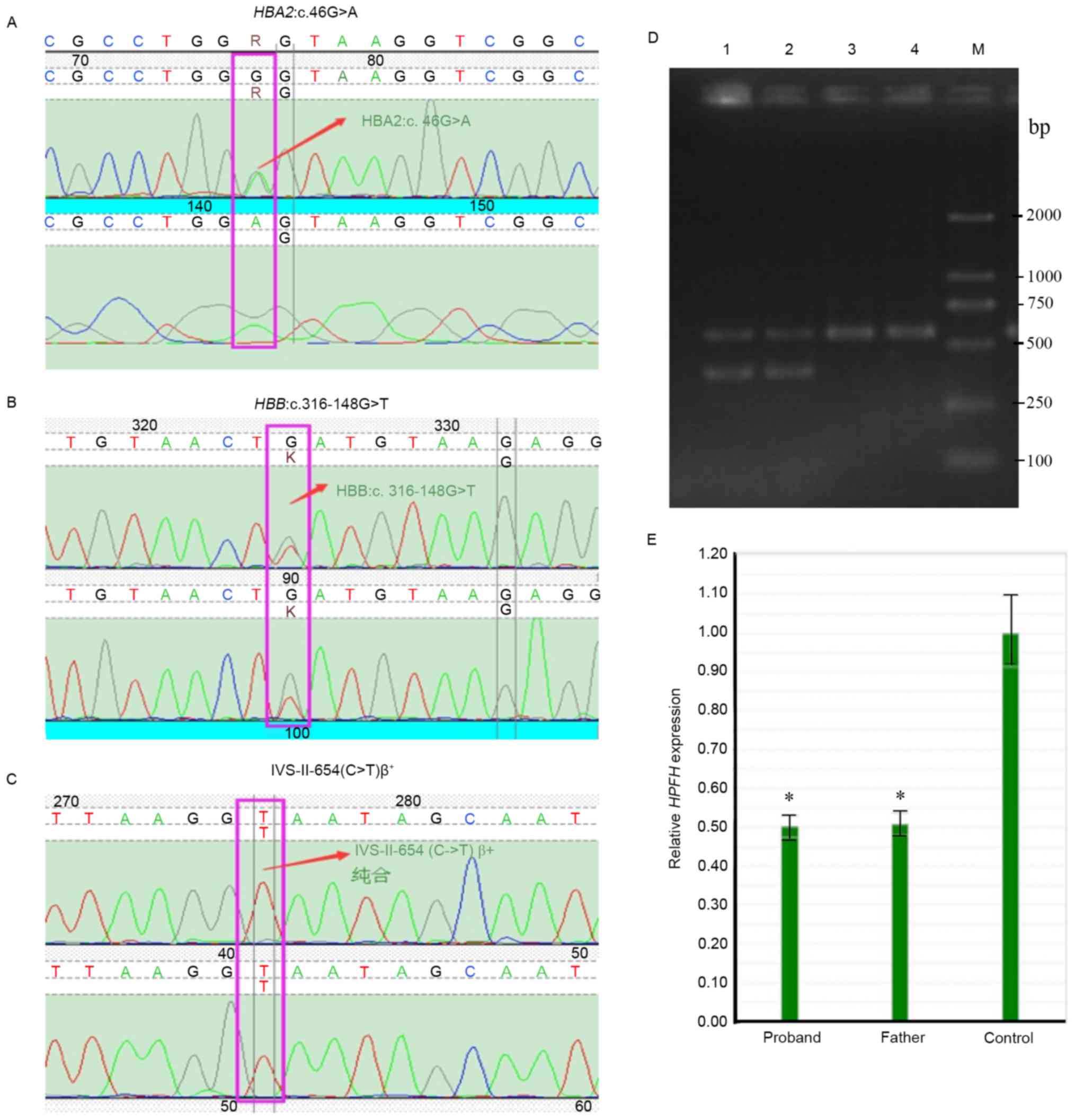
The data indicated that the proband had compound mutations of the
heterozygous IVS-II-654(C>T)β+, Southeast Asian
(SEA)-type-hereditary persistence of fetal hemoglobin
(SEA-HPFH) deletion (NC-000011.9:g.5222878-5250288del)
(10), HBA2:c.46G>A
heterozygous, and HBB:c.316-148G>T heterozygous (Fig. 1). Although Sanger sequencing
indicated that the father had a normal IVS-II-654 gene, results
from RT-qPCR (Fig. 1E) indicated
that he carried a SEA-HPFH mutation (a deletion of the β
chain), which was verified by Gap-PCR. Gap-PCR was applied to
detect 5α-thalassemia deletions including the SEA, α-3.7, α-4.2,
Filipino and Thailand deletions, and was performed as described
previously (11). The mother of
