Introduction
Previously, only a small fraction of all bacteria
had been isolated and characterized, and analysis of any bacterial
community was severely limited by the available technology and the
shortage of reference genomes (1,2). The
development of next generation sequencing provided an excellent
platform to explore the association between humans and microbiota
(3–6).
The human body harbors bacterial, viral and
eukaryotic communities in the skin, nasopharynx, oral cavity,
respiratory tract, gastrointestinal tract and female reproductive
tract (7–10), and the microbes have profound
implications on humanmetabolism, immunity and the gut-brain axis
(3–6). As it has been demonstrated, the human
intestine exists in symbiosis with hundreds of trillions of
microbes, and there is an increasing awareness that the bacteria
residing within the gut has a significant influence on host health
(7). In addition, microbial
dysbiosis caused by the altered intestinal microbes has been proved
to contribute to the onset of several disorders, and it is
important to identify the key bacteria that serve a direct or
indirect effect on human health.
Previously, analysis of microbial diversity in
specific environments was severely limited by available
technologies and referenced genomes. Technological advances in next
generation sequencing have enabled the elucidation of the
pleiotropic effects of microorganisms on the human host, and the
high-throughput sequencing can detect almost all the DNA signatures
of microbes within specific environments, even those bacteria
present in low numbers (2).
However, a number of studies have only focused on the examination
of microbial diversity using high-throughput sequencing technology,
which provided little information on the potential and limitations
of this approach in microbial ecology studies (11–13).
Sample handling, DNA extraction, amplification
efficiency, run processing and downstream analyses have seriously
affected the generation of high quality data (2,14–17).
Therefore, in the present study, a number of known bacteria were
mixed together at a certain percentage, and extracted DNAs were
subjected high throughput sequencing to evaluate the accuracy and
sensitivity of high-throughput sequencing technology, providing
basic data to help researchers to better investigate the
relationship between microorganisms and host health.
Materials and methods
Bacterial activation and culture
A total of seven common bacterial species, including
Escherichia coli (no. 44102; isolated from the donated human
feces to screen the bacteria in human intestines, and stored in the
authors' lab), Lactobacillus plantarum (no. HM218749;
isolated from sourdough and stored in the author's lab),
Streptococcus thermophilus [no. 19258; American Type Culture
Collection (ATCC), Manassas, VA, USA], Bifidobacterium
bifidum (no. WBIN03; isolated from yogurt and stored in the
authors' lab), Bacillus subtilis (no. 14416; ATCC),
Enterococcus faecalis (no. HM218543; isolated from the
donated human feces to screen the bacteria in human intestines, and
stored in the authors' lab) and Salmonella typhimurium (no.
14028; ATCC), were selected in the present studyand divided into
five groups (Table I). All
bacteria were propagated in corresponding media three times before
DNA extraction (Table I).
 | Table I.Bacterial composition in groups A, B,
C, D and E. |
Table I.
Bacterial composition in groups A, B,
C, D and E.
|
| Group, CFU/ml |
|---|
|
|
|
|---|
| Bacteria | A | B | C | D | E |
|---|
| E. coli
(44102) |
1×106 |
1×104 |
1×103 |
1×102 | 1×10 |
| L. plantarum
(HM218749) |
1×106 |
1×104 |
1×103 |
1×102 | 1×10 |
| S.
thermophilus (ATCC 19258) |
1×106 |
1×106 |
1×106 |
1×106 |
1×106 |
| B. bifidum
(WBIN03) |
1×106 |
1×106 |
1×106 |
1×106 |
1×106 |
| B. subtilis
(ATCC 14416) |
1×106 |
1×106 |
1×106 |
1×106 |
1×106 |
| E. faecalis
(HM218543) |
1×106 |
1×106 |
1×106 |
1×106 |
1×106 |
| S.
typhimurium (ATCC 14028) |
1×106 |
1×106 |
1×106 |
1×106 |
1×106 |
Extraction of genomic DNA and
high-throughput sequencing
Genomic DNA from each sample was extracted using a
TIANamp Genomic DNA kit (Tiangen Biotech Co., Ltd., Beijing, China)
combined with bead beating, as previously described (3). Subsequently, the genomic DNA was sent
to a high-throughput sequencing company (Biomarker Technologies
Corporation, Beijing, China) for high-throughput sequencing and
analysis.
