Introduction
Pulmonary arterial hypertension (PAH) is a serious
pulmonary vascular disease, which is defined by a mean pulmonary
arterial pressure ≥25 mmHg (1).
The pathological features of PAH include distal pulmonary artery
intimal hyperplasia, plexiform lesions, medial hypertrophy, muscle
infarction and thrombosis, gradually leading to occlusion of the
lumen and high pulmonary arterial pressure, eventually leading to
right heart failure (2,3). In previous years, several studies
have demonstrated that the structure, function and metabolic
changes of endothelial cells are important features in the
development of PAH, which has a marked effect on vessel homeostasis
(4–6).
A typical feature of endothelial injury is an
imbalance of nitric oxide (NO)-reactive oxygen species (ROS)
levels, which is caused by reductions in endothelium-derived NO
synthesis, release and activity, and is increased by ROS generation
and release (7,8), which is involved in the excessive
contraction and remodeling of pulmonary vessels. It has been
demonstrated that the small G proteins, Ras homolog family, member
A (RhoA) and Ras-related C3 botulinum toxin substrate 1 (Rac1), are
involved in changes of endothelial function through the Rho
kinase-endothelial NO synthase (eNOS) pathway or regular NADPH
oxidase, respectively (9).
Therefore, repairing endothelial function and reversing excessive
oxidative stress represent he pathophysiological foundation of PAH
therapy (7).
Previous studies have demonstrated that the
mevalonate pathway is involved in small G-protein activation
(10,11). The mevalonate pathway is an
important pathway of cholesterol synthesis at the cellular level.
Mevalonate is a precursor of cholesterol, in addition to several
non-steroid isoprenoid complexes, including farnesylpyrophosphate
(FPP) and geranylgeranylpyrophosphate (GGPP) (12). These non-steroid isoprenoids are
important in small G protein post-translational modification
(10). Studies have indirectly
demonstrated that the mevalonate pathway maybe involved in the
development of PAH. It has been suggested that mevalonate and other
mid products produced in the mevalonate pathway can regulate
signaling proteins, including those of Ras family (13), which are essential for cell
proliferation and other important features. The inhibitor of the
3-hydroxy-3-mehtylglutaryl-coenzyme A (HMGR) enzyme, a key enzyme
of the mevalonate pathway, decreases blood lipids, inhibits the
proliferation of smooth muscle cells, ameliorates endothelial cell
function and increases the expression of eNOS (14,15).
Animal experiments have shown that statins were able to mitigate
pulmonary pressure and right ventricular hypertrophy in
monocrotaline (MCT)-induced PAH rats, and this was associated with
an increased expression of eNOS (16,17).
At present, the effects of key enzymes, including
farnesyldiphosphate synthase (FDPS), farnesyltransferase α (FNTA),
farnesyltransferase β (FNTB) and geranylgeranyltransferase type I
(GGTase-I) on endothelial dysfunction have not been reported, with
the exception of the initial enzyme, HMGR. The pathway downstream
of FPP produced in the mevalonate pathway has three branches:
Asterol branch, which primarily contributes to cholesterol
synthesis, and two non-sterol branches, which regulate signal
transduction proteins, including those of the Ras and Rho families
(13,14). Therefore, the mevalonate pathway
may affect protein prenylation, vasoactive substance generation and
endothelial function, and regulate excessive vascular contraction
and remodeling in the pulmonary vascular tissues of PAH rats.
Based on previous studies, the present study aimed
to determine whether the expression of key enzymes, including HMGR,
FDPS, FNTA, FNTB and GGTase-I, in the mevalonate pathway are
altered in MCT-induced PAH rats, which may potentially serve as
novel therapeutic targets for PAH.
Materials and methods
Animal model
The present study was performed in adherence with
the National Institutes of Health Guidelines for the Care and Use
of Laboratory Animals andapproved by the ethics committee of
Zhejiang University (approval no. ZJU20170874; Hangzhou, China). A
total of 100 F344 male rats (6-weeks old; weighing 200±10 g) were
obtained from Shanghai Laboratory Animal Center (Chinese Academy of
Sciences, Shanghai, China). A total of 5 rats/cage, temperature of
22°C, light and dark cycle 12-h, free access to drinking water and
normal feed. The rat PAH model was induced by injecting a single
dose of MCT (60 mg/kg, dissolved in 1N HCl, neutralized with 1N
NaOH, diluted with saline), purchased from Sigma-Aldrich; Merck
Millipore (Darmstadt, Germany) and fed for 4 weeks.
The rats were randomly divided into two groups
(n=6); in the control group, each rat was injected with a single
dose of saline; in the PAH group, each rat was injected with a
single dose of MCT (60 mg/kg). On day 28, all rats were
sacrificed.
Hemodynamic parameters
The rats were injected with 8% chloral hydrate
following weighing (1 ml/200 g), and fixed on an autopsy table.
Right ventricular systolic pressure (RVSP) was measured by
insertion of a PE50 pipe through the jugular vein to the right
ventricle; the RVSP was transferred into an electric signal and
recorded using MedLab software (version 6.3; Nanjing Medease
Science and Technology, Nanjing, China).
Measurement of right ventricular
hypertrophy (RVH)
All rats were sacrificed following measurement of
pulmonary arterial pressure, and the hearts, lungs and pulmonary
arteries were harvested. The blood was removed in cold PBS. The
right ventricle (RV) was isolated from the left ventricle (LV) and
septum (S), and these two components were weighted separately. RVH
was determined as the ratio of RV weight to (LV+S) weight.
Histological analysis
A sample of lung tissue was fixed in prepared 4%
paraformaldehyde for 24 h, made into paraffinized sections (4-µM
thick), and then stained with hematoxylin and eosin. A fluorescence
microscope (Nikon Eclipse 80i; Nikon, Tokyo, Japan) was used to
observe the pulmonary arteries in the stained sections.
Western blot analysis
The pulmonary arteries were cleared in cold PBS and
frozen in liquid nitrogen prior to being stored at −80°C. The
pulmonary artery tissues were homogenized in lysis buffer
(radioimmunoprecipitation assay buffer, PMSF; 100:1) and then
centrifuged at 13,800 × g for 15 min at 4°C. The protein
concentrations were determined using a bicinchoninic protein assay
kit, and 30 µg were separated on a 10% SDS-PAGE gel, followed
transferal onto a polyvinylidene difluoride membrane. The membrane
was blocked in 5% skim milk (5 g skim milk dissolved in 100 ml
Tris-buffered saline Tween solution) at room temperature for 1 h.