the proband was a carrier of the compound heterozygous mutations
IVS-II-654(C>T)β+ and HBA2:c.46G>A, and his
sister inherited the IVS-II-654(C>T)β+ heterozygous
mutation from the mother (Figs. 1
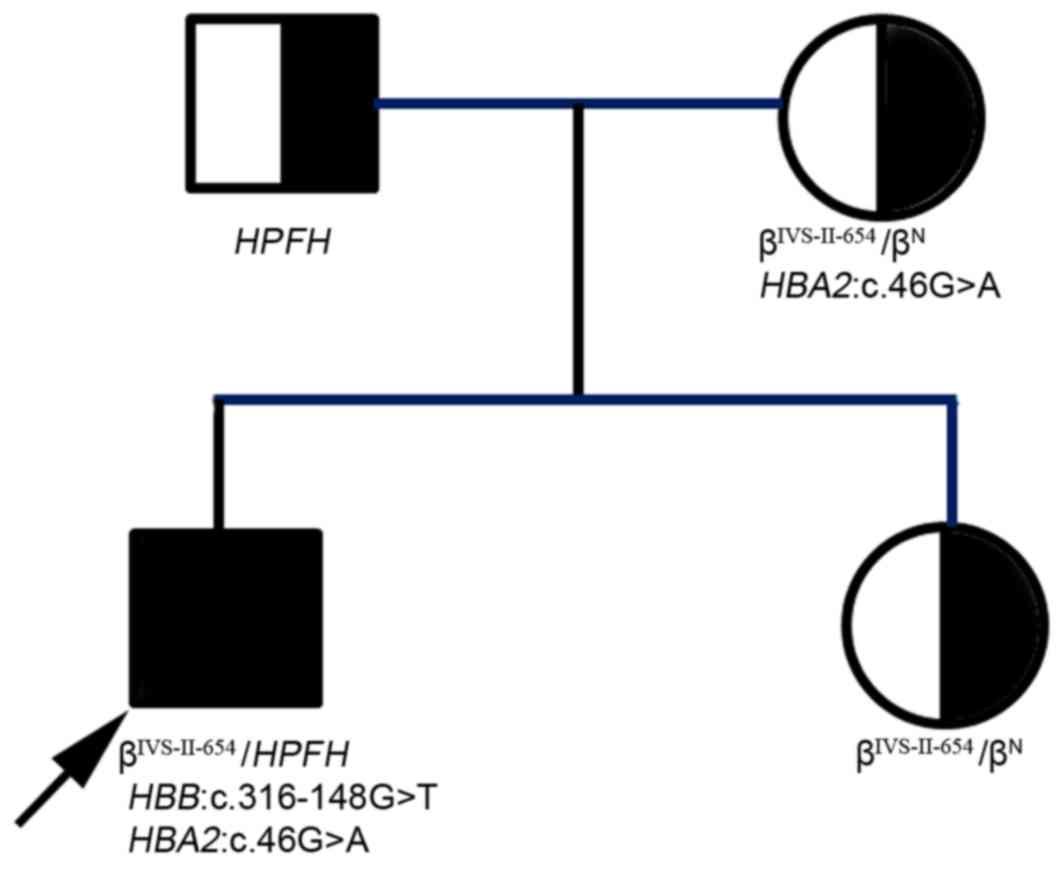
and 2; Table II). Based on the above data, the
proband was identified as having a somatic mutation
HBB:c.316-148G>T, and inherited the other thalassemia
mutations from his parents: That is, the SEA-HPFH mutation
from the father and the IVS-II-654(C>T)β+ allele and
HBA2:c.46G>A mutation from the mother (Fig. 2). Thus, the proband clinically
displayed heavy thalassemia, whereas his mother and sister
exhibited weaker manifestations of the disease.
| Table II.Thalassemia genotypes of the proband
and family members. |
Table II.