The extracted genomic DNAs extracted from these
seven common bacteria were used as the templates, and the universal
primer pair 338F/806R with the respective barcode (supplied by
BiomarkerTechnologiesCorporation, Beijing, China) for ease of
identification (Table II) were
used to to amplify the V3-V4 region of 16S ribosomal (r)RNA genes
of all samples. Polymerase chain reaction (PCR), pyrosequencing of
the PCR amplicons and quality control of raw data were performed as
described previously with minor modifications as presented in the
‘Bioinformatics and multivariate statistics’ section below
(18).
| Table II.Primer barcodes. |
Table II.
Primer barcodes.
| No. | Sequence |
|---|
| A1 |
AGGGTCAATGAACCTT |
| A2 |
AGGGTCAAAGTCAACA |
| A3 |
AGGGTCAACTCTCTAT |
| B1 |
AGGGTCAAAGAGTAGA |
| B2 |
AGGGTCAAGTAAGGAG |
| B3 |
AGGGTCAAACTGCATA |
| C1 |
AGGGTCAAAAGGAGTA |
| C2 |
AGGGTCAACTAAGCCT |
| C3 |
AGGAGTGGTGAACCTT |
| D1 |
AGGAGTGGAGTCAACA |
| D2 |
AGGAGTGGCTCTCTAT |
| D3 |
AGGAGTGGAGAGTAGA |
| E1 |
AGGAGTGGGTAAGGAG |
| E2 |
AGGAGTGGACTGCATA |
| E3 |
AGGAGTGGAAGGAGTA |
Bioinformatics and multivariate
statistics
Low-quality sequences were eliminated from the
analysis based on the following criteria: i) Raw reads shorter than
400 bp; ii) a sequence producing >8 homopolymers; iii) >2
mismatches in the primers; and iv) 1 or more mismatches in the
barcode. Pyrosequenced amplicons were removed using the PyroNoise
algorithm in Mothur (version 1.33.3) (19). Bioinformatics analysis was
implemented using the Quantitative Insights Into Microbial Ecology
(QIIME) platform (version 1.8.0) (20). Briefly, 16S rRNA operational
taxonomic units (OTUs) were clustered using an open-reference OTU
picking protocol based on 97% nucleotide similarity with the UCLUST
algorithm (21). ChimeraSlayer was
employed to remove chimeric sequences (22). The relative abundance of each OTU
was determined as a proportion of the sum of sequences for each
sample. Taxonomic relative abundance profiles (including at the
phylum, class, order, family and genus levels) were generated based
on OTU annotation. The microbial community structure (i.e., species
richness, evenness and between-sample diversity) of bacterial
samples was estimated by biodiversity. Shannon index, phylogenetic
diversity, chao1 index and the observed number of species were used
to evaluate α diversity, and the weighted and unweighted UniFrac
distances were used to evaluate β diversity.
All of these indices (α and β diversity) were
calculated via the QIIME pipeline.
Statistical analysis
Statistical analysis was implemented using the R
platform. Principal coordinate analysis (PCoA) was performed using
the ‘ape’ package based on the UniFrac distances between samples.
The difference among groups was further assessed using analysis of
similarities and multi-response permutation planning methodswith
Metastats software (http://metastats.cbcb.umd.edu/) as described
previously, and statistical significance was set at P<0.05 for
correction of multiple comparisons (23).
Results
Sequencing coverage
To compare the microbes in each sample, 16S rRNA
amplicon sequencing analysis was used to sequence the V3-V4
hypervariable region, the sequencing data was filtered to obtain
the valid data, and the effective tags of all samples were
clustered, and those sequences with >97% similarity were
considered to beone OTU. In total, 2,032,484 filtered clean tags
(135,498.9 tags/sample) and 1,840 OTUs were obtained from all the
samples with an average of 122.7 OTUs per group (Table III). The chao1 index was almost
saturated and the rarefaction curve of every sample could enter the
plateau phase (data not shown).
| Table III.Number of raw tags, clean tags,
average bp, OTUs and actual bacterial composition in groups A, B,
C, D and E by high-throughput sequencing. |
Table III.