The membrane was incubated with the following antibodies: HMGR
(cat. no. ab174830, 1:2,000), FDPS (cat. no. ab189874, 1:1,000),
FNTA (cat. no. ab109738, 1:1,000), and FNTB (cat. no. ab109748,
1:1,000) (all from Abcam, Cambridge, UK), GGT-I (cat. no. sc18996,
1:200; Santa Cruz Biotechnology Co., Ltd., Dallas, TX, USA),
phosphorylated (p)-eNOS (cat. no. 95719, 1:1,000), eNOS (cat. no.
9586, 1:1,000), and RhoA (cat. no. ARH04, 1:1,000) (all from CST
Biological Reagents Company Limited, Shanghai, China), Rac1 (cat.
no. ARC03, 1:1,000; BD Biosciences, Franklin Lakes, NJ, USA) at 4°C
for 16 h. The membranes were then incubated with the appropriate
secondary antibody: Goat-anti-rabbit immunoglobulin G (IgG) (cat.
no. 1268, 1:2,500), goat-anti-mouse IgG (cat. no. 1265, 1:2,500),
and rabbit-anti-goat IgG (cat. no. M1102, 1:2,500) (all from
Biovision, Inc., Milpitas, CA, USA) for 2 h at room temperature.
The target protein bands were visualized using chemiluminescence
and quantified using ImageJ software (version 1.47; National
Institutes of Health, Bethesda, MD, USA). GAPDH was used as an
endogenous control (cat. no. 377R, 1:5,000; Biovison, Inc.). All
antibodies were diluted in 5% BSA (HuaBio, China).
NADPH oxidase activation assay
The activation of NADPH oxidase was detected using a
Tissue NADPH Oxidase Activation Assay kit (Genmed Scientifics,
Inc., Arlington, MA, USA). The pulmonary arteries were homogenized
in lysis buffer and protein concentrations were determined using a
BCA protein assay kit. According to the manufacturer's
instructions, this was finally detected at 550 nm using a
microplate reader (Thermo Fisher Scientific, Inc., Waltham, MA,
USA.).
ROS kinase activation assay
A Tissue ROS Kinase Activation Assay kit (Genmed
Scientifics, Inc.) was used to measure the level of ROS in the
pulmonary artery. The pulmonary arteries were homogenized in lysis
buffer and protein concentrations were determined using a BCA
protein assay kit. The results were detected at 340 nm using a
microplate reader (Thermo Fisher Scientific, Inc.).
Measurement of serum NO levels
The blood samples were collected and centrifuged at
13,8000 × g, 4°C for 15 min. The serum was then removed into a new
EP tube, and serum NO levels were determined using a Nitric Oxide
Fluorometric Assay kit (Nanjing Jiancheng Bioengineering Institute,
Nanjing, China) according to the manufacturer's instructions. The
results were final detected using a microplate reader (Thermo
Fisher Scientific, Inc.).
Statistical analysis
All values are expressed as the mean ± standard
error of the mean. Statistical significance was measured using
one-way analysis of variance. Software used for analysis was
GraphPad Prism (version 6.0c; GraphPad Software, Inc., La Jolla,
CA, USA.). P<0.05 was considered to indicate a statistically
significant difference.
Results
MCT-induced PAH
The PAH model was induced by injection of a single
dose of MCT (60 mg/kg). After 4 weeks, RVSP was measured by
insertion of a PE50 pipe through the jugular vein to the right
ventricle. The pressures were transferred into electric signals and
collected using MedLab software. The RVSP of the PAH group was
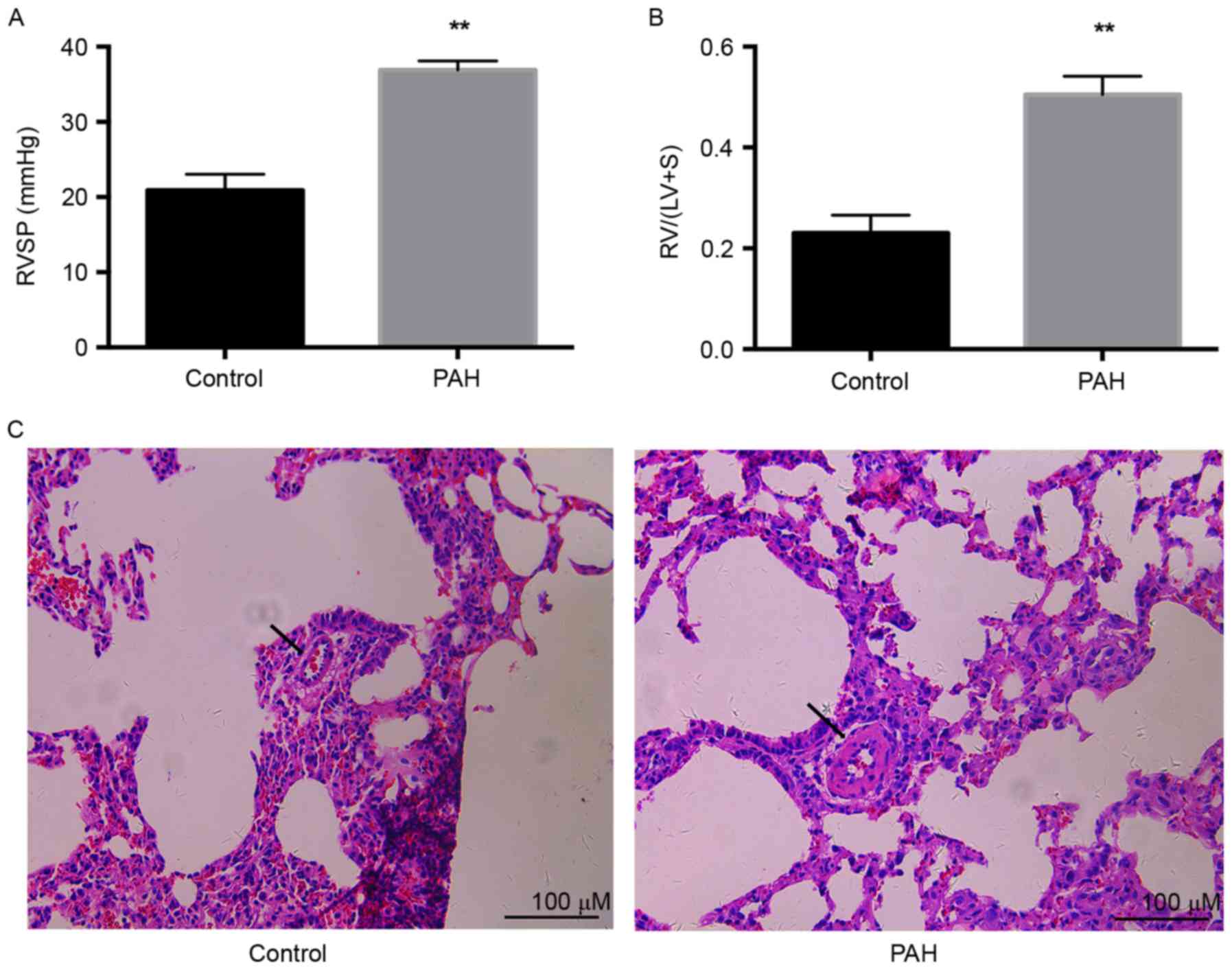
significantly increased, compared with that in the control
(36.9±1.1 vs. 20.9±1.1; P<0.01) as exhibited in Fig. 1A. RVH, calculated as the ratio of
RV weight to (LV+S) weight, was significantly increased by ~2-fold
in the PAH group compared with that in the control (0.50±0.02 vs.
