Introduction
The corticotropin-releasing factor (CRF) family is
well known for its role in the stress response. Psychosocial or
environmental stressors induce or aggravate the development of
inflammatory bowel disease (IBD) (1). During stress, CRF is released from
the neurons of the paraventricular nucleus and activates the
hypothalamic-pituitary-adrenal axis. Subsequently, glucocorticoids
are produced in response to stress. In addition to central
activation, CRF and associated peptides (urocortins) are released
from regional sensory and sympathetic nerves, immune cells, and gut
enteroendocrine cells to act locally. Along with its actions in the
brain, CRF receptor signalling is involved in the pathogenesis of
IBD by modulating gastrointestinal motility, sensitivity and
inflammation (2–4).
In recent years, numerous studies have focused on
the peripheral action of the CRF family on gastrointestinal
inflammation (5). CRF and its
receptors are located on immune cells in the blood, such as T
cells, B cells and macrophages, as well as on colonic mucosal
macrophages and enterochromaffin cells (6,7). In
a previous study, urocortin 1 and CRF receptor 1 (CRF-R1)
expression levels were increased in colon biopsies from patients
with ulcerative colitis (UC) compared with those of healthy
subjects, indicating a possible peripheral role for CRF-R1
signalling in UC (8). CRF induced
macrophages to release tumour necrosis factor (TNF)-α, interleukin
(IL)-1β and IL-6, and CRF-R1 antagonists blocked these effects
(9). Furthermore, CFR-R1 knockout
mice exhibited less inflammation than wild type mice in the dextran
sulphate sodium (DSS)-induced UC mouse model (10). Based on these results, CRF-R1 may
exert a pro-inflammatory role in intestinal inflammation. However,
certain contrasting evidence supports an anti-inflammatory role for
CRF-R1. CFR-R1 knockout did not reduce colon ulceration or edema
(11), and a CRF deficiency
increased the animals' susceptibility to developing colitis
(12). CRF-R1 activation
suppressed TNF-α release from lipopolysaccharide (LPS)-treated
macrophages and inducible nitric oxide synthase from endothelial
cells (13). Therefore, the pro-
and anti-inflammatory roles of CRF-R1 remain controversial and may
even be paradoxical. Studies are required to determine the role of
CRF-R1 in colonic inflammation.
Macrophages are an important component of the
pro-inflammatory response in murine models of colitis (14) and human IBD (15). Infiltration of activated
macrophages into the lamina propria contributes to the development
and progression of intestinal inflammation. CRF receptors are
co-localized with macrophages and involved in their apoptosis
(16). Furthermore, activation of
CRF receptors augments cytokine production, such as TNF-α and IL-6,
in LPS-induced macrophages (17),
indicating that macrophages are a possible target of the CRF family
to modulate inflammation.
Generally, nuclear factor (NF)-κB controls various
pro-inflammatory genes, chemokines and signalling enzymes,
including TNF-α, IL-6, IL-8 and IL-1β, which are involved in
regulating immune and inflammatory responses. NF-κB activation is
induced in macrophages isolated from inflamed tissue specimens from
patients with IBD (18), and the
expression was correlated with the severity of colonic inflammation
(19). CRF regulates the
expression of the Toll-like receptor (TLR)-4 gene in macrophages
and activates downstream effectors, such as NF-κB and
pro-inflammatory cytokines, to induce an inflammatory reaction
(12,20). Therefore, regulation of NF-κB
signalling may present as a potential therapeutic approach for
IBD.
In the present study, the correlation between CRF-R1
expression and the disease severity was evaluated in a DSS-induced
UC model. Subsequently, a CRF-R1 agonist and antagonist were used
to investigate the effects of CRF-R1 on colitis. Furthermore, the
modulatory effects of CRF-R1 on macrophages and the NF-κB
signalling pathway were evaluated.
Materials and methods
Animals
A total of 56 specific pathogen-free, male BALB/c
mice (weight, 18–22 g; Xi'an Jiaotong University Animal Centre,
Xi'an, China) were randomly allocated into the following eight
groups: DSS 0 group (n=6), mice were administered normal drinking
water; DSS 1 day group (n=6), mice were administered 3% DSS for 1
day; DSS 4 day group (n=6), mice were administered 3% DSS for 4
consecutive days; DSS 7 day group (n=6), mice were administered 3%
DSS for 7 consecutive days; normal group (n=8), mice received
saline (0.2 ml) intraperitoneal (ip) injection daily and normal
water drinking for 7 consecutive days; saline + DSS group (n=8),
mice received saline ip injection daily and 3% DSS for 7
consecutive days; CRF + DSS group (n=8), mice received CRF (30
µg/kg) ip injection daily and 3% DSS for 7 consecutive days;
CP154526 + DSS group (n=8), mice received CP154526 (10 mg/kg) ip
injection daily and 3% DSS for 7 consecutive days. All mice were
housed under standard conditions, 4 mice per cage (temperature,
20–22°C; 50–60% humidity, 12-h light/dark cycle), with free access
to standard rat chow and tap water. All experiments were performed
in accordance with the United Kingdom Animals Act 1986 and were
approved by the Ethical Committee of the Medical Faculty of
Medicine of Xi'an Jiaotong University.
