Introduction
Acute respiratory distress syndrome (ARDS) is acute
respiratory failure, which was the main cause of death among
critically ill patients, with a mortality rate as high as 40%
(1,2). ARDS has a close association with
sepsis in the intensive care unit (ICU). Sepsis is the most common
of ARDS (3), ~50% of ICU patients
with sepsis also have complications with ARDS (4). Additionally, clinical research
reveals that when sepsis-induced ARDS patients are compared with
non-sepsis ARDS patients, greater severity of the illness and
higher in-hospital mortality rates were observed (5). However, the mechanism of the sepsis
leading to the development of ARDS remains to be elucidated
(6).
Sepsis is the systematic response to infection;
excessive inflammation damages the lungs leading to ARDS. Previous
studies show inflammatory damage triggers a robust influx of
neutrophils and monocytes to the site of tissue injury (7), and the damaged or dead cells may
trigger the inflammasome-dependent responses, then alert the innate
immune system to the impending tissue damage, suggesting differing
roles for inflammasomes in the development of ARDS (8). The mitogen-activated protein kinase
(MAPK) signal transduction pathways of inflammatory cells modulate
a dysregulated, overly aggressive inflammatory response, which
promotes the development of sepsis (9). Inflammatory signals are initiated by
the recognition of inflammatory stimuli by specific transmembrane
and intracellular receptors, previous studies have found that
following inflammatory stimulation, some major MAPK subfamilies,
including extracellular signal-regulated kinase (ERK), p38 and Jun
N-terminal kinase (JNK) were activated (10), which induces the expression of
multiple genes that together regulate the inflammatory response
(11).
Through the phosphorylation of a range of downstream
substrates, different MAPKs have diverse roles in transmitting the
receptor-proximal signals to the transcriptional activation of
selected genes (12). Previous
studies found that MAPK pathways may provide drug targets in
inflammation to inhibit cytokine production (13,14).
Previous studies determined that in mice with cecal ligation and
puncture (CLP) the protein expression of Toll-like receptor 4
(TLR4), phosphorylated (p)-p38, p-JNK and p-ERK was increased,
whereas treatment with Compound 9a protected against septic injury
by suppressing MAPK-mediated inflammatory signaling (15). In a lipopolysaccharide
(LPS)-stimulated ARDS mouse model, some drugs or compounds
(including Decitabine, 5-azacitidine, Andrographolide sulfonate,
Hydroxy-Jolkinolide B-1 and Astilbin) alleviated LPS-induced ARDS
by suppressing LPS-induced activation of the MAPK signaling
pathways by blocking the phosphorylation of JNK, ERK and p38 in
lung tissues (16–19). Previous studies indicated that
glycyrrhizic acid and Losartan have a protective effect against
sepsis-induced acute lung injury by inhibiting the inflammatory
response, reducing damage from oxidative stress, and apoptosis via
inactivation of JNK and p38 MAPK (20,21).
The aforementioned studies indicated the effect of MAPK signaling,
particularly the JNK and p38 MAPK on sepsis-induced ARDS. The aim
of the current study was to identify the effect of MAPK signaling
on sepsis-induced acute lung injury in ARDS rats, to further
clarify the mechanism of sepsis leading to the development of
ARDS.
Materials and methods
Animals
A total of 72 adult male Sprague-Dawley (SD) rats
(6–8 weeks old; weight, 220–270 g) were obtained from Laboratorial
Animal Center of Shandong University (Jinan, China). All animals
were kept in a standard environment with ~23°C room temperature,
30–60% humidity and a 12-h light/dark cycle, allowed free access to
standard rodent chow and drink, and adapted to laboratory
conditions for a minimum of 3 days. The study protocols conformed
to the Guide for the Care and Use of Laboratory Animals (National
Institutes of Health, Bethesda, MD, USA) and were approved by the
Institutional Animal Care and Use Committee of Qingdao University
(Qingdao, China).
Rat model of sepsis-induced ARDS
(22)
All rats were anesthetized with 10% chloral hydrate
(300 mg/kg) by intra-peritoneal injection before surgical
procedures with 8 h preoperative fasting food. After a 2-cm
incision through midline, the cecum was carefully isolated and
ligated at distal to the ileocecal valve with a 4-0 silk suture to
avoid intestinal obstruction. The cecum was punctured twice with a
sterile 20-gauge needle and gently squeezed to extrude a small
amount of feces from the perforation sites. The quantity of
extruded feces was limited (small droplet, ~1 mm in diameter) and
consistent in all rats. Next, the abdominal cavity was closed in
two layers with continuous suture of 3–0 silk after the cecum was
returned. In the sham group, the abdomen was opened and the cecum
manipulated, but no cecal ligation or cecal puncture was performed
and the abdomen was closed. Following surgery each rat received 1
ml normal saline by subcutaneous injection for fluid resuscitation
and no antibiotics were administrated.
Experimental protocol
Rats were randomly divided into 6 groups according
to a random number table (n=12 for each group): i) Sham group
(group A); ii) ARDS group (group B), in which rats were challenged
with CLP to induce the ARDS model; iii) dimethyl sulfoxide (DMSO) +
ARDS group (group C), rats received an equal amount of 10% DMSO 4 h
before induction of ARDS (16);
iv) SP600125 + ARDS group (group D), rats received SP600125 (JNK
inhibitor) at 30 mg/kg 4 h before induction of ARDS, SP600125 was
dissolved in 10% DMSO; v) SB203580 + ARDS group (group E), rats
received SB203580 (p38 MAPK inhibitor) at 10 mg/kg 4 h before
induction of ARDS, SB203580 being dissolved in DMSO; and vi)
SP600125 + SB203580 + ARDS group (group F), rats received SP600125
(30 mg/kg) and SB203580 (10 mg/kg), 4 h before induction of ARDS.
DMSO (Amresco, LLC, Solon, OH, USA), SP600125 (Selleck Chemicals,
Houston, TX, USA) and SB203580 (Selleck Chemicals) were all
administered by intragastric injection, in group A and group B rats
received an equal amount of normal saline. Rats were sacrificed at
1, 6 or 24 h after CLP challenge, and samples were collected from
each rat for histological evaluation, lung water content (LWC) t
and biochemical analyses.