0.23±0.02; P<0.01), as exhibited in Fig. 1B. Pulmonary vascular remodeling was
a notable characteristic of PAH. Significant thickening of
micro-pulmonary arteries was demonstrated in the PAH group,
compared with the control group (Fig.
1C). These results indicated that injection with a single dose
of MCT (60 mg/kg) successfully induced PAH 4 weeks later.
Expression of key enzymes in the
mevalonate pathway are altered in MCT-induced PAH
The present study aimed to determine whether the
expression levels of key enzymes in the mevalonate pathway are
altered in PAH. This involved comparing the expression levels of
key enzymes in the pulmonary artery between the PAH and control
groups.
HMGR is an initial enzyme in the mevalonate pathway,
and the present study found no significant difference in its
expression between the PAH and control group (Fig. 2A and B). A significant increase in
the expression of FDPS was detected in the PAH group, compared with
that in the control group (P<0.05), as exhibited in Fig. 2C. As FTase and GGTase-I catalyze
the farnesylation for several small G-proteins, including the Ras
and Rho family respectively (18,19),
the levels of FNTA and GGTase-I were significantly increased in the
PAH group, compared with those in the control group P<0.01),
whereas no significant difference in FNTB was demonstrated
(Fig. 2D-F). Overall, the
expression levels of enzymes FDPS, FNTA and GGTase-I were elevated
in the PAH rats.
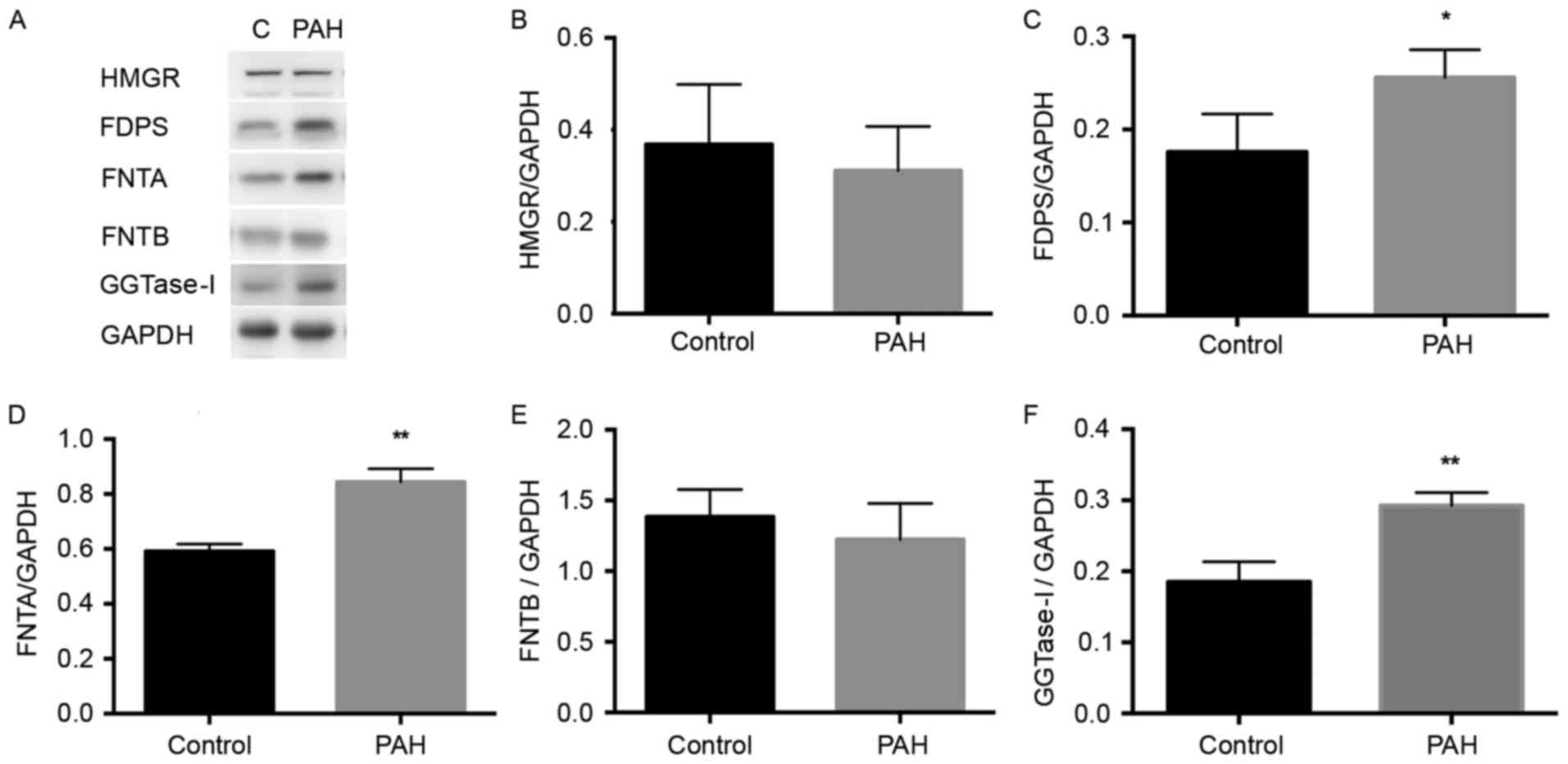 | Figure 2.Expression levels of HMGR, FDPS,
FNTA, FNTB and GGTase-I in the MCT-induced PAH rat pulmonary
artery. The proteins were extracted from pulmonary arteries of each
group. (A) Western blot analyses demonstrate the expression of
HMGR, FDPS, FNTA, FNTB and GGTase-I in the control and MCT-induced