Animal model of UC
Mice were administered 3% DSS (9011-18-1; molecular
weight, 40,000; MP Biomedicals Inc., Santa Ana, CA, USA) as their
sole source of drinking water for 7 consecutive days to develop UC
model. Control mice received normal drinking water. Mice of normal
group, saline + DSS group, CRF + DSS group and CP154526 + DSS group
were sacrificed 7 days following the administration of DSS. Colon
samples (1.5 cm) were collected from the anus. One third of the
colon sample were rinsed in 4% paraformaldehyde for haematoxylin
and eosin (H&E) and immunohistochemical staining, and two
thirds of the colon sample were rinsed in liquid nitrogen for
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR), ELISA and western blot analysis. CRF (1151; R&D
Systems Inc., Minneapolis, MN, USA) was ip-injected into the
DSS-treated mice at a dose of 30 µg/kg from the first day to the
seventh day. CP154526 (PZ0100; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was ip-injected into the DSS-treated mice at a
dose of 10 mg/kg from the first day to the seventh day. Saline was
ip-injected into the DSS-treated mice daily to serve as the control
group. The CRF and CP154526 doses were determined according to our
pilot experiment and previous studies (21–23).
In order to evaluate alterations in expression
levels of CRF-R1 with the number of days of DSS administration,
mice of DSS 0, DSS 1, DSS 4 and DSS 7 day groups were sacrificed on
day 0, 1, 4 and 7 days, respectively. Colon samples were collected
for H&E staining and RT-qPCR analysis as previously
aforementioned.
Disease activity index (DAI)
assessment
The DAI has been used to evaluate the severity of
colitis, which is well correlated with the pathological findings in
DSS-treated mice (24). The DAI is
the combined score of weight loss, stool consistency and bleeding.
Scores were defined for weight loss as follows: 0, no loss; 1,
1–5%; 2, 6–10%; 3, 11–15%; and 4, >15%. Scores for stools were
as follows: 0, normal; 2, loose stool; 4, diarrhoea. Bleeding
scores were defined as follows: 0, no blood; 2, presence; 4, gross
hematochezia. The DAI was scored from days 0 to 7 of DSS
treatment.
Histopathological analysis
Colon sections were fixed overnight with 4%
paraformaldehyde at 4°C, and were then embedded in paraffin and
serially sectioned. Samples were sliced into 4-µm sections. Tissue
sections were stained with hematoxylin (MHS16; Sigma-Aldrich; Merck
KGaA; at room temperature for 20 min) and eosin (230251;
Sigma-Aldrich; Merck KGaA; at room temperature for 30 sec), and
examined by a pathologist who was blinded to the experimental
groups. The colonic histopathological score was assigned based on
the extent and range of oedema, ulceration and infiltration of
inflammatory cells, as described in a previous study (25). For immunohistochemistry, 4%
paraformaldehyde-fixed, paraffin-embedded tissues were sliced into
4-µm slices. Non-specific antigens were blocked with 10% goat serum
(SP-9001; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.,
Beijing, China) at room temperature for 15 min following antigen
retrieval. Antigen retrieval was performed in 0.01 M citrate buffer
(pH 6.0) for 10 min in boiling water in a microwave oven. Sections
were then incubated overnight at 4°C with a cluster of
differentiation (CD)68 primary antibody (BA3638; 1:200; Wuhan
Boster Biological Technology, Ltd., Wuhan, China). A SPlink
detection kit (SP-9001; Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd.) was used as a ready-to-use secondary
antibody. The sections were incubated at 37°C for 30 min. Positive
binding was detected using diaminobenzidine (ZLI9033; Beijing
Zhongshan Golden Bridge Biotechnology Co., Ltd.) staining for 1–3
min. Counterstaining with hematoxylin was performed at room
temperature for 1 min. The sections were examined using an optical
microscopy (Olympus Corporation, Tokyo, Japan).
Measurement of CRF-R1 expression
levels with RT-qPCR
Total RNA was extracted from the colon tissue using
TRIzol™ reagent (15596026; Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) according to the manufacturer's
instructions. RT was performed using a First Strand cDNA Synthesis
kit (K1612; Thermo Fisher Scientific, Inc.). cDNA (1 µl) served as
the template and was amplified by qPCR using the 2X SYBR-Green qPCR
mix (PC3302; Aidlab Biotechnologies, Co., Ltd., Beijing, China) and
1 µl sense and antisense primers in a final reaction volume of 25
µl to quantify the cDNA contents. The primer sequences were as
follows: CRF-R1 sense, TCT ATG AGG AAG GAG TGG and antisense, GGC
TTA GTT AGT TAG TTG TC; and β-actin sense, ACC ACA CCT TCT ACA ATG
AG and antisense, ACGACCAGAGGCATACAG. The size of the amplified
products was expected to be 196 bp. The reactions were performed in
triplicate to allow for statistical evaluation, and RT-qPCR was
performed using an ABI-Prizm 7000 Real-Time PCR, DNA Thermal Cycler
using the following cycling parameters: Pre-amplification cycle
(denaturation at 95°C for 5 min), 40 cycles of amplification
(denaturation at 95°C for 15 sec, annealing at 60°C for 30 sec and
extension at 72°C for 30 sec) and a final extension step at 55°C
for 30 sec. No by-products were present in the reaction, as
indicated by the dissociation curve provided at the end of the
reaction. The 2−∆∆Cq method was used to calculate the
relative gene expression levels.