Number of raw tags, clean tags,
average bp, OTUs and actual bacterial composition in groups A, B,
C, D and E by high-throughput sequencing.
| Sample ID | Raw tags | Clean tags | Average length,
bp | OTU | Actual bacterial
number |
|---|
| A1 | 180,452 | 163,786 | 467 | 120 | 7 |
| A2 | 167,355 | 147,971 | 467 | 82 | 7 |
| A3 | 159,440 | 144,734 | 467 | 120 | 7 |
| B1 | 135,586 | 118,439 | 467 | 131 | 7 |
| B2 | 88,832 |
71,781 | 469 | 84 | 7 |
| B3 | 134,956 | 117,370 | 468 | 136 | 7 |
| C1 | 139,230 | 123,136 | 468 | 122 | 7 |
| C2 | 163,421 | 144,130 | 467 | 130 | 7 |
| C3 | 188,147 | 167,154 | 468 | 131 | 7 |
| D1 | 187,451 | 162,980 | 468 | 132 | 7 |
| D2 | 139,141 | 120,711 | 468 | 128 | 7 |
| D3 | 147,408 | 129,402 | 467 | 129 | 7 |
| E1 | 147,574 | 125,254 | 468 | 135 | 7 |
| E2 | 166,163 | 147,835 | 468 | 131 | 7 |
| E3 | 165,512 | 147,801 | 468 | 129 | 7 |
| Average |
154,044.5 |
135,498.9 |
467.7 |
122.7 | 7 |
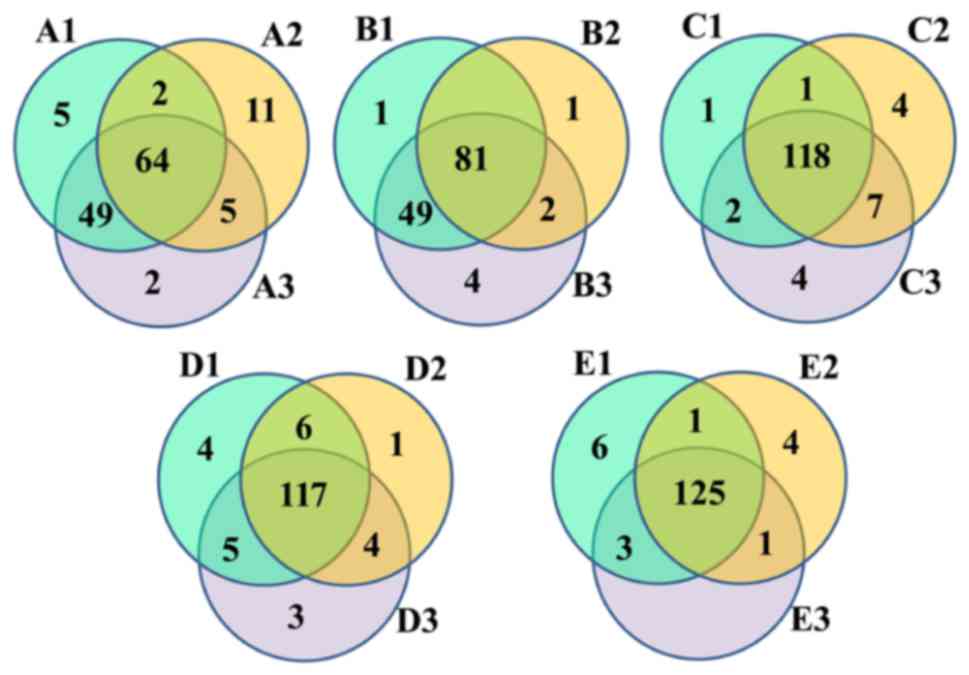
Shared genera in each sample
The Venn diagrams reflected the differences between
all groups. As presented in Fig.
1, there were 138 (A), 138 (B), 137 (C), 140 (D) and 140 (E)
OTUs in each group, and the percentage of common OTUs was 46.38
(64/138), 58.70 (81/138), 86.13 (118/137), 83.57 (117/140) and
89.29% (125/140), respectively.