PAH rats. GAPDH was used as the endogenous loading control. Graphs
demonstrate the relative changes in (B) HMGR, (C) FDPS, (D) FNTA,
(E) FNTB and (F) GGTase-I in the control and MCT groups. Data are
expressed as the mean ± standard deviation. *P<0.05 and
**P<0.01 compared with the control group. PAH, pulmonary
arterial hypertension; MCT, monocrotaline; HMGR,
3-hydroxy-3-methylglutaryl-coenzyme A; FDPS, farnesyldiphosphate
synthase; FNTA, farnesyltransferase α; FNTB, farnesyltransferase β;
GGTase-I, geranylgeranyltransferase type I. |
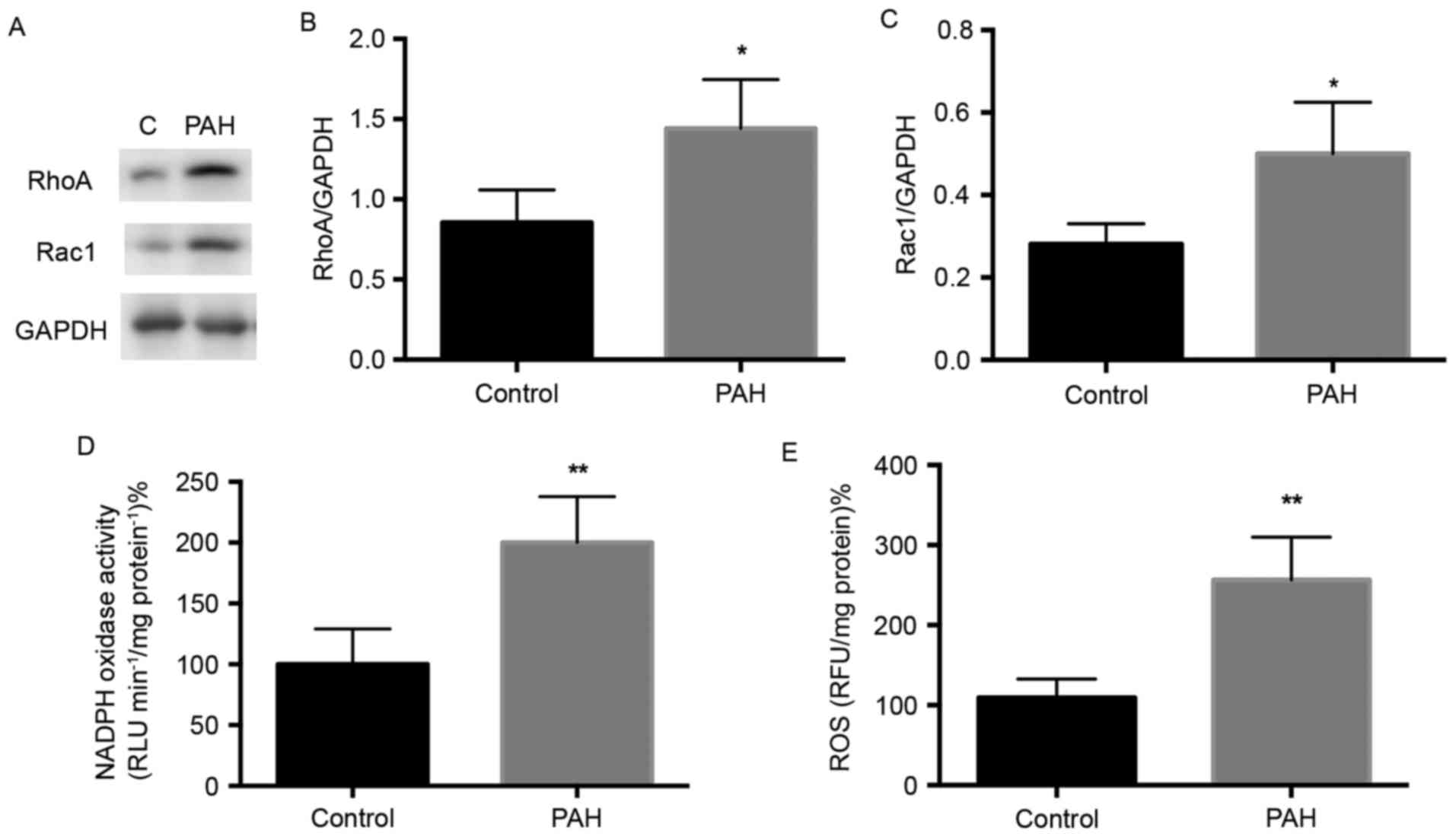
Expression of small G-protein RhoA and
Rac1 in MCT-induced PAH
Small G-proteins are important in regular specific
cell function and gene expression (11), and evidence indicates that the
prenylation and activation of small G-proteins are regulated by the
enzymes GGTase-I and FTase (18).
The expression levels of small G-protein RhoA and Rac1 in the
present study were determined using western blot analysis (Fig. 3A). The level of RhoA was
significantly elevated in the PAH group, compared with that in the
control group (Fig. 3A and B;
P<0.05), and Rac1 was also significantly increased in the PAH
group (Fig. 3A and C; P<0.05).
These results indicated that the expression of small G-proteins
were increased in MCT-induced PAH rats.
NADPH activity is increased in
MCT-induced PAH
NADPH oxidase is the downstream effector of small
G-protein, and its activation depends on the Rac protein (20). The present study detected whether
the expression of NADPH oxidase, the downstream effector, was
altered in PAH. Following the treatment of protein lysates
according to the manufacturer's protocol of the NADPH Oxidase
Activation Assay kit, it was found that NADPH oxidase activity was
increased by ~2-fold in the PAH group, compared with that in the
control (Fig. 3D).