Measurement of CRF-R1 and p65 NF-κB
phosphorylation using western blot analysis
Total protein was extracted from the colon tissue
samples via radio immunoprecipitation assay (RIPA) lysis buffer
(WB-0071; Beijing Dingguo Changsheng Biotechnology Co., Ltd.,
Beijing, China). Protein samples were mixed with sample buffer (5X)
and denatured by boiling. For immunoblots, proteins were separated
by sodium dodecyl sulphate polyacrylamide gel electrophoresis on a
10% polyacrylamide gel. Samples were electrophoresed at 80–100 V
for 2 h until the dye migrated to the bottom of the gel. The
proteins were transferred to polyvinylidene difluoride membranes
and the membranes were blocked in blocking buffer (5% non-fat dry
milk) for 2 h at room temperature, and incubated with primary
antibodies against CRF-R1 (ab59023; 1:500; Abcam, Cambridge, MA,
USA) or p-p65 NF-ΚB (3031; 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA) and β-actin (wl01845; 1:1,000;
Wanlei Biotechnology, Co., Ltd., Shenyang, China) overnight at 4°C.
Membranes were then incubated with a horseradish peroxidase
(HRP)-conjugated anti-goat (SA00001-4; 1:7,000; Proteintech Group,
Inc., Rosemont, IL, USA) and anti-rabbit (Wla023a; 1:8,000; Wanlei
Biotechnology, Co., Ltd.) secondary antibody for 1 h at room
temperature. The proteins were visualized with a chemiluminescent
substrate (WBULS0100; EMD Millipore, Billerica, MA, USA) using an
imaging system (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Measurement of the TNF-α and IL-6
expression levels using ELISA
A murine cytokine ELISA kit (K00164 and K00041;
Shanghai Yeyuan Biology Science & Technology, Shanghai, China)
was used to measure the production of cytokines, TNF-α and IL-6 in
the colon tissue samples. The samples were homogenized with saline
(1:9) in a digital homogenizer, and the supernatant was obtained by
centrifugation at 1,500 × g, 4°C for 10 min. All reagents were
brought to room temperature. The plate was set for standard wells
(6 wells) and testing sample wells (31 wells). Subsequently, each
standard well was added 50 µl standard sample; each testing sample
well was added 10 µl testing sample and then 40 µl sample diluent.
The plate was gently agitated for 1 min and was then incubated at
room temperature for 2 h. Subsequently, 100 µl HRP-labeled antibody
was added to each well and was incubated at 37°C for 1 h. After
washing, chromogen solution A (50 µl) and chromogen solution B (50
µl) were added to each well and the reaction was terminated with 50
µl stop solution. The TNF-α and IL-6 concentrations were calculated
based on the optical density at a wavelength of 450 nm using a
microtiter plate reader (Bio-Rad Laboratories, Inc.).
Statistical analysis
Data processing and statistical analysis were
performed using SPSS 18.0 statistical software (SPSS, Inc.,
Chicago, IL, USA) and all data are expressed as the means ±
standard error of the means. The data from multiple groups were
compared using one-way analysis of variance, and comparisons
between two groups were performed with the least significant
difference (LSD) test. P<0.05 was considered to indicate a
statistically significant difference.