Western blot analysis
Total protein was extracted by using
radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime
Institute of Biotechnology, Haimen, China). Protein was separated
by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and electro-transferred to polyvinylidene fluoride
(PVDF) membranes (0.45 mm; EMD Millipore, Bedford, MA, USA). The
membranes were blocked in Tris-buffered saline (TBS; pH 7.4)
containing 0.1% Tween-20 (Shanghai Chemical Reagent Company of
China Pharmaceutical Group, Shanghai, China) and 5% bovine serum
albumin (Thermo Fisher Scientific, Inc, Waltham, MA, USA) for 1 h
at room temperature, then incubated at 4°C overnight with primary
antibodies for p-JNK (cat. no. sc-135642; 1:100), total JNK (cat.
no. sc-571; 1:100), p-p38 MAPK (cat. no. sc-101759; 1:100), total
p38 (cat. no. sc-7149; 1:100) and β-actin (cat. no. sc-130656;
1:500; all from Santa Cruz Biotechnology, Inc, Dallas, TX, USA).
Subsequently, membranes were incubated for 1 h at room temperature
with a goat anti-rabbit IgG-horseradish peroxidase (HRP)-conjugated
secondary antibody (cat. no. 93974; 1:10,000; OriGene Technologies,
Beijing, China). Immunoreactive bands were detected with Pierce ECL
western blotting substrate (Thermo Fisher Scientific, Inc.).
Hematoxylin and eosin (H&E)
staining and lung injury scoring
The right upper lobe of the lung was embedded in
paraffin (Thermo Fisher Scientific, Inc.) and sectioned at 5 µm.
The sections were stained with H&E (Beyotime Institute of
Biotechnology). The severity of lung injury was determined as
previously described (23). Lung
injury was graded from 0 (normal) to 4 (severe) for the following:
Edema, alveolar and interstitial inflammation, alveolar and
interstitial hemorrhage, atelectasis and hyaline membrane
formation. The total lung injury score per mouse was determined as
sum of the aforementioned scores. Two investigators blinded to the
experimental protocol analyzed ten randomly selected high-power
fields in each slide at a magnification ×400.
Evaluation of the LWC
The right lower lobe of the lung from the rats was
weighed immediately and subsequently dried at 80°C for 48 h to
calculate the wet/dry weight ratio (W/D). The LWC was calculated
using the following formula: LWC = (1 - W/D) ×100.
Cytokine detection
Blood samples (5 ml) were collected by cardiac
puncture and centrifuged at 1,500 × g for 5 min at 4°C. The
concentration of interleukin-6 (IL-6), IL-10, and tumor necrosis
factor-α (TNF-α) proteins in the supernatant were detected using a
commercial rat cytokine-specific enzyme-linked immunosorbent assay