Thalassemia genotypes of the proband
and family members.
| Individual | Genotype |
|---|
| Proband |
βIVS-II-654/βN;
HBA2:c.46G>A; |
|
|
HBB:c.316-148G>T;
SEA-HPFH |
| Father |
SEA-HPFH/βN |
| Mother |
βIVS-II-654/βN;
HBA2:c.46G>A |
| Sister |
βIVS-II-654/βN |
Based on previous reports (12,13),
it was determined that HBA2:c.46G>A and
HBB:c.316-148G>T were novel alterations located near the
known mutations in the thalassemia mutation databases (12) (www.globin.bx.psu.edu/hbvar; www.ncbi.nlm.nih.gov/clinvar/?term=HBB[gene] AND
single_gene[prop]) (Table III).
HBA2:c.46G>A leads to an amino acid replacement
(p.Gly16Ser) that causes a hemoglobin disease, whereas
HBB:c.316-148G>T is a mutation in intron 2 of HBB,
the pathogenic effect of which is unclear.
| Table III.Adjacent positions of novel mutations
in the thalassemia mutation database. |
Table III.
Adjacent positions of novel mutations
in the thalassemia mutation database.
| Novel
mutations | Known adjacent
mutations |
|---|
|
IVS-II-654(C>T) |
AAGGCAATA→AAG^GTAATA,
HBB:c.316-197C>T |
|
IVS-II-705(T>G) |
GATGTAAGA→GAG^GTAAGA,
HBB:c.316-146T>G |
|
IVS-II-726(A>G) | (adjacent sequence
unavailable) HBB:c.316-125A>G |
|
IVS-II-745(C>G) | CAGCTACCAT→CAG^G',
HBB:c.316-106C>G |
|
IVS-II-661(A>G) | (adjacent sequence
unavailable) HBB:c.316-90A>G |
| Hb Ottawa, α2 or
α1 | 15(A13)Gly>Arg,
HBA2:c.46G>C |
The HbF level of the proband was considerably high
(18%; Table I), which was
consistent with previous reports (1,2,14–16).
Small-scale deletions or single-base mutations in the HBB
gene may result in low HbF levels (0.5–6.0%), whereas large-scale
deletions may cause high levels of HbF (6.0–15.0%) (16,17).
HbF levels >15% may result from compound mutations, such as a
large-scale deletion combined with other mutations (16,17).
Upon determination of the genetic background of the proband, it was
decided that a BMT should be performed. Firstly, the HLA types of
the proband and other family members were determined through
high-resolution HLA testing by PCR-sequence based typing. Briefly,
DNA was extracted from the proband and family members then
amplified by PCR using HLA Class I (exons 1–8) and HLA Class II
(exons 2–4) primers (PCR primer sequences were patented and
withheld by the Beijing Genomics Institute-Shenzhen), and the PCR
product was purified for sequencing via the aforementioned Sanger
sequencing methodology. The sequencing results indicated that the
HLA type of the proband was fully matched with their sister
(Table IV), thus the sister was
selected as the bone marrow donor, and BMT was conducted involving
the proband and sister. To monitor the results of the BMT,
follow-up testing was performed using fluorescence in situ
hybridization (FISH). Peripheral blood (~2 ml) were collected from
the proband and cultured in the presence of a mitogen
[KaryoMAX®Phytohemagglutinin (M-Form; PHA); cat. no.