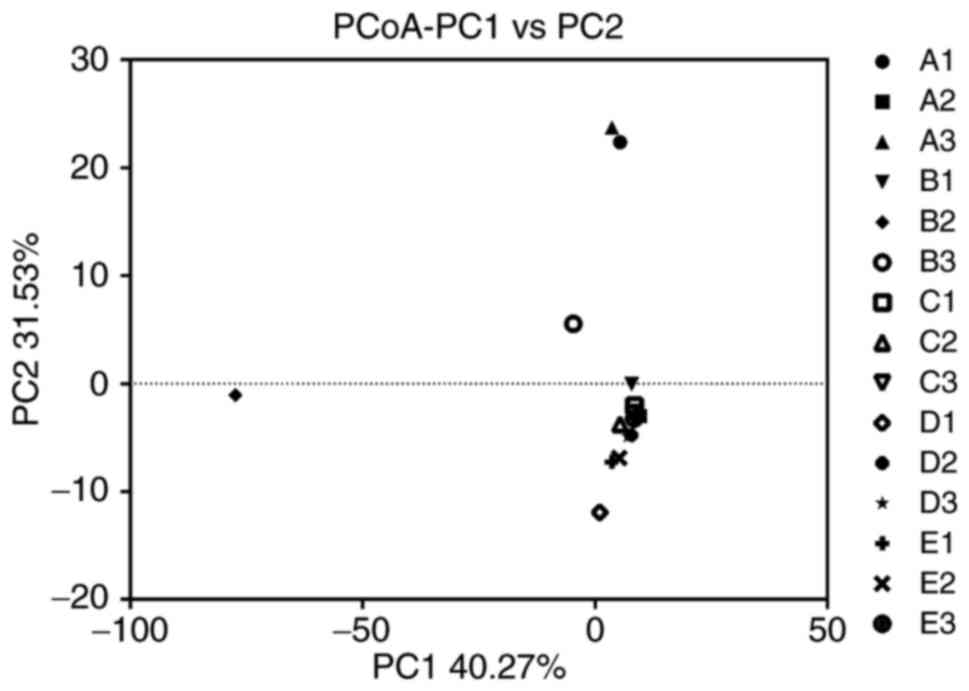
β diversity of the microbial
community
The overall graph of the microbial composition of
the samples was obtained by using PCoA, based on the relative
abundance profiles of bacterial taxa. As presentedin Fig. 2, the samples A1 and A3 clustered
together on the right upper corner of the coordinate axis, samples
A2, B1, B3, C1, C2, C3, D1, D2, D3, E1, E2 and E3 gathered together
on the lower right corner of the coordinate axis and sample B2
scattered on the bottom left corner.
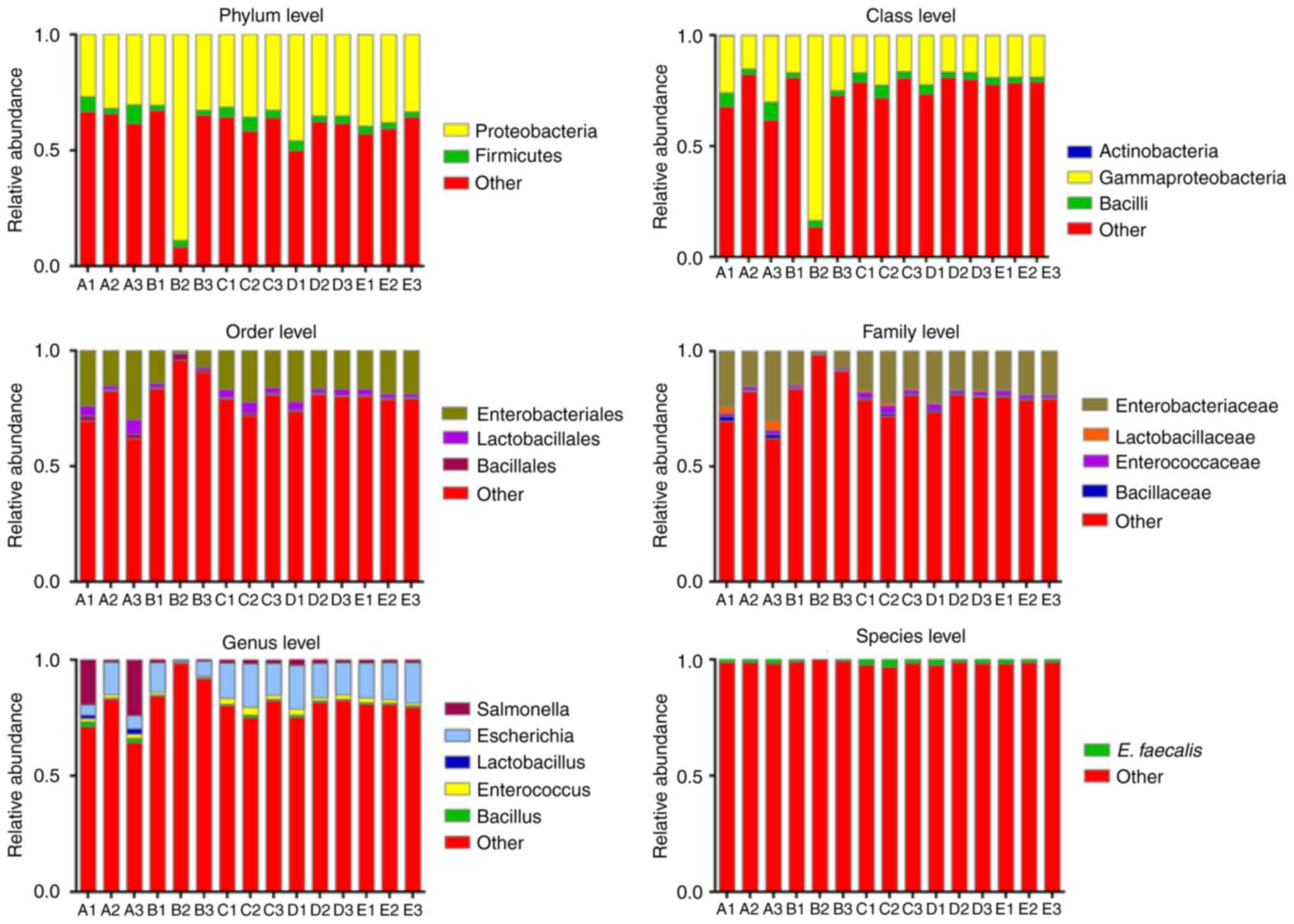
Scientific classification of bacterial
communities in each sample
To further investigate the relative abundance of the
known bacteria in each group, the identified bacterial abundance
was compared at the phylum, class, order, family, genus and species
levels. Fig. 3 demonstrated that
all the known bacteria were detected at the phylumand class levels,
although their percentages only occupied 41.66 and 28.10% of the
total bacteria, respectively. In addition, certain bacteria, for
example, Bifidobacteriales at the order level,
Streptococcaceae and Bifidobacteriaceae at the family
level, Streptococcus and Bifidobacterium at the genus
level, and E. coli, L. plantarum, S.
thermophilus, B. bifidum, B. subtilis and S.
typhimurium at the species level, failed to be detected, and
the identified bacteria only occupied 20.84, 20.98, 19.31 and 1.62%
at the order, family, genus and species levels, respectively
(Fig. 3).
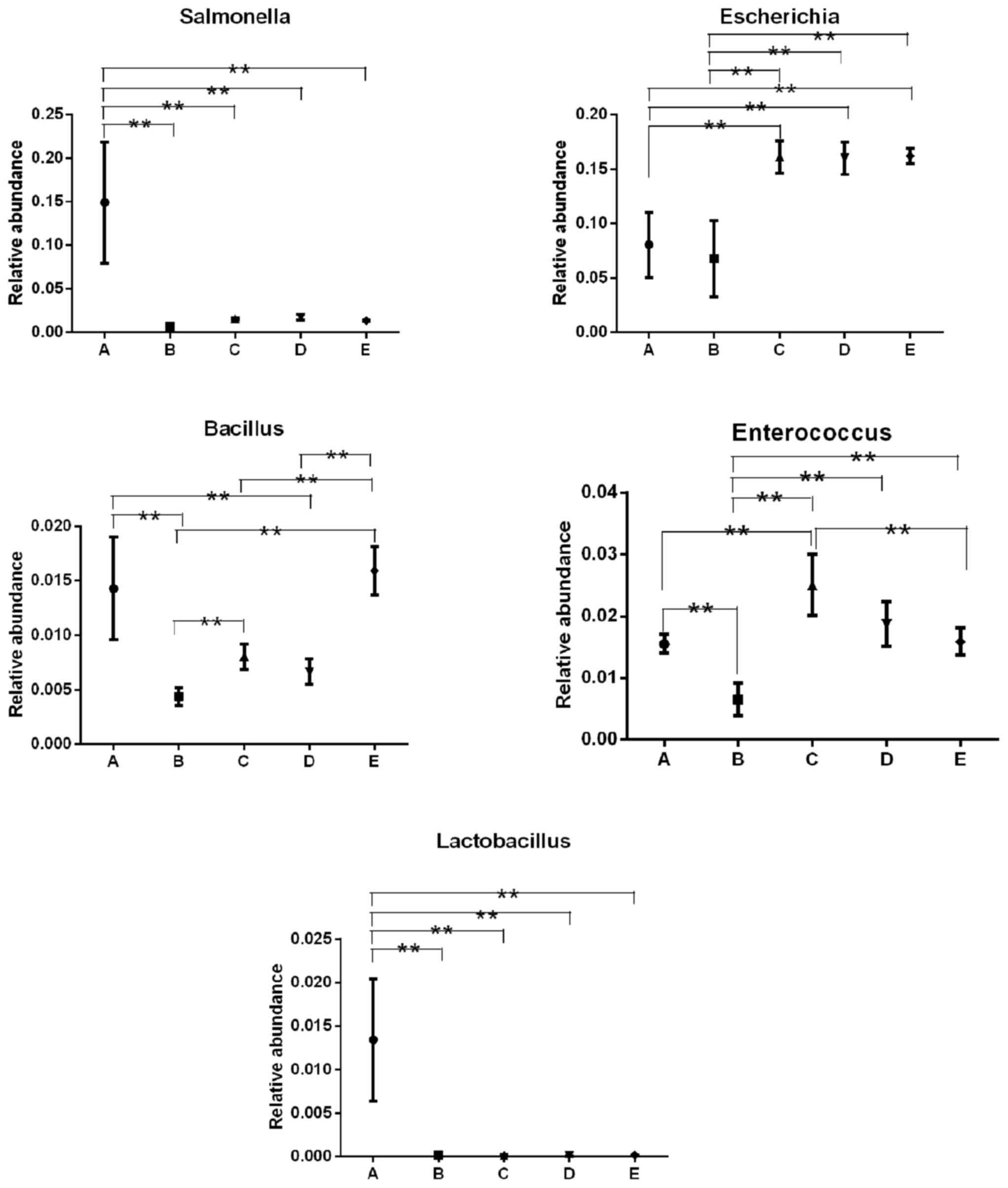
Relative abundance of the bacterial
communities in each sample
To compare the relative abundances among different