Results
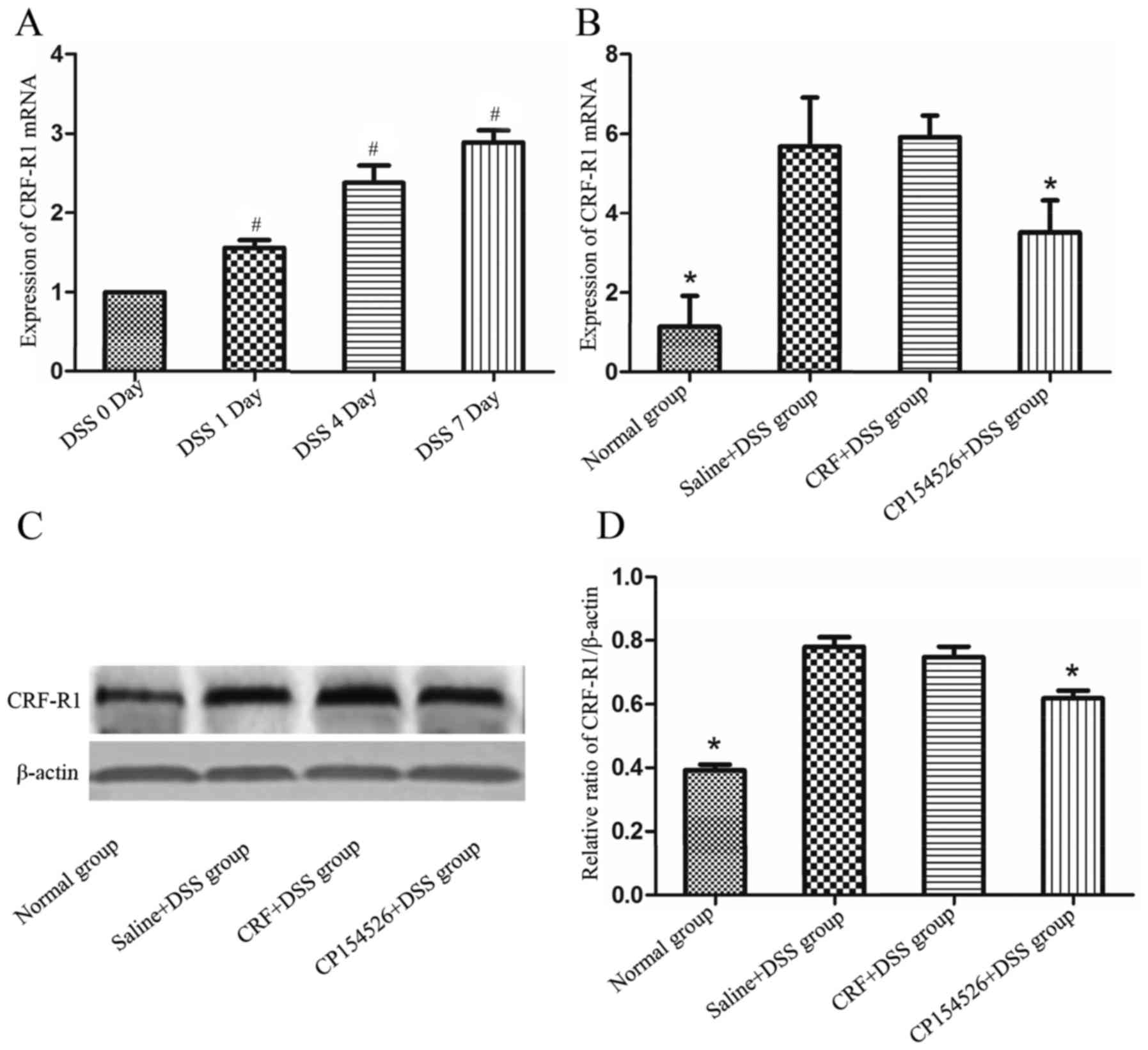
CRF-R1 expression level was increased
in the colon samples from DSS-treated mice
To observe the expression level of CRF-R1 at
different days of DSS administration, mice of DSS 0, DSS 1, DSS 4
and DSS 7 day groups were sacrificed on day 0, 1, 4 and 7
respectively, and samples from the colons were obtained to detect
CRF-R1 expression levels and mucosal inflammation. As the number of
days that the animals received DSS-containing water increased, the
mucosal inflammation was significantly aggravated, which manifested
as increased mucosa oedema, ulceration and infiltration of
inflammatory cells. The expression level of the CRF-R1 mRNA
increased as the number of days the animals were treated with DSS
increased (Fig. 1A), and was
correlated with the degree of mucosal inflammation.
To evaluate the effect of CRF-R1 agonist and
antagonist on expression level of CRF-R1 of DSS induced UC mice,
mice of normal group, saline + DSS group, CRF + DSS group and
CP154526 + DSS group were sacrificed 7 days after commencing DSS
administration. CP154526, a CRF-R1 antagonist, was ip-injected at a
dose of 10 mg/kg for 7 consecutive days and significantly decreased
the expression levels of the CRF-R1 mRNA and protein in the
DSS-treated mice compared with that in the controls. Furthermore,
CRF, a CRF-R1 agonist, was ip-injected at a dose of 30 µg/kg for 7
consecutive days and marginally increased the expression level of
the CRF-R1 mRNA and protein in the DSS-treated mice compared with
that in the controls (Fig.
1B-D).