10576; Thermo Fisher Scientific, Inc.] for 68 h at 37°C, then
0.05–0.1 µg/ml KaryoMAXColcemid® Solution (mitotic
inhibitor; cat. no. 15210; Thermo Fisher Scientific, Inc.) was
added to the culture at room temperature for 20 min. Cells were
subsequently treated with 5 ml hypotonic solution (0.068 M KCl) at
room temperature for 15 min, then 1 ml fresh ice cold fixative
(absolute methanol:glacial acetic acid, 3:1) was added, and the
cells were spun down at 500 × g at room temperature for 7 min. The
supernatant was then removed, and 5 ml of fresh, ice-cold fixative
was added drop by drop (with continuous vortexing) at 4°C for 20
min. This fixation step was repeated until the supernatant became
clear, then the cell pellet was resuspended in 1.5 ml fixation
solution and placed onto slides; the slides were left at 55°C
overnight, then they were kept at 4°C for subsequent use. For FISH,
the prepared slides were incubated at 50°C for 2.5 h, then
denatured in 70% formamide at 70°C for 2 min, followed by
dehydration in a series of ethanol solutions (70, 80, 90 and 100%)
at room temperature for 2 min each. The slides were subjected to
hybridization using a denatured CSPX/CSPY probe mixture (Beijing GP
Medical Technologies, Ltd., Beijing, China; denatured at 75°C for 5
min then 0°C for 5–10 min) at 37°C for 15–17 h. Following
hybridization, the slides were washed three times (10 min each) in
50% (v/v) formamide containing 2X SSC, then once in 2X SSC for 10
min and once in 2X SSC containing 0.1% NP-40 (Amresco LLC, Solon,
OH, USA) for 5 min. The slides were then dehydrated in 70% ethanol
for 3 min and allowed to air dry at room temperature. A total of 12
µl DAPI (Beijing GP Medical Technologies, Ltd.) was added to each
slide and incubated at room temperature for 20 min in the dark
followed by analysis using a fluorescence microscope (OLYMPUS-BX51;
Olympus Corporation, Tokyo, Japan). The results demonstrated that
the donor cells (karyotype: 46, XX) accounted for 99% of the blood
cells in the proband 30 days post-treatment (Fig. 3), and the proband's blood was
normal 60 days following BMT; four months post-BMT, the blood type
of the proband was transformed from type O to type A, which was the
same as the donor (the proband's sister). Six months following BMT,
the proband exhibited a thalassemia genotype that was consistent
with his sister; thus, no further blood transfusions were
required.
| Table IV.HLA type matching results of the
proband with his parents and sister. |
Table IV.
HLA type matching results of the
proband with his parents and sister.
| Case | HLA-A | HLA-B | HLA-C | HLA-DRB1 | HLA-DQB1 |
|---|
| Proband | 0301,1101 | 2705,4001 | 0202,0702 | 1001,1202 | 0501,0301 |
| Father | 0301.3303 | 2705,5801 | 0202,0302 | 0301,1001 | 0501,0201 |
| Sister | 0301,1101 | 2705,4001 | 0202,0702 | 1001,1202 | 0501,0301 |
| Mother | 1101,1102 | 4001,4601 | 0102,0702 | 0901,1202 | 0301,0303 |
Discussion
Thalassemia is an inherited, autosomal recessive
blood disorder that is characterized by an abnormal formation of
hemoglobin. Patients with thalassemia make less hemoglobin, and
this hemoglobin is abnormal; patients also have fewer circulating
red blood cells, which results in mild to severe microcytic anemia
(1,2,15,18).
Currently, the diagnosis of thalassemia relies on routine blood
testing combined with blood hemoglobin electrophoresis and
thalassemia gene detection (17).
Conventional thalassemia gene screening methods only detect known
point mutations. In the present study, the single-gene screening
method was noted to miss some potential genetic changes; for
example, Sanger sequencing failed to detect the SEA-HPFH
deletion mutation of the father, and prior to RT-qPCR testing the
proband was initially misdiagnosed as having a
IVS-II-654(C>T)β+ homozygous mutant. These findings
suggested that multiple thalassemia gene screening methods may be
required for precise genotyping of the disease.
The mother of the proband carried the
IVS-II-654(C>T) mutation located on chromosome 11, which is a
commonly identified β-thalassemia mutation in Chinese people
(14,19), and which results in a splicing
error that produces abnormal mRNA and hemoglobin protein,
eventually causing β-thalassemia (20). Although the
IVS-II-654(C>T)β+ heterozygous mutation may cause
‘light’ β-thalassemia that does not require special treatment,
homozygous and compound heterozygous mutations may lead to severe
disease and the affected patient usually requires regular blood
transfusions. In the present case, the proband inherited the
IVS-II-654(C>T)β+ mutation from his mother and the
SEA-HPFH deletion from his father, which constituted a
compound heterozygous mutation that led to heavy β-thalassemia. The
SEA-HPFH mutation is a rare β-thalassemia genotype in the
Chinese population; it was first identified in Vietnamese and
Cambodian patients (21,22). SEA-HPFH mutations involve a
large DNA deletion that includes the β-globin gene cluster
(18,21,23).