(ELISA) kits (cat. nos. MM-0190R1, MM-0195R1 and MM-0180R1; Jingmei
Biotech, Beijing, China) following the manufacturer's protocol. All
samples were tested in duplicate.
Statistical analysis
The data were presented as the mean ± standard
deviation. Statistical analyses were performed using SPSS version
19.0 (IBM, Armonk, NY, USA). Comparisons among multiple groups were
performed using one-way ANOVA followed by a Bonferroni's post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Regulation of pulmonary JNK and p38
MAPK signalling after CLP challenge
The protein expression of total JNK, total p38 MAPK,
p-JNK and p38 MAPK (p-p38 MAPK) in lung tissues 1, 6, and 24 h
after CLP challenge were evaluated using western blotting analysis.
The p-JNK/total JNK and p-p38 MAPK/total p38 MAPK protein were
increased in the sepsis-induced lung injury group (group B)
compared with the shame control group (group A) (P<0.05). The
p-JNK/total JNK protein was downregulated in the groups D and F
compared with group B (P<0.05; Fig.
1A) and the p-p38 MAPK/total p38 MAPK protein was downregulated
in the group E and F compared with group B (P<0.05; Fig. 1B). The same results were found when
compared with the DMSO control (group C) (P<0.05; Fig. 1).
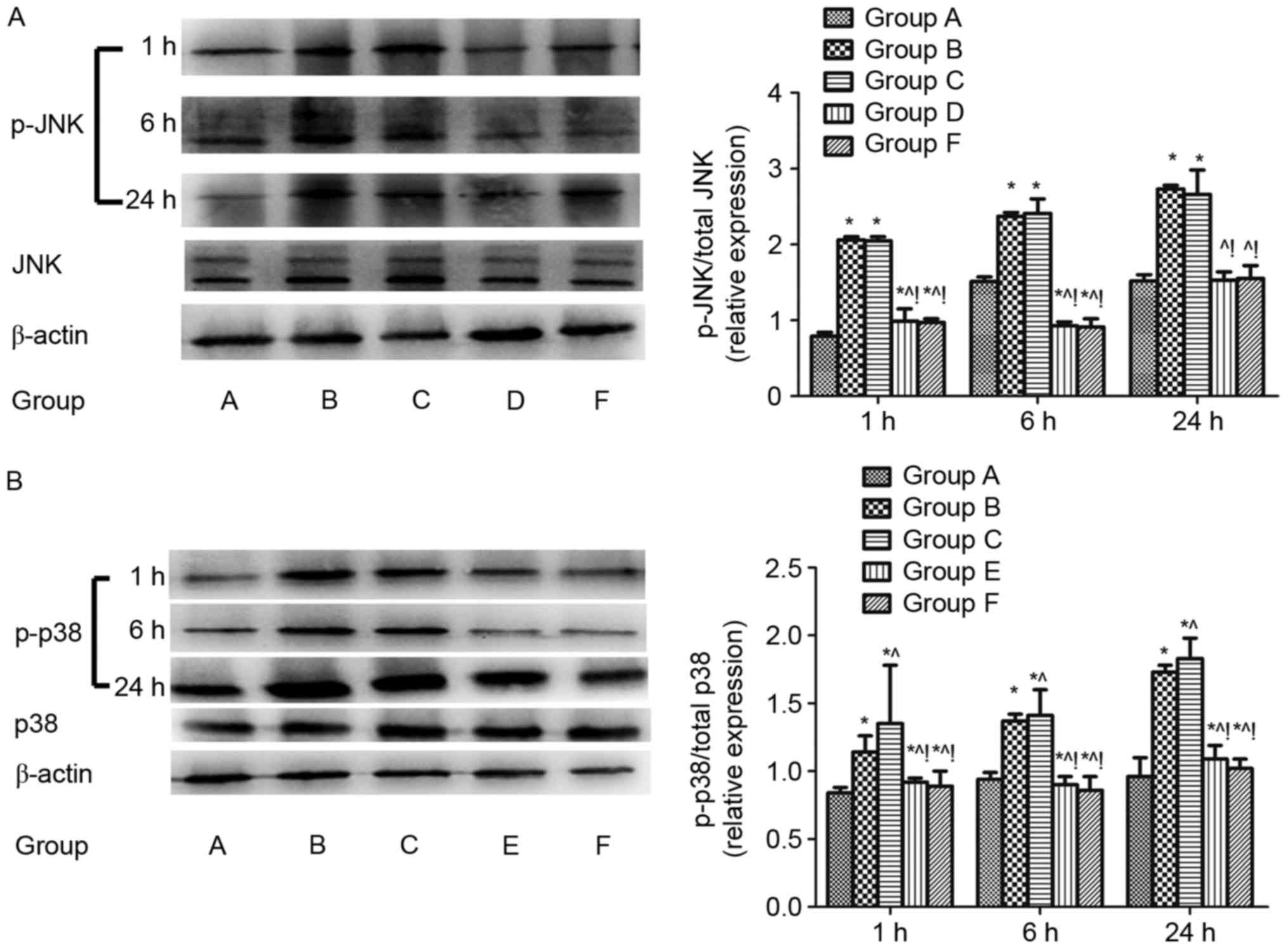 | Figure 1.Regulation of JNK and p38 MAPK
signalling in lungs after cecal ligation and puncture challenge.
The regulation of pulmonary JNK and p38 MAPK signalling were
assayed by ratio of (A) phosphorylated JNK/total JNK and (B)
phosphorylated p38/total p38 in the lung tissue 1, 6, and 24 h
after CLP challenge analyzed by western blotting. β-actin was used
as an internal control, and the results were expressed as mean ±
standard deviation. n=4 at each time point for each group.
*P<0.05 vs. group A, ^P<0.05 vs. B,
!P<0.05 vs. C, D, E and F. ARDS, acute respiratory
distress syndrome; MAPK, mitogen activated protein kinase; JNK, Jun
N-terminal kinase; A, sham; B, ARDS; C, DMSO + ARDS; D, SP600125 +
ARDS; E, SB203580 + ARDS; F, SP600125 + SB203580 + ARDS. |
Effect of MAPK signalling on pulmonary
histopathology of sepsis-induced acute lung injury rats. The
thickening of the alveolar wall, alveolar and interstitial
inflammatory cell infiltration, haemorrhaging, alveolar exudates
and the edema were increased in the lung tissue of rats after
sepsis-induced lung injury (group B), and the lung injury score for
quantification of the lung injury was also increased compared with
group A (P<0.05). However, these histopathological
characteristics and the lung injury score were alleviated at 1, 6,