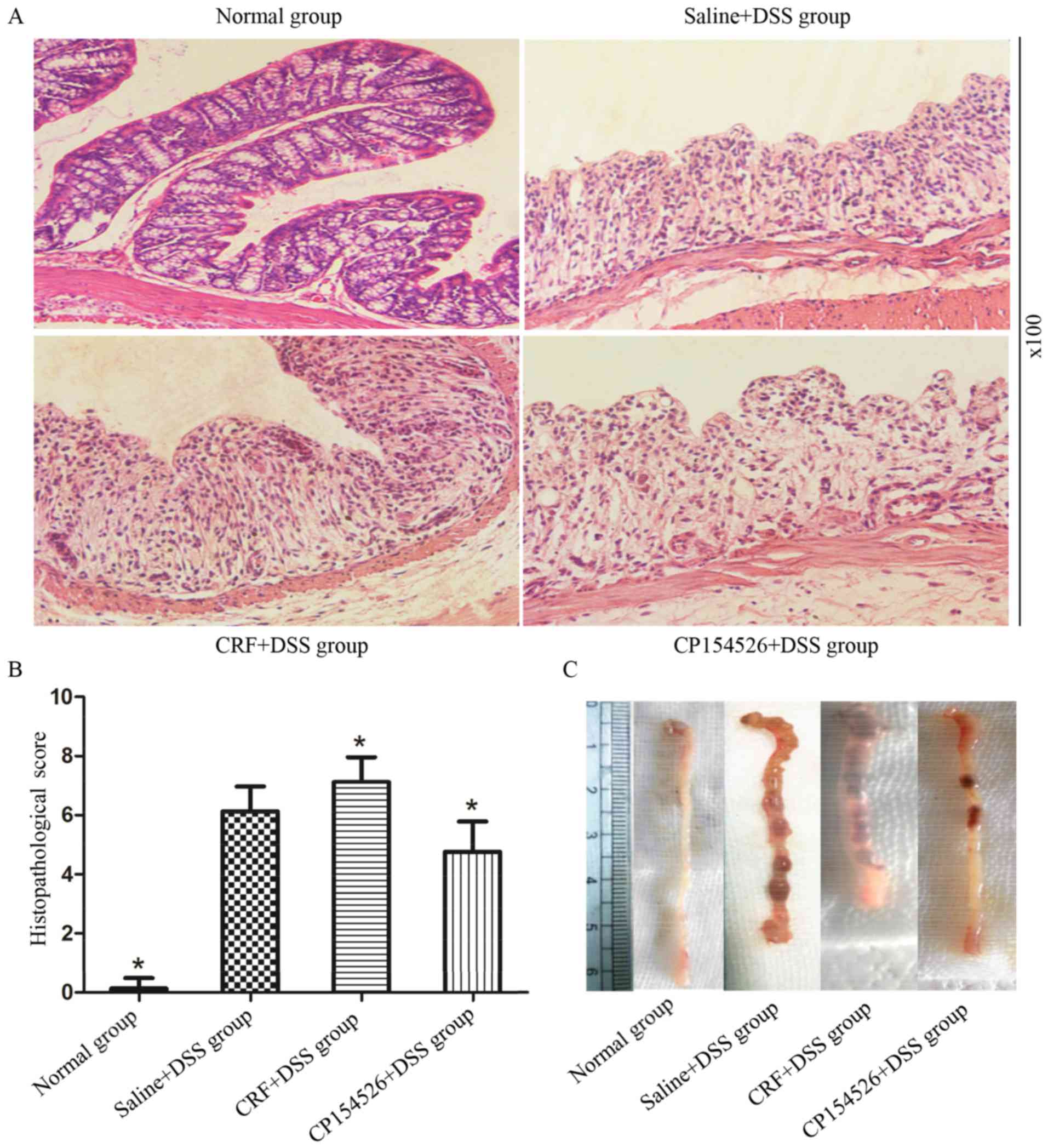
Effect of CRF-R1 on DSS-induced
colitis
The ip injections of CRF aggravated the colonic
mucosal inflammation induced by DSS on the seventh day compared
with the controls. By contrast, the ip injections of CP154526
alleviated the colonic mucosal inflammation. The DAI (Table I) and histopathological score
(Fig. 2) were significantly
increased in the CRF-treated group and decreased in the
CP154526-treated group compared with those in the saline + DSS
controls. One mouse from the CRF-treated group succumbed on the
seventh day of DSS treatment.
 | Table I.Effect of CRF-R1 on DAI of DSS-induced
mice. |
Table I.
Effect of CRF-R1 on DAI of DSS-induced
mice.