groups, the Metastats method was applied in the presentstudy. At
the genus level, the relative abundances of Salmonella,
Bacillus and Enterococcus were each lowered or
enhanced in the groups, although their actual number in each group
was the same (Table I). For E.
coli, actual bacterial number in groups A, B, C, D and E, was
1×106, 1×104, 1×103,
1×102 and 1×101 CFU/ml, respectively, while
the OTU numbers in groups C, D and E were significantly higher
compared with groups A and B (Fig.
4). In addition, though the OTU number of L. plantarum
in groups B, C, D and E was significantly decreased compared with
group A (Fig. 4), there was no
significant difference observed among groups B, C, D and E, and the
actual bacterial number of L. plantarum in groups B, C, D
and E was 1×104, 1×103, 1×102 and
1×101, respectively (Table
I).
Discussion
The world is dominated by prokaryotes. The total
number of microbial cells on Earth is estimated to be
1×1030 and, in the human body alone, there are up to 100
trillion organisms, which approximately equates to ten times the
number of our own human cells (24,25),
and there are millions of prokaryotic species which may not be
cultivated (15,26). In the humanbody, bacteria serve
important roles in the modulation of digestive, endocrineand immune
functions. With the discovery of more recent
culture-independentsequencing-based methods, the composition and
diversity of the human microbiome is being uncovered. However,
sample handling, DNA extraction, amplification efficiency, run
processing and downstream analyses may seriously affect the
generation of high quality data (2,14–17)