The deletion range covers NC-000011.9:g. 5222878-5250288del,
missing the entire β-globin gene and its 3′hyper sensitive site 1
(10,24,25),
which may not be detected by the common β-thalassemia detection
assays. The average SEA-HPFH carrier exhibits no clinical
symptoms and their peripheral blood cells appear completely normal.
However, when this mutation coincides with another β-gene mutation
and/or constitute compound heterozygous mutations, it may lead to
severe or heavy β-thalassemia, which is what occurred in the
present case study.
To the best of our knowledge, the present study was
the first to identify the HBA2:c.46G>A mutation in α
chain on chromosome 16. This mutation leads to a Gly16Ser amino
acid replacement; however, whether it is causative of thalassemia
requires further analysis. The HBB:c.316-148G>T mutation
is located in intron 2 of the β-globin gene, and was determined to
be a novel somatic alteration identified in the proband. The
alteration does not change the protein structure; however, whether
it affects the generation of mRNA and causes the disease also
merits further investigation. These novel mutations enriched the
thalassemia mutation spectrum in the Chinese population, which may
be helpful in future genetic counseling and clinical diagnosis.
Currently, hematopoietic stem cell transplantation
(HSCT) is the only effective way to cure severe β-thalassemia in
the clinical practice (15,26,27).
The influence on transplant success rate of donor mainly depends on
the HLA typing on chromosome 6 (28). In the present case, the HLA type of
the sister completely matched that of the proband, which led to
successful BMT and cured the patient of the disease.
Acknowledgements
The authors thank the many clinicians, data managers
and research staff who participated in this project.
References
|
1
|
Higgs DR, Engel JD and Stamatoyannopoulos
G: Thalassaemia. Lancet. 379:373–383. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Muncie HL Jr and Campbell J: Alpha and
beta thalassemia. Am Fam Physician. 80:339–344. 2009.PubMed/NCBI
|
|
3
|
Cai R, Li L, Liang X, Liu Z, Su L, Li W,
Zhu Q, Mo Q, Pan L, Ouyang H, et al: Prevalence survey and
molecular characterization of alpha and beta thalassemia in Liuzhou
city of Guangxi. Zhonghua Liu Xing Bing Xue Za Zhi. 23:281–285.
2002.(In Chinese). PubMed/NCBI
|
|
4
|
Xu XM, Zhou YQ, Luo GX, Liao C, Zhou M,
Chen PY, Lu JP, Jia SQ, Xiao GF, Shen X, et al: The prevalence and
spectrum of alpha and beta thalassaemia in Guangdong Province:
Implications for the future health burden and population screening.
J Clin Pathol. 57:517–522. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang J, Zhu BS, He J, Zeng XH, Su J, Xu
XH, Li SY, Chen H and Zhang YH: The spectrum of a- and
b-thalassemia mutations in Yunnan Province of Southwestern China.
Hemoglobin. 36:464–473. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu ZH, Liu YL, Zeng ZY, Zhang XL and Zhu
QY: Beta-thalassemia majar caused by compound heterozygosity for
+40 to −43(−AAAC), IVS-2-654 (C to T) and codon 41/42 (−TCTT).
Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 25:418–420. 2008.(In Chinese).
PubMed/NCBI
|
|
7
|
Babashah S, Jamali S, Mahdian R, Nosaeid
MH, Karimipoor M, Alimohammadi R, Raeisi M, Maryami F, Masoudifar M
and Zeinali S: Detection of unknown deletions in beta-globin gene
cluster using relative quantitative PCR methods. Eur J Haematol.
83:261–269. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fallah MS, Mahdian R, Aleyasin SA, Jamali
S, Hayat-Nosaeid M, Karimipour M, Raeisi M and Zeinali S:
Development of a quantitative real-time PCR assay for detection of
unknown alpha-globin gene deletions. Blood Cells Mol Dis. 45:58–64.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Z, Zhong X, Li Z and Xu X: Rapid
detection of an HPFH deletion by PCR amplification with three
primers bridging the breakpoint. Zhonghua Yi Xue Yi Chuan Xue Za
Zhi. 16:41–43. 1999.(In Chinese). PubMed/NCBI
|
|
11
|
Chong SS, Boehm CD, Cutting GR and Higgs
DR: Simplified multiplex-PCR diagnosis of common southeast asian
deletional determinants of alpha-thalassemia. Clin Chem.