and 24 h in the groups D, E and F compared with group B
(P<0.05). The same effect was found when compared with the DMSO
control (group C) (P<0.05; Fig.
2).
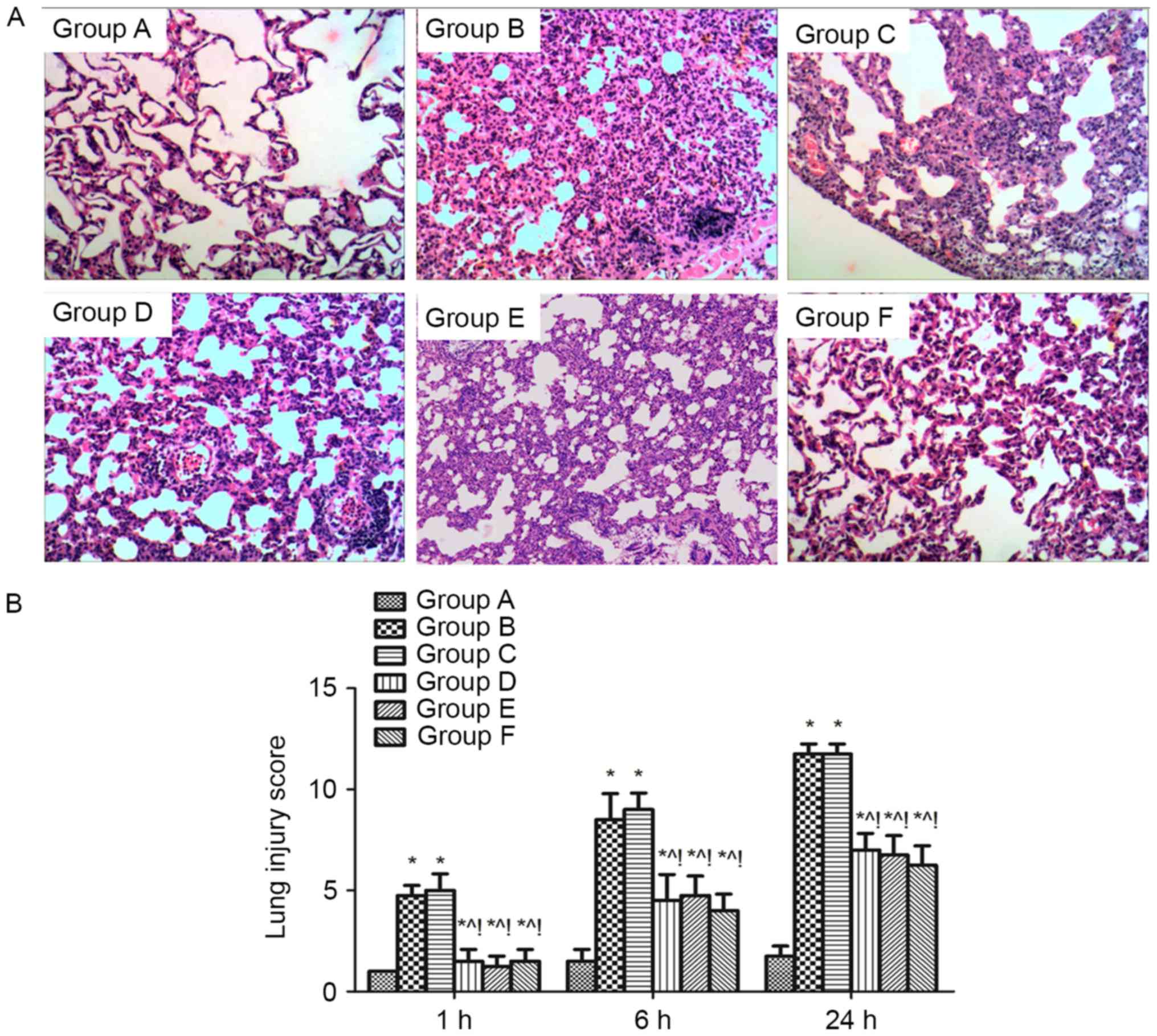 | Figure 2.Effect of MAPK signalling on the
histopathology of sepsis-induced lung injury. (A) Histopathological
analysis of lung tissues was performed at 1, 6 and 24 h after the
cecal ligation and puncture challenge. Magnification, ×100. (B)
Pathological lung injury scores were expressed as mean ± standard
deviation. The results showed a significant reduction in the
severity of lung injury in group D, E and F mice compared with
groups B and C. There was no difference between group B and C. n=4
at each time point for each group *P<0.05 vs. group A,
^P<0.05 vs. B, !P<0.05 vs. C, D, E, and
F. ARDS, acute respiratory distress syndrome; A, sham; B, ARDS; C,
DMSO + ARDS; D, SP600125 + ARDS; E, SB203580 + ARDS; F, SP600125 +
SB203580 + ARDS. |
Effect of MAPK signalling in sepsis-induced lung
permeability. The LWC was calculated to evaluate lung edema. LWC
was significantly reduced at 1, 6, and 24 h in the groups D, E and
F compared with group B (P<0.05). The same effect was found when
compared with the DMSO control (group C) (P<0.05; Fig. 3).
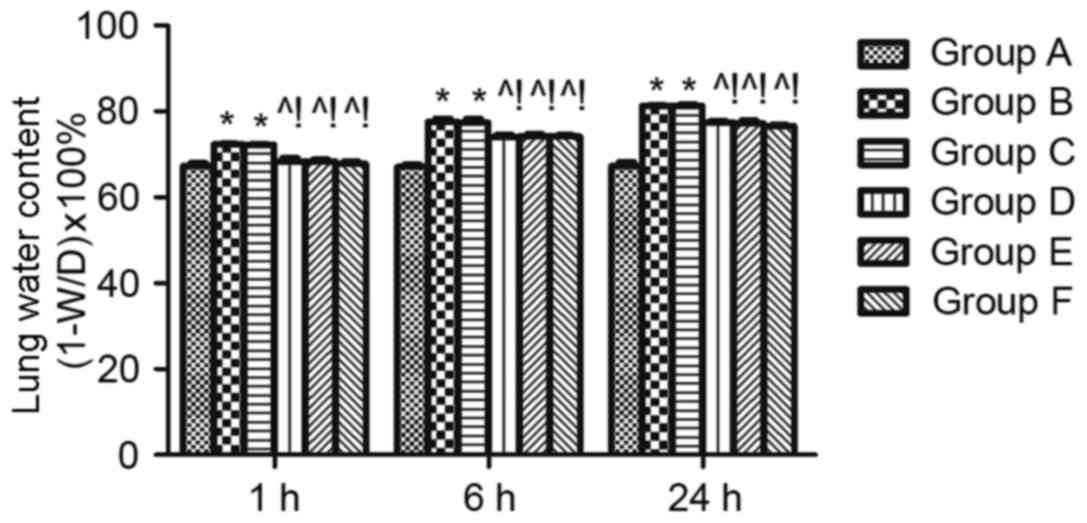 | Figure 3.Effect of mitogen activated protein
kinase signalling on sepsis-induced lung permeability. Lung edema
was measured using the lung water content. W/D indicates the ratio
of the lung wet weight to dry weight, which was detected at 1, 6
and 24 h after cecal ligation and puncture. n=4 at each time point
for each group. The results are expressed as the mean ± standard
deviation. *P<0.05 vs. group A, ^P<0.05 vs. B,
!P<0.05 vs. C, D, E, and F. ARDS, acute respiratory
distress syndrome; A, sham; B, ARDS; C, DMSO + ARDS; D, SP600125 +
ARDS; E, SB203580 + ARDS; F, SP600125 + SB203580 + ARDS. |
Effect of MAPK signalling on the serum levels of
inflammatory factors in sepsis-induced acute lung injury rats. The
levels of the pro-inflammatory cytokines IL-6 and TNF-α and the
anti-inflammatory cytokine IL-10 were measured in the serum of rats
1, 6, and 24 h after CLP challenge. Levels of all three cytokines
were significantly higher at 6, and 24 h when group B is compared
with group A, while IL-6 and TNF-α were increased at 1 h in group B
compared with group A (P<0.05; Fig.
4). IL-6 and TNF-α were reduced at 1, 6, and 24 h in the groups
D, E and F compared with group B (Fig.
4A and B), whereas IL-10 was increased (P<0.05; Fig. 4C). The same effects were found when
compared with the DMSO control (group C) (P<0.05; Fig. 4).