ROS kinase activity is increased in
MCT-induced PAH
NADPH oxidase is a resource for generating ROS
(21). The overexpression of ROS
is involved in endothelial injury and vessel remodeling (8). The present study measured ROS kinase
activity and found a significant increase in the PAH group,
compared with that in the control (Fig. 3E; P<0.01). This indicated that
oxidative stress was elevated in the pulmonary artery of rats with
MCT-induced PAH.
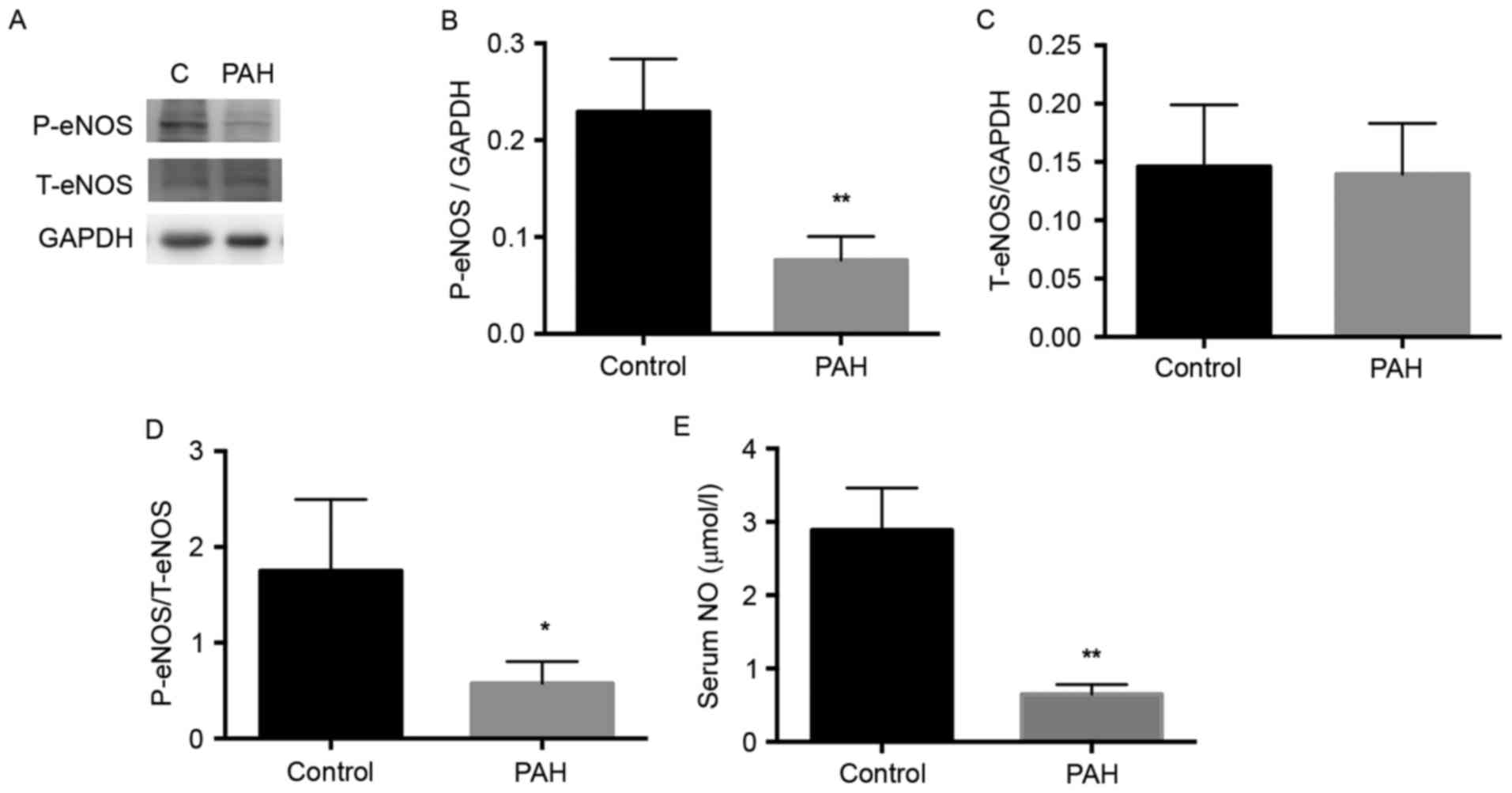
Activation of eNOS is decreased in
MCT-induced PAH
The eNOS enzyme catalyzes the biosynthesis of NO,
and NO in its molecular form has been reported to be important for
the development of PAH (22).
Therefore, the present study detected the expression of eNOS
(Fig. 4A). The level of p-eNOS was
significantly decreased in the PAH group, compared with that in the
control group (Fig. 4B;
P<0.01), whereas no significant difference in total NOS was
observed between the PAH and the control group (Fig. 4C). Therefore, the activity of eNOS,
represented by the ratio of p-eNOS to total eNOS, was significantly
decreased in the PAH group, compared with the control (Fig. 4C; P<0.05).
Serum levels of NO are decreased in
MCT-induced PAH
NO is an important molecule for cardiovascular
health. It is released as an endothelium-derived relaxing factor
(23). The present study detected
the serum levels of NO using a Nitric Oxide Fluorometric Assay kit.
The serum level of NO in the PAH group was significantly lower,
compared with that in the control (Fig. 4D; P<0.01). This may by due to
the decreased activity of eNOS in the pulmonary artery of
MCT-induced rats.
Discussion
PAH is a serious pulmonary vascular disease, which
can lead to right heart failure following qualitative changes in
the artery and lumen, blood flow and pressure, and cardiac muscle
(2,24). In the present study, changes in the
expression of key enzymes of the mevalonate pathway were detected
in PAH rats. The MCT-induced PAH model is an established method
generally used in rat experiments (25). In the present study, initial
measurements of RVSP revealed a significantly increase in the PAH
group 4 weeks following MCT injection. This result was accompanied
by a high ratio of RV/(LV+S) in the PAH group. These results
indicated that the PAH model and right ventricular remodeling had
been successfully established.
In the present study, it was demonstrated that the
expression of key enzymes in the mevalonate pathway, including
FDPS, FNTA and GGTase-I, were significantly increased in the
pulmonary artery of MCT-induced PAH rats. The expression of small
G-protein Rac1 and RhoA were also increased, which was consistent
with the results of GGTase-I and FTase. Small G-proteins downstream
effectors, including NADPH and ROS, were also examined; significant
increases in ROS and NADPH oxidase activity were demonstrated,
whereas the protein expression of eNOS and release of serum NO
decreased.