and, therefore, it is important to evaluate the accuracy and
sensitivity of high-throughput sequencing technology.
In the present study, seven common bacteria were
mixed together to make a known microbiota, of which S.
thermophilus, B. bifidum, B. subtilis, E.
faecalis and S. typhimurium sustained a constant number
of 1×106 CFU/ml in each sample, and the typical
gram-negative bacteria E. coli and gram-positive bacteria
L. plantarum were decreased between groups A and E.
Following mixing of the known bacteria, DNA extraction was
performed and the extracted genomic DNA was used as a template to
amplify the V3-V4 region of 16S rRNA genes using primers of
338F/806R (18), and the results
suggested that the DNA quality and PCR amplicons met the
requirements for pyrosequencing (data not shown).
To evaluate tag quality, the raw tags and clean
tags, average bp and OTUs per sample were compared. The mean number
of 135,498.9 clean tags, average length of 467.7 bp, and saturated
chao1 index and rarefaction curves ensured their reliability for
future analysis. However, the average OTUs (122.7) in all groups
indicated a 17.5-fold (122.7/7) increase compared with the actual
microbial number in each group (7). To evaluate the consistency of the
high-throughput sequencing, the Venn and PCoA methods were utilized
and the results suggested that only 46.38% (64/138), 58.70%
(81/138), 86.13% (118/137), 83.57% (117/140) and 89.29% (125/140)
common OTUs were identified in groups A, B, C, D and E, of which
the majority of OTUs did not belong to the added bacteria. However,
the PCoA results demonstrated that the majority of samples
clustered together, except for sample B2.