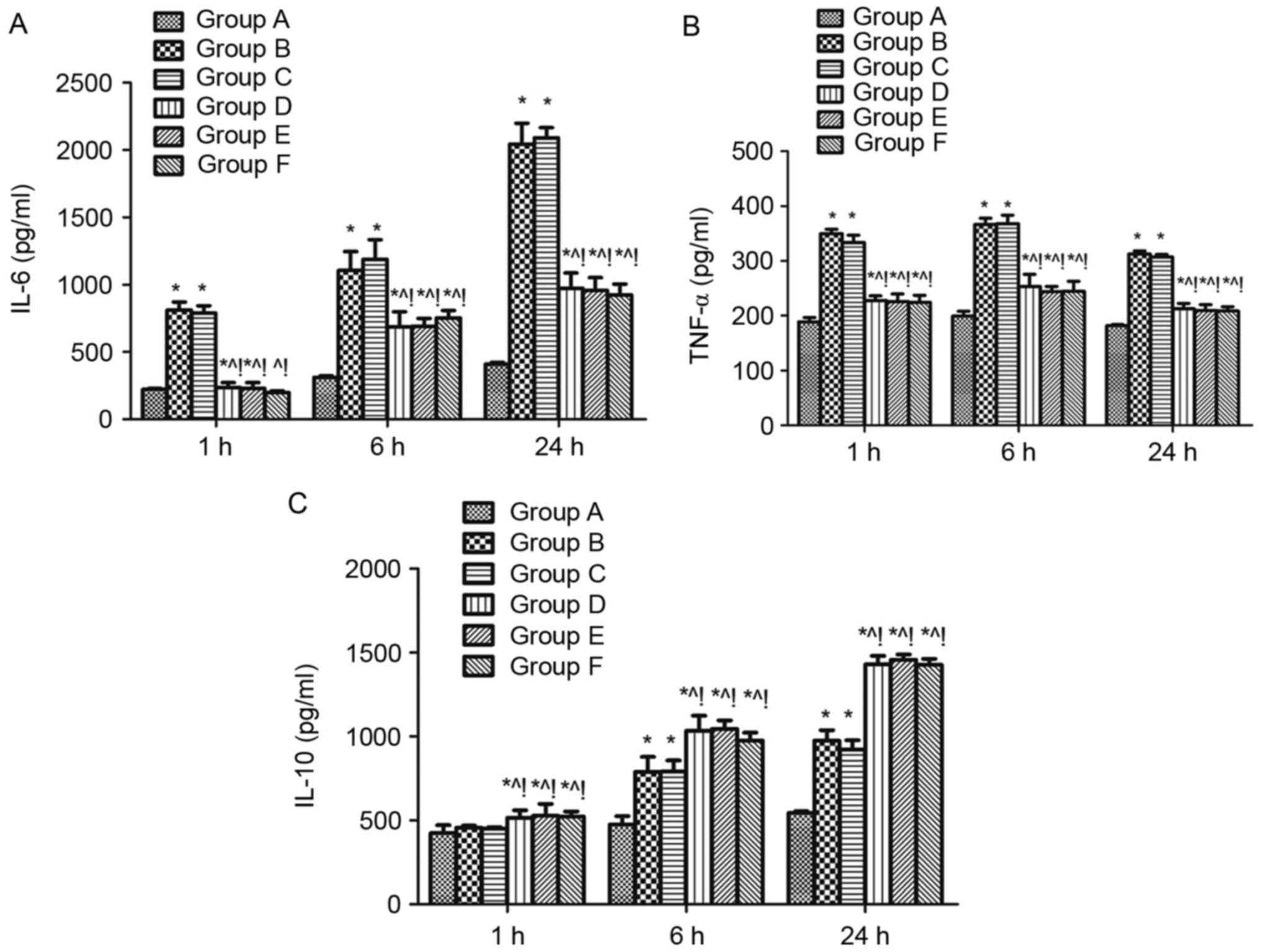 | Figure 4.Effect of mitogen activated protein
kinase signalling on the serum levels of inflammatory factors in
sepsis-induced acute lung injury rats. Levels of the
proinflammatory cytokines (A) IL-6, (B) TNF-α and anti-inflammatory
cytokine (C) IL-10 in serum of rats 1, 6, and 24 h after cecal
ligation and puncture challenge were measured using ELISA. n=4 at
each time point for each group. Data are expressed as the mean ±
standard deviation. *P<0.05 vs. group A, ^P<0.05
vs. B, !P<0.05 vs. C, D, E, and F. IL-6, −10,
interleukin-6, −10; TNF-α, tumor necrosis factor-α; ARDS, acute
respiratory distress syndrome; A, sham; B, ARDS; C, DMSO + ARDS; D,
SP600125 + ARDS; E, SB203580 + ARDS; F, SP600125 + SB203580 +
ARDS. |
Discussion
ARDS is regarded as part of a systemic inflammatory
response, particularly systemic sepsis (24), which is usually accompanied by
excessive inflammatory cell infiltration, cascade release of
inflammatory factors, and extravasation of protein-rich fluid
(25). Previous studies have
determined that MAPK signaling pathways may have an important
pathogenic role in the inflammatory process associated with
sepsis-induced ARDS (19,26,27).
As the current study demonstrated, ARDS triggered by sepsis after
CLP challenge may induce phosphorylation of JNK and p38 MAPK in the
lung tissue and modulation of MAPK signaling by JNK or/and p38
MAPK-specific inhibitor may significantly improve the pulmonary
histopathology and lung permeability, increasing the serum levels
of anti-inflammatory factors and reducing the serum levels of
pro-inflammatory factors in sepsis-induced acute lung injury
rats.
Some of MAPKs, including ERK1, ERK2, p38α, JNK1 and
JNK2 have been confirmed to be involved in innate immunity
(10), Germline-encoded pattern
recognition receptors (PRRs) recognize invariant microbial
components, termed pathogen-associated molecular patterns and then
stimulate the innate immune response after infection (28,29).
PRRs activate both MAPK and nuclear factor-κB (NF-κB) pathways then
activate the immune responses (30). Previous studies have revealed that
in sepsis-induced ARDS mice after CLP the phosphorylation of p38
MAPK, ERK, and JNK increased significantly in lung tissue (15,21).
As the links between inflammation and ARDS (13), another study found that SB203580, a
selective p38 inhibitor inhibited TNF production (14), and SP600125, a specific JNK
inhibitor, reduced CLP-induced activation of JNK and modulated the
early and late steps of the inflammatory cascade in a murine model
of CLP-induced sepsis (31).