HMGR is the initial enzyme in cholesterol synthesis.
The therapeutic effect of statins on pulmonary hypertension is
controversial in clinical trails. In the present study, no
significant change was demonstrated in HMGR in the PAH rats,
consistent with a previous meta-analysis, which reported that HMGR
inhibitor statins had no benefit in patients with PAH (26). Isoprenoid is the intermediate of
the mevalonate pathway, and is important for diverse cellular
functions (12). The three known
end-products of isoprenoid are cholesterol, dolichol and the
polyisoprene side chain of ubiquinone (27). Steroidal and non-steroidal
isoprenoids partially regulate the expression of enzymes in the
mevalonate pathway (28). FDPS is
an important enzyme in the mevalonate pathway, which catalyzes the
synthesis of FPP and GPP from the substrate mevalonate (28). In the present study, an increase in
the enzyme FDPS was found in the pulmonary artery tissue of the PAH
rats. This increased expression of FDPS may be responsible for
pulmonary vessel remodeling and right ventricular hypertrophy.
Therefore, FDPS inhibitors are being investigated as a treatment
option for certain diseases in which FDPS is overexpressed. A
previous study demonstrated inhibiting FDPS by
N6-isopentenyladenosine, an adenosine and isoprenoid derivative,
was demonstrated to inhibit the proliferation of tumor cells
(29). In addition,
bisphosphonates are widely used FDPS inhibitor in the treatment of
osteoporosis (30). The chronic
inhibition of FDPS can also attenuate cardiac hypertrophy and
fibrosis (31), although the
specific signaling mechanism remains to be elucidated. It was
hypothesized that the abnormal expression of FDPS may be a
potential therapeutic target for the treatment of PAH.
The higher level of FDPS induced the accumulation of
downstream products, including GPP and FPP. As an intermediate
product, FPP is an important precursor in the synthesis of sterols,
dolichols and ubiquinones (32).
It is well known that the activation of proteins, including Ras and
Rab, require farnesylation by FTase with FPP as substrate in the
transmembrane, which responds to cellular signaling. Therefore, Ras
can activate genes involved in cell growth, proliferation, and
differentiation, and lead to abnormal vessel growth and remodeling
(33). The present study
demonstrated that the expression levels of FTase and GGTase-I were
increased in the PAH group. The enzyme GGTase-I catalyzes the
gernalygernalation of proteins, including the Rho family and Rac.
The elevated expression level of GGTase-I has been reported in
several human diseases, including renal fibrosis, spontaneous
hypertension and cancer (34).
Previous studies have attempted to use GGTase-I inhibitor to treat
diseases, including renal fibrosis and cancer, and have achieved
promising results (35). In order
to localize in correct subcellular membranes, small G-proteins of
the Rac and RhoA family require post-translational prenylation by
FTase and GGTase-I, and then transducing signals to downstream
effectors (36). Unlike certain
cardiovascular diseases, including pressure overload-induced
cardiac hypertrophy and spontaneous hypertension, in which the
expression of FNTB is elevated (37), the present study found no
significant change in the expression of FNTB in the PAH model. This
may be due to differences in tissue expression and disease models,
indicating that FNTA is more important in the development of PAH
than FNTB.
The Rho family, including Rac and Rho, can regulate
the function of the cytoskeleton (38). Rho proteins are involved in the
activation of extracellular signal-regulated kinase (ERK) in
response to angiotensin II or stretch-induced hypertrophy (39). Certain studies have demonstrated
that Rac can provoke thec-Jun N-terminal kinase/p38 subgroup of
nuclear mitogen-activated protein kinases (MAPKs) (40). Activated Rac can stimulate the
activity of p21-activated kinase (PAK) (41), and activated PAK can affect the
activation of ERK via the phosphorylation of the serine/threonine
protein kinase Raf (42). The
transcription of nuclear genes associated with cell proliferation
and growth can be regulated following the activation of Raf, MAPK
and ERK proteins (43,44).
NADPH oxidase is a membrane-bound enzyme complex.
Several studies have shown that the activation of NADPH oxidase
depends on the Rac protein and two other cytosolic proteins,
p47phox and p67phox (10). Rac-GDP
is converted into Rac-GTP and translocated to the correct membrane,
and NADPH oxidase is activated (45). The present study measured the
activity of NADPH oxidase and found it was markedly increased in
the PAH rats, consistent with the increase of Rac1. This result was
supported by a previous study, which demonstrated the same
increased activity of NADPH oxidase in an MCT-induced PAH model
(46).
NADPH oxidase is a source of superoxides, and
superoxides undergo further reactions to generate ROS (47). According to the present study, the
PAH rats generated more NADPH-derived ROS, compared with the
control rats, which was consistent with previous studies (46,48).
ROS is an essential regulator of normal cell physiology. The
overproduction of ROS is associated with cardiovascular disorders,
metabolic dysfunction and other diseases (47,49),
therefore, it was hypothesized that generated ROS contributes to
the development of PAH. Accumulated ROS may function through
reacting with cellular components, affecting the function of
endothelial cells, inducing the proliferation of smooth muscle
cells, and vascular remodeling (50,51).
In several heart diseases, NO is an important
molecule in vasodilation. The present study found decreased
activity of eNOS and serum levels of NO in the PAH group. Although
the bio-activity of eNOS was weaker in PAH, no differences in the
expression of eNOS were demonstrated between PAH and the control
group. This contradicted the results of a previous study, which
reported that the expression level of eNOS decreased in the PAH
model (52). However, previous
studies commonly used endothelial cells, including human umbilical
vein endothelial cells, whereas the experiments in the present
study used whole pulmonary artery tissue. This may be the reason
for the difference in results. Reductions in NO have not been
observed in previous studies (53). The superoxide-like ROS can react
with NO to reduce NO bioavailability (9), and the imbalance of ROS-NO levels may
lead to endothelial dysfunction, consequently stimulating the
development of PAH.
The present study detected changes in the expression
of key enzymes of the mevalonate pathway in an MCT-induced PAH
model, which may serve as drug targets for PAH treatment. The
inhibitors of certain enzymes, including FDPS and GGTase-I, have
been used in the treatment of certain human diseases. However,
whether treatment with the inhibitors of altered enzymes in PAH can
attenuate persistent pulmonary artery remodeling and high pressure
remains to be elucidated, for which further investigations are
required.