| Groups | Day 1 | Day 3 | Day 5 | Day 6 | Day 7 |
|---|
| Normal group |
0.000±0.000 |
0.042±0.118 |
0.083±0.154 |
0.042±0.118a |
0.083±0.153a |
| Saline + DSS
group |
0.125±0.173 |
0.750±0.636b |
1.833±1.141 |
2.917±1.318 |
3.417±0.886 |
| CRF + DSS
group |
0.167±0.252 |
0.916±0.463b |
2.082±0.635a |
3.500±0.535a |
3.917±0.155a |
| CP154526 + DSS
group |
0.124±0.171 |
0.750±0.344b |
1.417±0.497a |
2.417±0.683a |
3.084±0.463a |
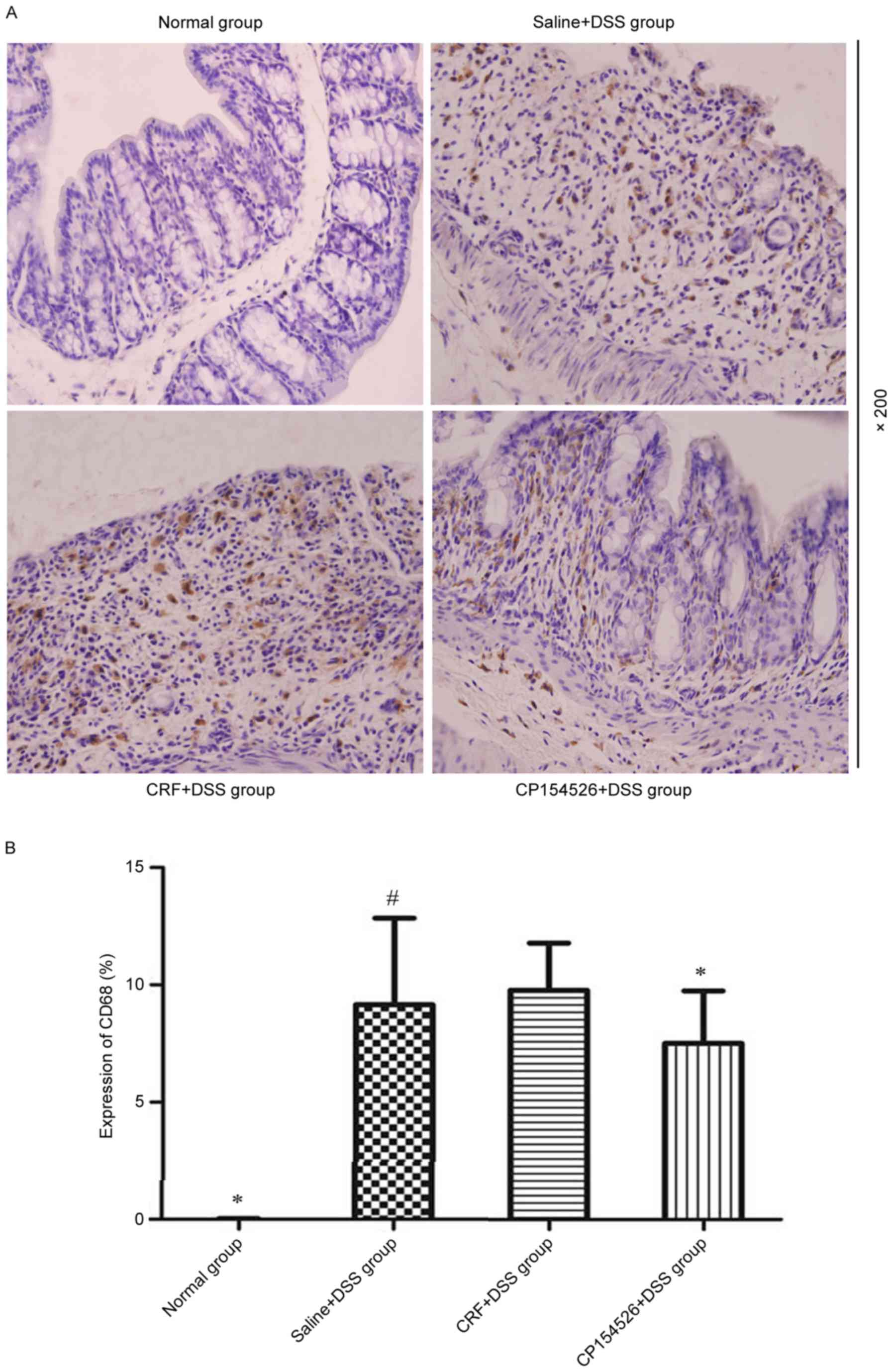
Macrophages were diffusely located throughout the
lamina propria and submucosal layer in the colon samples of the
DSS-treated mice, which appeared as brown cytoplasmic staining. The
DSS treatment significantly increased the number of CD68-positive
macrophages compared with that in the controls, particularly on the
seventh day. CP154526 decreased the infiltration of macrophages in
the colons of the DSS-treated mice; however, CRF induced a marginal
increase in the number of macrophages (Fig. 3).