According to these findings, the present study identified a robust
phosphorylation of JNK and p38 MAPK in lung tissue of
sepsis-induced ARDS rats after CLP challenge. The specific JNK or
p38MAPK inhibitor, SP600125 or SB203580 administered by
intragastric injection may significantly reduce the phosphorylation
of JNK and p38 MAPK in lung tissues. This suggested that in
sepsis-induced ARDS, the JNK and p38 MAPK signalling in lung tissue
were stimulated, which could be downregulated by oral
administration of their specific inhibitor.
Previous studies showed that inflammatory injuries
trigger a robust influx of neutrophils and monocytes to the site of
tissue injury (7) and the damaged
or dead cells are thought to trigger the inflammasome-dependent
responses, then alert the innate immune system to the impending
tissue damage (8). Previous
studies confirmed that the mechanism of some drugs, including
Decitabine, 5-azacitidine, Losartan and Andrographolide sulfonate,
protect lungs against injury induced by sepsis via the inhibition
of the phosphorylation of the ERK, JNK and p38 MAPK, which may
result in the suppression of the proinflammatory cytokine
expression (16,17,21).
Inhibition of the p38 MAPK and JNK, but not ERK could alleviate
inflammatory cell infiltration and microvascular permeability in
sepsis-induced ARDS mice (32). It
is possible that JNK and p38 MAPK have an important role in
sepsis-induced lung injury. In the current study, lung injury after
CLP increased the expression of phosphorylated JNK and p38MAPK in
lung tissue and administration of the JNK and p38 MAPK inhibitor
may be able to reduce the severity of lung injury in mice. The
beneficial effects of the JNK and p38MAPK inhibitor achieved in
this model confirmed that JNK and p38MAPK are essential for the
development of ARDS after sepsis and the potential therapeutic
effects of JNK and p38MAPK inhibitor in such pathological
conditions were elucidated.
A previous study determined that p38 MAPK activation
was essential for CXCR3-mediated endothelial cell apoptosis and was
associated with the increase of the leakage of protein-rich fluid
and inflammatory cells in ARDS-induced lung by CLP (33). Inhibition of LPS-induced activation
of JNK, ERK, and p38MAPK pathways in lung tissues may decrease the
indices of pulmonary edema, lung wet-to-dry weight ratios markedly
(19). Inhibition of the p38 MAPK
and JNK, but not ERK may alleviate inflammatory cell infiltration
and microvascular permeability in sepsis-induced ARDS mice
(32). All of these findings
suggested that the effect of JNK and p38 MAPK on sepsis-induced
lung permeability. The present study found that after JNK and p38
MAPK inhibitor administration the lung edema in ARDS rats induced
by sepsis after CLP was significantly improved, therefore it is
possible that JNK and p38 MAPK were involved the deterioration of
lung permeability following sepsis.
MAPKs also have an important role in inducing
cytokine production. It has been previously established that
inflammatory stimuli may lead to the activation of MAPK and the
transcription factor NF-κB, which mediates the expression of
several pro-inflammatory cytokines, including TNF-α, IL-1β, and
IL-6 which have an important role in many inflammatory disease
processes (34–37). Previous studies indicated that p38
MAPK inhibitors can suppress IL-6 and TNF-α expression in monocytes
and mast cells (38,39). Additional studies have suggested
roles for p38 MAPK in the in vitro production of
inflammatory factors, such as TNF-α (40), IL-6 (41), somewhat paradoxically, the
anti-inflammatory factor IL-10 (42,43).
A previous study determined that the suppression of the activation
of the JNK and p38 MAPK would significantly reduce TNF-α content in
the plasma of LPS-induced ARDS mice (44). Another previous study revealed that
berberine inhibited LPS-induced expression of proinflammatory genes
including IL-1β, IL-6 via suppression of the phosphorylation of
p38, JNK and ERK in peritoneal macrophages (45). Hesperidin downregulated the
LPS-induced expression of pro-inflammatory cytokines, including
TNF-α, IL-1β, IL-6 and enhanced the production of anti-inflammatory
IL-10, IL-4, IL-12, which may be controlled by JNK and p38 MAPK
pathways (46). Glutamine
treatment inhibited phosphorylation of p38 MAPK and ERK pathways
critical for cytokine release, meanwhile, significantly attenuated
TNF-α and IL-6 after CLP (47). A
previous study found that the regulation of TLR4-mediated induction
of TNF production is ERK1 and ERK2 independent (48). In the present study, the JNK and
p38 MAPK inhibitor was able to significantly reduce the
pro-inflammatory cytokines IL-6 and TNF-α. Meanwhile, an increase
the anti-inflammatory cytokine IL-10 ws observed in the serum of
rats after CLP challenge. It provides the direct evidence that the
JNK and p38 MAPK have an important role in the system inflammation
response induced by CLP in rats. This confirmed that JNK and p38
MAPK may be essential for the development of acute lung injury
induced by sepsis.
It is of note noted that the present study did not
identify a difference between the group D or E (single
administration of SB203580 or SP600125) and the group F (combined
application of SB203580 and SP600125) on pulmonary histopathology,
lung permeability and the serum levels of inflammatory factors.
This suggested that there may be a common downstream pathway
between JNK and p38 MAPK and further investigation is required to
confirm this.
In conclusion, JNK and p38 MAPK inhibitor improved
the lung permeability, attenuated system inflammation and
alleviated the lung injury induced by sepsis. JNK and p38 MAPK
signaling are essential for the development of ARDS following
sepsis, further investigations are required to elucidate the
detailed mechanisms of JNK and p38 MAPK signaling in sepsis-induced
ARDS.
Acknowledgements
The current study was supported by the Youth
Scientific Research Foundation of the Affiliated Hospital of
Qingdao University (grant no. 2384).