46:1692–1695. 2000.PubMed/NCBI
|
|
12
|
Giardine B, Borg J, Viennas E, Pavlidis C,
Moradkhani K, Joly P, Bartsakoulia M, Riemer C, Miller W, Tzimas G,
et al: Updates of the HbVar database of human hemoglobin variants
and thalassemia mutations. Nucleic Acids Res. 42(Database Issue):
D1063–D1069. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Carlice-Dos-Reis T, Viana J, Moreira FC,
Cardoso GL, Guerreiro J, Santos S and Ribeiro-Dos-Santos Â:
Investigation of mutations in the HBB gene using the 1,000 genomes
database. PLoS One. 12:e01746372017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Long G and Zhang J: Hemoglobin &
Hemoglobinopathies. Guangxi Science & Technology Press;
Guangxi: pp. 233–235. 2003
|
|
15
|
Cao A and Galanello R: Beta-thalassemia.
Genet Med. 12:61–76. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mosca A, Paleari R, Leone D and Ivaldi G:
The relevance of hemoglobin F measurement in the diagnosis of
thalassemias and related hemoglobinopathies. Clin Biochem.
42:1797–1801. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu G and Sun S: The laboratory diagnosis
of thelassemia: Selection and evaluation of tests and methods. Chin
J Lab Med. 35:385–387. 2012.
|
|
18
|
Bollekens JA and Forget BG: Delta beta
thalassemia and hereditary persistence of fetal hemoglobin. Hematol
Oncol Clin North Am. 5:399–422. 1991.PubMed/NCBI
|
|
19
|
Zhang J, He J, Zeng XH, Ge SJ, Huang Y, Su
J, Ding XM, Yang JQ, Cao YJ, Chen H, et al: Genetic heterogeneity
of the β-globin gene in various geographic populations of Yunnan in
southwestern China. PLoS One. 10:e01229562015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang SZ, Zeng FY, Ren ZR, Lu ZH, Rodgers
GP, Schechter AN and Zeng YT: RNA transcripts of the
beta-thalassaemia allele IVS-2-654 C->T: A small amount of
normally processed beta-globin mRNA is still produced from the
mutant gene. Br J Haematol. 88:541–546. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Changsri K, Akkarapathumwong V, Jamsai D,
Winichagoon P and Fucharoen S: Molecular mechanism of high
hemoglobin F production in Southeast Asian-type hereditary
persistence of fetal hemoglobin. Int J Hematol. 83:229–237. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng W, Liu Y, Chen D, Rong K, Ge Y, Gong
C and Chen H: Complex interaction of Hb Q-Thailand and Hb E with
alpha(0)-thalassemia and hereditary persistence of fetal hemoglobin
in a Chinese family. Ann Hematol. 89:883–888. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xiu X: The molecular basis of the
δβ-thalassemia for HPFH and deletion seen in the Chinese. Chin J
Med Genet. 15:315–317. 1998.
|
|
24
|
Bhardwaj U and McCabe ER: Multiplex-PCR
assay for the deletions causing hereditary persistence of fetal
hemoglobin. Mol Diagn. 9:151–156. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu XM, Li ZQ, Liu ZY, Zhong XL, Zhao YZ
and Mo QH: Molecular characterization and PCR detection of a
deletional HPFH: Application to rapid prenatal diagnosis for
compound heterozygotes of this defect with beta-thalassemia in a
Chinese family. Am J Hematol. 65:183–188. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Smiers FJ, Krishnamurti L and Lucarelli G:
Hematopoietic stem cell transplantation for hemoglobinopathies:
Current practice and emerging trends. Pediatr Clin North Am.
57:181–205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Caocci G, La Nasa G, d'Aloja E, Vacca A,
Piras E, Pintor M, Demontis R and Pisu S: Ethical issues of
unrelated hematopoietic stem cell transplantation in adult
thalassemia patients. BMC Med Ethics. 12:42011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Valcárcel D, Sierra J, Wang T, Kan F,
Gupta V, Hale GA, Marks DI, McCarthy PL, Oudshoorn M, Petersdorf
EW, et al: One-antigen mismatched related versus HLA-matched
unrelated donor hematopoietic stem cell transplantation in adults
with acute leukemia: Center for International blood and marrow
transplant research results in the era of molecular HLA typing.
Biol Blood Marrow Transplant. 17:640–648. 2011. View Article : Google Scholar : PubMed/NCBI
|