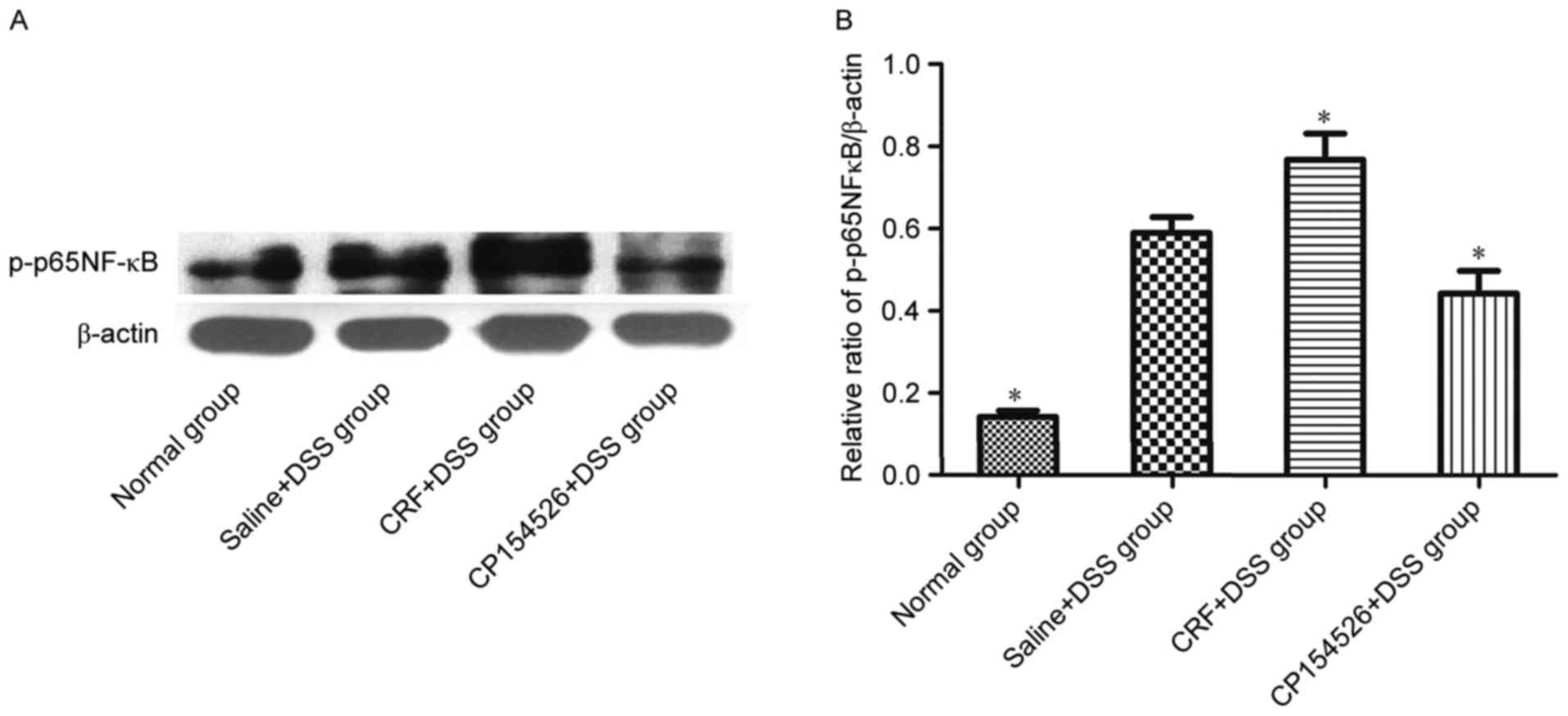
Phosphorylation of p65 NF-κB was
involved in the effect of CRF-R1 on DSS-induced mucosal
inflammation
The effect of CRF-R1 on NF-κB activation was
examined by western blot analysis to investigate the mechanism by
which CRF-R1 modulates inflammation in the DSS-treated mice. CRF,
the CRF-R1 agonist, increased the level of p-p65 NF-κB in the colon
compared with that in the saline-injected controls. However,
CP154526, the CRF-R1 antagonist, decreased the level of p-p65 NF-κB
in the colon (Fig. 4). Based on
the results, NF-κB activation was involved in the effect of CRF-R1
on DSS-induced colitis.