Glossary
Abbreviations
Abbreviations:
|
ARDS
|
acute respiratory distress
syndrome
|
|
MAPK
|
mitogen-activated protein kinase
|
|
JNK
|
Jun N-terminal kinase
|
|
CLP
|
cecal ligation and puncture
|
|
ICU
|
intensive care unit
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
TLR4
|
Toll-like receptor 4
|
|
SD
|
Sprague-Dawley
|
|
DMSO
|
dimethyl sulfoxide
|
|
LWC
|
lung water content
|
|
RIPA
|
radioimmuneprecipitation assay
|
|
SDS-PAGE
|
sodium dodecyl sulfate-polyacrylamide
gel electrophoresis
|
|
PVDF
|
polyvinylidene fluoride
|
|
TBS
|
Tris-buffered saline
|
|
HRP
|
horseradish peroxidase
|
|
H&E
|
hematoxylin and eosin
|
|
W/D
|
wet/dry weight ratio
|
|
IL-6
|
interleukin-6
|
|
IL-10
|
interleukin-10
|
|
TNF-α
|
tumor necrosis factor-α
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
References
|
1
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goss CH, Brower RG, Hudson LD and
Rubenfeld GD; ARDS Network, : Incidence of acute lung injury in the
United States. Crit Care Med. 31:1607–1611. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hudson LD, Milberg JA, Anardi D and
Maunder RJ: Clinical risks for development of the acute respiratory
distress syndrome. Am J Respir Crit Care Med. 151:293–301. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sevransky JE, Martin GS, Shanholtz C,
Mendez-Tellez PA, Pronovost P, Brower R and Needham DM: Mortality
in sepsis versus non-sepsis induced acute lung injury. Crit Care.
13:R1502009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
de Luis Cabezon N, Sánchez Castro I,
Uriarte UX Bengoetxea, Casanova MP Rodrigo, García Peña JM and
Celorrio L Aguilera: Acute respiratory distress syndrome: A review
of the Berlin definition. Rev Esp Anestesiol Reanim. 61:319–327.
2014.(In Spanish). PubMed/NCBI
|
|
7
|
Kono H and Rock KL: How dying cells alert
the immune system to danger. Nat Rev Immunol. 8:279–289. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Iyer SS, Pulskens WP, Sadler JJ, Butter
LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA,
Leemans JC and Sutterwala FS: Necrotic cells trigger a sterile
inflammatory response through the Nlrp3 inflammasome. Proc Natl
Acad Sci USA. 106:pp. 20388–932. 2009; View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Strassheim D, Park JS and Abraham E:
Sepsis: Current concepts in intracellular signaling. Int J Biochem
Cell Biol. 34:1527–1533. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arthur JS and Ley LC: Mitogen-activated
protein kinases in innate immunity. Nat Rev Immunol. 13:679–692.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kyriakis JM and Avruch J: Mammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammation. Physiol Rev. 81:807–869.
2001.PubMed/NCBI
|
|
12
|
Ahmed AU, Williams BR and Hannigan GE:
Transcriptional Activation of Inflammatory Genes: Mechanistic
insight into selectivity and diversity. Biomolecules. 5:3087–3111.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cohen P: Targeting protein kinases for the
development of anti-inflammatory drugs. Curr Opin Cell Biol.
21:317–324. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dar AC and Shokat KM: The evolution of
protein kinase inhibitors from antagonists to agonists of cellular
signaling. Annu Rev Biochem. 80:769–795. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim SJ, Baek KS, Park HJ, Jung YH and Lee
SM: Compound 9a, a novel synthetic histone deacetylase inhibitor,
protects against septic injury in mice by suppressing MAPK
signalling. Br J Pharmacol. 173:1045–1057. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang X, Kong G, Li Y, Zhu W, Xu H, Zhang
X, Li J, Wang L, Zhang Z, Wu Y, et al: Decitabine and 5-azacitidine
both alleviate LPS induced ARDS through
anti-inflammatory/antioxidant activity and protection of glycocalyx
and inhibition of MAPK pathways in mice. Biomed Pharmacother.
84:447–453. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peng S, Hang N, Liu W, Guo W, Jiang C,
Yang X, Xu Q and Sun Y: Andrographolide sulfonate ameliorates
lipopolysaccharide-induced acute lung injury in mice by
down-regulating MAPK and NF-κB pathways. Acta Pharm Sin B.
6:205–211. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu X, Liu N, Zhang YX, Cao J, Wu D, Peng
Q, Wang HB and Sun WC: The protective effects of HJB-1, a
derivative of 17-Hydroxy-Jolkinolide B, on LPS-induced acute
distress respiratory syndrome mice. Molecules. 21:772016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kong G, Huang X, Wang L, Li Y, Sun T, Han
S, Zhu W, Ma M, Xu H, Li J, et al: Astilbin alleviates LPS-induced
ARDS by suppressing MAPK signaling pathway and protecting pulmonary
endothelial glycocalyx. Int Immunopharmacol. 36:51–58. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao H, Zhao M, Wang Y, Li F and Zhang Z:
Glycyrrhizic acid prevents sepsis-induced acute lung injury and
mortality in rats. J Histochem Cytochem. 64:125–137. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shen L, Mo H, Cai L, Kong T, Zheng W, Ye
J, Qi J and Xiao Z: Losartan prevents sepsis-induced acute lung
injury and decreases activation of nuclear factor kappaB and
mitogen-activated protein kinases. Shock. 31:500–506. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Otero-Antón E, González-Quintela A,
López-Soto A, López-Ben S, Llovo J and Pérez LF: Cecal ligation and
puncture as a model of sepsis in the rat: Influence of the puncture
size on mortality, bacteremia, endotoxemia and tumor necrosis
factor alpha levels. Eur Surg Res. 33:77–79. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mrozek JD, Smith KM, Bing DR, Meyers PA,
Simonton SC, Connett JE and Mammel MC: Exogenous surfactant and
partial liquid ventilation: Physiologic and pathologic effects. Am
J Respir Crit Care Med. 156:1058–1065. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dolinay T, Kim YS, Howrylak J, Hunninghake
GM, An CH, Fredenburgh L, Massaro AF, Rogers A, Gazourian L,
Nakahira K, et al: Inflammasome-regulated cytokines are critical
mediators of acute lung injury. Am J Respir Crit Care Med.