Compared with the relative abundance of known
microbiota, all known bacteria were identified at the phylum and
class levels; one or more bacteria was missed at the order, family
and genus levels, and the known bacteria only occupied ~20% of the
total OTUs at these levels. As microbiota are generally analyzed at
the genus level, the statistical analysis in the present study was
performed at the genus level, and Salmonella,
Bacillus and Enterococcus, which existed at the same
number in each group, exhibited a significant decreased or increase
using high-throughput sequencing technology; whereas, the ten-fold
dilution of Escherichia and Lactobacillus among
groups C, D and E exhibited little alteration.
Microbial genomic DNA extraction and purification is
the first step for library preparation however researchers
indicated that there were significant differences in microbial
composition when comparing microbiota diversity obtained from the
same samples using different DNA extraction methods (27). To avoid the influence of DNA
extraction, all the DNA samples were simultaneously extracted by
the same researcher using the same DNA extraction kit. Therefore,
the DNA extraction method is not the key factor for the
misidentification of the known microbiota. In addition, all the DNA
was amplified using the same primers of 338F/806R, as a result
ofthe potential for amplification bias during PCR amplification
reactions and the generation of chimeric amplification products
that may exaggerate the bacterial number (28). Furthermore, the chimeric sequences,
which are not identified by computational filtering software will
lead to incorrect taxonomic identifications and an overestimated
bacterial richness in the final microbiota profiling results
(24,28).
In conclusion, the results of the present study
suggested that the actual bacterial number in a specific
environment maybe greatly exaggerated due to run processing and
downstream analyses, and that DNA extraction and amplification
efficiency may cause a reduction or exaggeration of certain
bacteria. Therefore, certain measures, for example, adding the
indicated bacteria/microbiota and analysing using more than three
types of calculation software, are required to provide reasonable
results. However, this result was only based on the present study,
and the small size and lack of the comparison among various
sequencing companies may not allowthe results of the present study
to fully reflect the actual drawbacks of high-throughput
sequencing. In the authors' future work, the microbial diversity in
various environments will be tested using enlarged sample sizes and
comparing the data quality among different sequencing companies.
This will provide basic data for the improvement of high-throughput
sequencing technology and benefit its applications in the
monitoring of bacterial alterations during various diseases.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 31560264,
81503364, 91639106, 81270202, and 91339113), the National Basic
Research Program of China (grant no. 2013CB531103) and grants from
Jiangxi Province (grant nos. 20171BCB23028 and 20175526).
References
|
1
|
Shokralla S, Spall JL, Gibson JF and
Hajibabaei M: Next-generation sequencing technologies for
environmental DNA research. Mol Ecol. 21:1794–1805. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yue-Xin M, Holmstrom C and Webb J:
Application of denaturing gradient gel electrophoresis (DGGE) in
microbial ecology. Acta Ecologica Sinica. 23:1561–1569. 2003.
|
|
3
|
Yu X, Wu X, Qiu L, Wang D, Gan M, Chen X,
Wei H and Xu F: Analysis of the intestinal microbial community
structure of healthy and long-living elderly residents in Gaotian
village of Liuyang City. Appl Microbiol Biot. 99:9085–9095. 2015.
View Article : Google Scholar
|
|
4
|
Sivan A, Corrales L, Hubert N, Williams
JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B,
Alegre ML, et al: Commensal Bifidobacterium promotes
antitumor immunity and facilitates anti-PD-L1 efficacy. Science.
350:1084–1089. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhernakova A, Kurilshikov A, Bonder MJ,
Tigchelaar EF, Schirmer M, Vatanen T, Mujagic Z, Vila AV, Falony G,
Vieira-Silva S, et al: Population-based metagenomics analysis
reveals markers for gut microbiome composition and diversity.
Science. 352:565–569. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Derrien M and van Hylckama Vlieg JE: Fate,
activity, and impact of ingested bacteria within the human gut
microbiota. Trends Microbiol. 23:354–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mimee M, Citorik RJ and Lu TK: Microbiome
therapeutics-Advances and challenges. Adv Drug Deliver Revi.
105:44–54. 2016. View Article : Google Scholar
|
|
8
|
Chen T, Shi Y, Wang X, Wang X, Meng F,
Yang S, Yang J and Xin H: High-throughput sequencing analyses of
oral microbial diversity in healthy people and patients with dental
caries and periodontal disease. Mol Med Rep. 16:127–132. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fang X, Wang X, Yang S, Meng F, Wang X,
Wei H and Chen T: Evaluation of the microbial diversity in
amyotrophic lateral sclerosis using high-throughput sequencing.