Acknowledgements
This study was financially supported by the National
Natural Sciences Foundation of China (grant no. 81400277) and the
Natural Sciences Foundation of Zhejiang Province (grant no.
LY17H020006). The abstract was presented at the 15th meeting of
China Interventional Therapeutics March 30 - April 2, 2017 in
Beijing, China (abstract no. AS-0341).
References
|
1
|
Hoeper MM, Bogaard HJ, Condliffe R, Frantz
R, Khanna D, Kurzyna M, Langleben D, Manes A, Satoh T, Torres F, et
al: Definitions and diagnosis of pulmonary hypertension. J Am Coll
Cardiol. 62 Suppl 25:D42–D50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Malenfant S, Margaillan G, Loehr JE,
Bonnet Sb and Provencher S: The emergence of new therapeutic
targets in pulmonary arterial hypertension: From now to the near
future. Expert Rev Respir Med. 7:43–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Houtchens J, Martin D and Klinger JR:
Diagnosis and management of pulmonary arterial hypertension.
Pulmonary Med. 2011:1–13. 2011. View Article : Google Scholar
|
|
4
|
Mam V, Tanbe AF, Vitali SH, Arons E,
Christou HA and Khalil RA: Impaired vasoconstriction and nitric
oxide-mediated relaxation in pulmonary arteries of hypoxia- and
monocrotaline-induced pulmonary hypertensive rats. J Pharmacol Exp
Ther. 332:455–462. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Giaid A and Saleh D: Reduced expression of
endothelial nitric oxide synthase in the lungs of patients with
pulmonary hypertension. N Engl J Med. 333:214–221. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Klinger JR, Abman SH and Gladwin MT:
Nitric oxide deficiency and endothelial dysfunction in pulmonary
arterial hypertension. Am J Respir Crit Care Med. 188:639–646.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cai H and Harrison DG: Endothelial
dysfunction in cardiovascular diseases: The role of oxidant stress.
Circ Res. 87:840–844. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dorfmüller P, Chaumais MC, Giannakouli M,
Durand-Gasselin I, Raymond N, Fadel E, Mercier O, Charlotte F,
Montani D, Simonneau G, et al: Increased oxidative stress and
severe arterial remodeling induced by permanent high-flow challenge
in experimental pulmonary hypertension. Respir Res. 12:119–130.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rinckel LA, Faris SL, Hitt ND and
Kleinberg ME: Rac1 disrupts p67phox:p40phox binding: A novel role
for rac in NADPH oxidase activation. Biochem Biophys Res Commun.
263:118–122. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takai Y, Sasaki T and Matozaki T: Small
GTP-binding proteins. Physiol Rev. 81:154–185. 2001.
|
|
12
|
Allal C, Favre G, Couderc B, Salicio S,
Sixou S, Hamilton AD, Sebti AM, Lajoie-Mazenc I and Pradines A:
RhoA prenylation is required for promotion of cell growth and
transformation and cytoskeleton organization but not for induction
of serum response element transcription. J Biol Chem.
275:31001–31008. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Singh RP, Kumar R and Kapur N: Molecular
regulation of cholesterol biosynthesis: Implications in
carcinogenesis. J Environ Pathol Toxicol Oncol. 22:75–92. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kinlay S: Potential vascular benefits of
statins. Am J Med. 118 Suppl 12A:S62–S67. 2005. View Article : Google Scholar
|
|
15
|
Koh KK: Effects of statins on vascular
wall: Vasomotor function, inflammation and plaque stability.
Cardiovasc Res. 47:648–657. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Murata T, Kinoshita K, Hori M, Kuwahara M,
Tsubone H, Karaki H and Ozaki H: Statin protects endothelial nitric
oxide synthase activity in hypoxia-induced pulmonary hypertension.
Arterioscler Thromb Vasc Biol. 25:2335–2342. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guerard P, Rakotoniaina Z, Goirand Fo,
Rochette L, Dumas M, Lirussi F and Bardou M: The HMG-CoA reductase
inhibitor, pravastatin, prevents the development of
monocrotaline-induced pulmonary hypertension in the rat through
reduction of endothelial cell apoptosis and overexpression of eNOS.
Naunyn Schmiedebergs Arch Pharmacol. 373:401–414. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang FL and Casey PJ: Protein
prenylation: Molecular mechanisms and functional consequences. Annu
Rev Biochem. 65:241–269. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moores SL, Schaber MD, Mosser SD, Rands E,
O'Hara MB, Garsky VM, Marshall MS, Pompliano DL and Gibbs JB:
Sequence dependence of protein isoprenylation. J Biol Chem.
266:14603–14610. 1991.PubMed/NCBI
|
|
20
|
Lőrincz ÁM, Szarvas G, Smith SM and Ligeti
E: Role of Rac GTPase activating proteins in regulation of NADPH
oxidase in human neutrophils. Free Radic Biol Med. 68:65–71. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Seno T, Inoue N, Gao D, Okuda M, Sumi Y,
Matsui K, Yamada S, Hirata KI, Kawashima S, Tawa R, et al:
Involvement of NADH/NADPH oxidase in human platelet ROS production.
Thromb Res. 103:399–409. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Machado RF, Nerkar MV Londhe, Dweik RA,
Hammel J, Janocha A, Pyle J, Laskowski D, Jennings C, Arroliga AC
and Erzurum SC: Nitric oxide and pulmonary arterial pressures in
pulmonary hypertension. Free Radic Biol Med. 37:1010–1017. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Crosswhite P and Sun Z: Mol mechanisms of
pulmonary arterial remodeling. Mol Med. 20:191–201. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yerly P, Prella M and Aubert JD: Current
management of pulmonary arterial hypertension. Swiss Med Wkly.
146:w143052016.PubMed/NCBI
|
|
25
|
Guzowski DE and Salgado ED: Changes in
main pulmonary artery of rats with monocrotaline-induced pulmonary
hypertension. Arch Pathol Lab Med. 111:741–745. 1987.PubMed/NCBI
|
|
26
|
Anand V, Garg S, Duval S and Thenappan T:
A systematic review and meta-analysis of trials using statins in
pulmonary arterial hypertension. Pulm Circ. 6:295–301. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Faust JR, Goldstein JL and Brown MS:
Synthesis of ubiquinone and cholesterol in human fibroblasts:
Regulation of a branched pathway. Arch Biochem Bioph. 192:86–99.