185:1225–1234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matthay MA, Zimmerman GA, Esmon C,
Bhattacharya J, Coller B, Doerschuk CM, Floros J, Gimbrone MA Jr,
Hoffman E, Hubmayr RD, et al: Future research directions in acute
lung injury: summary of a National Heart, Lung and Blood Institute
working group. Am J Respir Crit Care Med. 167:1027–1035. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu H, Zhao G, Jiang K, Chen X, Zhu Z, Qiu
C, Li C and Deng G: Plantamajoside ameliorates
lipopolysaccharide-induced acute lung injury via suppressing NF-κB
and MAPK activation. Int Immunopharmacol. 35:315–322. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee JH, Ko HJ, Woo ER, Lee SK, Moon BS,
Lee CW, Mandava S, Samala M, Lee J and Kim HP: Moracin M inhibits
airway inflammation by interrupting the JNK/c-Jun and NF-κB
pathways in vitro and in vivo. Eur J Pharmacol. 783:64–72. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Newton K and Dixit VM: Signaling in innate
immunity and inflammation. Cold Spring Harb Perspect Biol. 4:pii:
a0060492012. View Article : Google Scholar
|
|
29
|
Kawai T and Akira S: The role of
pattern-recognition receptors in innate immunity: Update on
Toll-like receptors. Nat Immunol. 11:373–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Medzhitov R and Horng T: Transcriptional
control of the inflammatory response. Nat Rev Immunol. 9:692–703.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pizzino G, Bitto A, Pallio G, Irrera N,
Galfo F, Interdonato M, Mecchio A, De Luca F, Minutoli L, Squadrito
F and Altavilla D: Blockade of the JNK signalling as a rational
therapeutic approach to modulate the early and late steps of the
inflammatory cascade in polymicrobial sepsis. Mediators Inflamm.
2015:5915722015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou H, Bian D, Jiao X, Wei Z, Zhang H,
Xia Y, He Y and Dai Y: Paeoniflorin protects against
lipopolysaccharide-induced acute lung injury in mice by alleviating
inflammatory cell infiltration and microvascular permeability.
Inflamm Res. 60:981–990. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu X, Zou Y, Wang B, Zhu J, Chen Y, Wang
L, Li J and Deng X: Blockade of CXC chemokine receptor 3 on
endothelial cells protects against sepsis-induced acute lung
injury. J Surg Res. 204:288–296. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rahman A and Fazal F: Blocking NF-κB: An
inflammatory issue. Proc Am Thorac Soc. 8:pp. 497–503. 2011;
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
McKenna S and Wright CJ: Inhibiting
IκBβ-NFκB signaling attenuates the expression of select
pro-inflammatory genes. J Cell Sci. 128:2143–2155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee IT and Yang CM: Inflammatory
signalings involved in airway and pulmonary diseases. Mediators
Inflamm. 2013:7912312013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lai JL, Liu YH, Liu C, Qi MP, Liu RN, Zhu
XF, Zhou QG, Chen YY, Guo AZ and Hu CM: Indirubin Inhibits
LPS-induced inflammation via TLR4 abrogation mediated by the NF-kB
and MAPK signaling pathways. Inflammation. 40:1–12. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guo X, Gerl RE and Schrader JW: Defining
the involvement of p38alpha MAPK in the production of anti- and
proinflammatory cytokines using an SB 203580-resistant form of the
kinase. J Biol Chem. 278:22237–22242. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jeong HJ, Na HJ, Hong SH and Kim HM:
Inhibition of the stem cell factor-induced migration of mast cells
by dexamethasone. Endocrinology. 144:4080–4086. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schafer PH, Wang L, Wadsworth SA, Davis JE
and Siekierka JJ: T cell activation signals up-regulate p38
mitogen-activated protein kinase activity and induce TNF-alpha
production in a manner distinct from LPS activation of monocytes. J
Immunol. 162:659–568. 1999.PubMed/NCBI
|
|
41
|
Beyaert R, Cuenda A, Berghe W Vanden,
Plaisance S, Lee JC, Haegeman G, Cohen P and Fiers W: The p38/RK
mitogen-activated protein kinase pathway regulates interleukin-6
synthesis response to tumor necrosis factor. EMBO J. 15:1914–1923.
1996.PubMed/NCBI
|
|
42
|
Foey AD, Parry SL, Williams LM, Feldmann
M, Foxwell BM and Brennan FM: Regulation of monocyte IL-10
synthesis by endogenous IL-1 and TNF-alpha: Role of the p38 and
p42/44 mitogen-activated protein kinases. J Immunol. 160:920–928.
1998.PubMed/NCBI
|
|
43
|
Koprak S, Staruch MJ and Dumont FJ: A
specific inhibitor of the p38 mitogen activated protein kinase
affects differentially the production of various cytokines by
activated human T cells: Dependence on CD28 signaling and
preferential inhibition of IL-10 production. Cell Immunol.
192:87–95. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yu X, Yu S, Chen L, Liu H, Zhang J, Ge H,
Zhang Y, Yu B and Kou J: Tetrahydroberberrubine attenuates
lipopolysaccharide-induced acute lung injury by down-regulating
MAPK, AKT, and NF-κB signaling pathways. Biomed Pharmacother.
82:489–497. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jeong HW, Hsu KC, Lee JW, Ham M, Huh JY,
Shin HJ, Kim WS and Kim JB: Berberine suppresses proinflammatory
responses through AMPK activation in macrophages. Am J Physiol
Endocrinol Metab. 296:E955–E964. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yeh CC, Kao SJ, Lin CC, Wang SD, Liu CJ
and Kao ST: The immunomodulation of endotoxin-induced acute lung
injury by hesperidin in vivo and in vitro. Life Sci. 80:1821–1831.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Singleton KD, Beckey VE and Wischmeyer PE:
Glutamine prevents activation of NF-kappaB and stress kinase
pathways, attenuates inflammatory cytokine release, and prevents
acute respiratory distress syndrome (ARDS) following sepsis. Shock.
24:583–589. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang HT, Papoutsopoulou S, Belich M,
Brender C, Janzen J, Gantke T, Handley M and Ley SC: Coordinate
regulation of TPL-2 and NF-κB signaling in macrophages by NF-κB1
p105. Mol Cell Biol. 32:3438–3451. 2012. View Article : Google Scholar : PubMed/NCBI
|