Front Microbiol. 7:14792016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang X, Hu X, Deng K, Cheng X, Wei J,
Jiang M, Wang X and Chen T: High-throughput sequencing of microbial
diversity in implant-associated infection. Infect Genet Evol.
43:307–311. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Planý M, Šoltys K, Budiš J, Mader A,
Szemes T, Siekel P and Kuchtaa T: Potential of high-throughput
sequencing for broad-range detection of pathogenic bacteria in
spices and herbs. Food Control. 70:360–370. 2016.
|
|
12
|
Ishaq SL and Wright AD: High-throughput
DNA sequencing of the ruminal bacteria from moose (Alces alces) in
Vermont, Alaska, and Norway. Microb Ecol. 68:185–195. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hamilton MJ, Weingarden AR, Unno T,
Khoruts A and Sadowsky MJ: High-throughput DNA sequence analysis
reveals stable engraftment of gut microbiota following
transplantation of previously frozen fecal bacteria. Gut Microbes.
4:125–135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moorthie S, Mattocks CJ and Wright CF:
Review of massively parallel DNA sequencing technologies. Hugo J.
5:1–12. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kircher M, Heyn P and Kelso J: Addressing
challenges in the production and analysis of illumina sequencing
data. BMC Genomics. 12:3822011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kircher M: Understanding and improving
high-throughput sequencing data production and analysis: 225.
2011.
|
|
17
|
Hall N: Advanced sequencing technologies
and their wider impact in microbiology. J Exp Biol. 210:1518–1525.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu J, Lian F, Zhao L, Zhao Y, Chen X,
Zhang X, Guo Y, Zhang C, Zhou Q, Xue Z, et al: Structural
modulation of gut microbiota during alleviation of type 2 diabetes
with a Chinese herbal formula. Isme J. 9:552–562. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schloss PD, Westcott SL, Ryabin T, Hall
JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH,
Robinson CJ, et al: Introducing mothur: Open-source,
platform-independent, community-supported software for describing
and comparing microbial communities. Appl Environ Microb.
75:7537–7541. 2009. View Article : Google Scholar
|
|
20
|
Caporaso JG, Kuczynski J, Stombaugh J,
Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich
JK, Gordon JI, et al: QIIME allows analysis of high-throughput
community sequencing data. Nat Methods. 7:335–336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Davenport M, Poles J, Leung JM, Wolff MJ,
Abidi WM, Ullman T, Mayer L, Cho I and Loke P: Metabolic
alterations to the mucosal microbiota in inflammatory bowel
disease. Inflamm Bowel Dis. 20:723–731. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Haas BJ, Gevers D, Earl AM, Feldgarden M,
Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren
E, et al: Chimeric 16S rRNA sequence formation and detection in
Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21:494–504.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu K, Ryan PA, Schlieper KA, Graffam ME,
Levine S, Wishnok JS, Swenberg JA, Tannenbaum SR and Fox JG:
Arsenic exposure perturbs the gut microbiome and its metabolic
profile in mice: An integrated metagenomics and metabolomics
analysis. Environ Health Perspect. 122:2842014.PubMed/NCBI
|
|
24
|
Boers SA, Jansen R and Hays JP: Suddenly
everyone is a microbiota specialist. Clin Microbiol Infec.
22:581–582. 2016. View Article : Google Scholar
|
|
25
|
Di Bella JM, Bao Y, Gloor GB, Burton JP
and Reid G: High throughput sequencing methods and analysis for
microbiome research. J Microbiol Meth. 95:401–414. 2013. View Article : Google Scholar
|
|
26
|
Van Vliet AH: Next generation sequencing
of microbial transcriptomes: Challenges and opportunities. Fems
Microbiol Lett. 302:1–7. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kennedy NA, Walker AW, Berry SH, Duncan
SH, Farquarson FM, Louis P, Thomson JM UK IBD Genetics Consortium,
Satsangi J, Flint HJ, et al: The impact of different DNA extraction
kits and laboratories upon the assessment of human gut microbiota
composition by 16S rRNA gene sequencing. PLoS One. 9:e889822014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boers SA, Hays JP and Jansen R: Micelle
PCR reduces chimera formation in 16S rRNA profiling of complex
microbial DNA mixtures. Sci Rep. 5:141812015. View Article : Google Scholar : PubMed/NCBI
|