1979. View Article : Google Scholar
|
|
28
|
Brown MS and Goldstein JL: Multivalent
feedback regulation of HMG CoA reductase, a control mechanism
coordinating isoprenoid synthesis and cell growth. J Lipid Res.
21:505–517. 1980.PubMed/NCBI
|
|
29
|
Laezza C, Notarnicola M, Caruso MG, Messa
C, Macchia M, Bertini S, Minutolo F, Portella G, Fiorentino L,
Stingo S and Bifulco M: N6-isopentenyladenosine arrests tumor cell
proliferation by inhibiting farnesyl diphosphate synthase and
protein prenylation. FASEB J. 20:412–418. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park SB, Park SH, Kang YK and Chung CK:
The time-dependent effect of ibandronate on bone graft remodeling
in an ovariectomized rat spinal arthrodesis model. Spine J.
14:1748–1757. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li L, Chen GP, Yang Y, Ye Y, Yao L and Hu
SJ: Chronic inhibition of farnesyl pyrophosphate synthase
attenuates cardiac hypertrophy and fibrosis in spontaneously
hypertensive rats. Biochem Pharmacol. 79:399–406. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun S and McKenna CE: Farnesyl
pyrophosphate synthase modulators: A patent review (2006–2010).
Expert Opin Ther Pat. 21:1433–1451. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bajaj A, Zheng Q, Adam A, Vincent P and
Pumiglia K: Activation of endothelial ras signaling bypasses
senescence and causes abnormal vascular morphogenesis. Cancer Res.
70:3803–3812. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wiemer AJ, Wiemer DF and Hohl RJ:
Geranylgeranyl diphosphate synthase: An emerging therapeutic
target. Clin Pharmacol Ther. 90:804–812. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Philips MR and Cox AD:
Geranylgeranyltransferase I as a target for anti-cancer drugs. J
Clin Invest. 117:1223–1225. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dunford JE, Rogers MJ, Ebetino FH, Phipps
RJ and Coxon FP: Inhibition of protein prenylation by
bisphosphonates causes sustained activation of Rac, Cdc42, and Rho
GTPases. J Bone Miner Res. 21:684–694. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han J, Jiang DM, Du CQ and Hu SJ:
Alteration of enzyme expressions in mevalonate pathway: Possible
role for cardiovascular remodeling in spontaneously Hypertensive
Rats. Circ J. 75:1409–1417. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hall A: Small GTP-binding proteins and the
regulation of the actin cytoskeleton. Ann Rev Cell Biol. 10:31–54.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aikawa R, Komuro I, Yanazaki T, Zou Y,
Kudoh S, Zhu W, Kadowaki T and Yazaki Y: Rho family small G
proteins play critical roles in mechanical stress-induced
hypertrophic responses in cardiac myocytes. Circ Res. 84:485–466.
1999. View Article : Google Scholar
|
|
40
|
Bagrodia S, Dérijard B, Davis RJ and
Cerione RA: Cdc42 and PAK-mediated Signaling Leads to Jun kinase
and p38 mitogen-activated protein kinase activation. J Biol Chem.
270:27995–27998. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bagrodia S and Cerione RA: PAK to the
future. Trends Cell Biol. 9:350–355. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
King AJ, Sun H, Diaz B, Barnard D, Miao W,
Bagrodia S and Marshall MS: The protein kinase Pak3 positively
regulates Raf-1 activity through phosphorylation of serine 338.
Nature. 396:180–183. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Klemke RL, Cai S, Giannini AL, Gallagher
PJ, de Lanerolle P and Cheresh DA: Regulation of cell motility by
mitogen-activated protein kinase. J Cell Biol. 137:481–492. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hunter JJ, Tanaka N, Rockman HA, Ross J
and Chien KR: Ventricular expression of a MLC-2v-ras fusion gene
induces cardiac hypertrophy and selective diastolic dysfunction in
transgenic mice. J Biol Chem. 270:23173–23178. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Norton CE, Broughton BR, Jernigan NL,
Walker BR and Resta TC: Enhanced depolarization-induced pulmonary
vasoconstriction following chronic hypoxia requires EGFR-dependent
activation of NAD(P)H oxidase 2. Antioxid Redox Signal. 18:1777–88.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kameshima S, Kazama K, Okada M and
Yamawaki H: Eukaryotic elongation factor 2 kinase mediates
monocrotaline-induced pulmonary arterial hypertension via reactive
oxygen species-dependent vascular remodeling. Am J Physiol Heart
Circ Physiol. 308:H1298–H1305. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Manea A: NADPH oxidase-derived reactive
oxygen species: Involvement in vascular physiology and pathology.
Cell Tissue Res. 342:325–339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sutendra G, Dromparis P, Bonnet S, Haromy
A, McMurtry MS, Bleackley RC and Michelakis ED: Pyruvate
dehydrogenase inhibition by the inflammatory cytokine TNFalpha
contributes to the pathogenesis of pulmonary arterial hypertension.
J Mol Med (Berl). 89:771–783. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Aziz SM, Toborek M, Hennig B, Mattson MP,
Guo H and Lipke DW: Oxidative stress mediates monocrotaline-induced
alterations in tenascin expression in pulmonary artery endothelial
cells. Int J Biochem Cell Biol. 29:775–787. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen MJ, Chiang LY and Lai YL: Reactive
oxygen species and substance P in monocrotaline-induced pulmonary
hypertension. Toxicol Appl Pharmacol. 171:165–73. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Herbert JM, Bono F and Savi P: The
mitogenic effec of H2O2 for vascular smooth
muxcle cells is mediated by an increase of the affinity of basid
fibroblast growth factor for its receptor. FEBS Lett. 395:43–47.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xu W, Kaneko FT, Zheng S, Comhair SA,
Janocha AJ, Goggans T, Thunnissen FB, Farver C, Hazen SL, Jennings
C, et al: Increased arginase II and decreased NO synthesis in
endothelial cells of patients with pulmonary arterial hypertension.
FASEB J. 18:1746–1748. 2004.PubMed/NCBI
|
|
53
|
Sahara M, Sata M, Morita T, Hirata Y and
Nagai R: Nicorandil attenuates monocrotaline-induced vascular
endothelial damage and pulmonary arterial hypertension. PLoS One.
7:e333672012. View Article : Google Scholar : PubMed/NCBI
|