Activation of CRF-R1 increased the
expression levels of inflammatory cytokines in the DSS-treated
mice
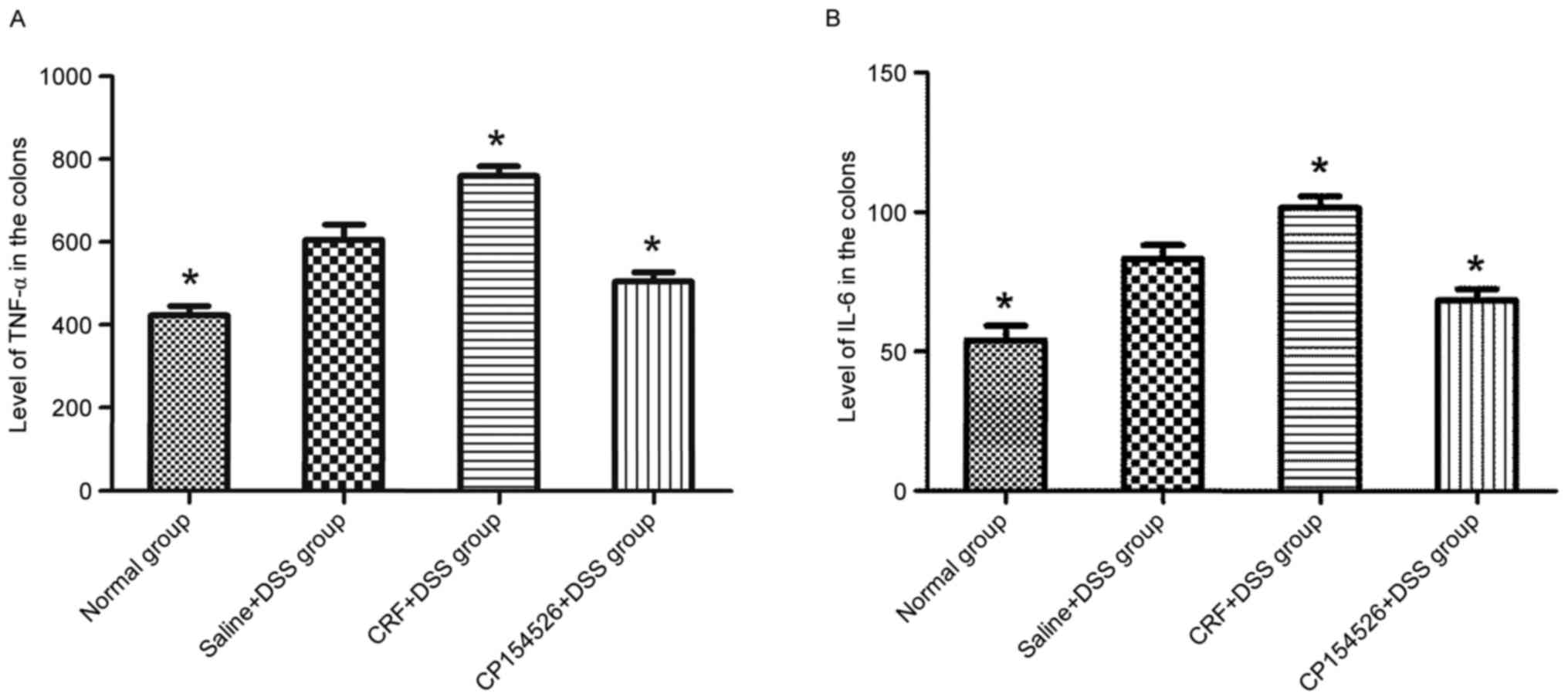
The ip injections of CRF increased the expression
levels of TNF-α and IL-6 in the colon compared with that in the
saline + DSS controls. By contrast, the ip injections of CP154526
decreased the expression levels of TNF-α and IL-6. Furthermore, the
TNF-α and IL-6 expression levels were identified to be
significantly different between these two groups (Fig. 5).
Discussion
Emerging evidence links the activation of CRF
receptors with intestinal inflammation. As demonstrated in the
present study, CRF-R1 expression levels were increased in the
colons of the DSS-treated mice compared with those in the control
mice and was proportional to the severity of colonic mucosal
inflammation. CRF-R1 is closely associated with inflammatory
disease. In the study of clostridium difficile toxin-A-induced
colonic inflammation by Wlk et al (26), the expression levels of CRF-R1 mRNA
were increased in the mouse ileum. The synthesis and secretion of
CRF-R1 from the colon is increased in patients with UC compared
with in healthy subjects (27). In
the current study, a DSS-induced UC model was used to further
verify that the increased expression level of CRF-R1 was
accompanied by aggravated ulcers, oedema and inflammatory cell
infiltration in the colon. Furthermore, when colitis was
alleviated, CRF-R1 expression levels decreased.
CRF is the predominant endogenous agonist that
activates CRF-R1, which has a greater affinity for CRF-R1 than for
CRF-R2. The CRF injections aggravated colitis in the DSS-treated
mice compared with in the controls, as demonstrated by the
increased DAI, colonic histopathology score and survival rate (one
mouse succumbed). By contrast, injection of the specific CRF-R1
antagonist, CP154526 decreased the DAI, colonic histopathology
score and expression level of CRF-R1 in the colon samples of the
DSS-treated mice. These results are consistent with previous
results obtained from CRF-R1−/− mice and the application
of antalamin (another CRF-R1 antagonist) in DSS-treated mice
(10,28), confirming the pro-inflammatory role
of CRF-R1 in the development of colitis. However, this finding
contrasts with results from a recent study of rats expressing
siCRF-R1, in which the histological scores and inflammatory
cytokine levels were unchanged in the trinitrobenzene sulfonic
acid-induced colitis model (11).
The differences in these results may be attributed to the
differences in the animal species, gene silencing methods and
colitis models used.
Macrophages are important in modulating the
inflammatory response in UC. According to the present study,
macrophages were located in the mucosal and submucosal layers of
the colon, and the level of CRF-R1 expression was proportional to
macrophage infiltration in the colon samples of the DSS-treated
mice. CRF-R1 is located in immune cells, such as lymphocytes,
monocytes, and macrophages, in the lamina propria of the colon. The
current results are similar to the data presented by Yuan et
al (8) and Liu et al
(10) who identified that the
majority of CRF-R1 is co-localized with macrophages in an animal
model of and patients with UC. Activation of CRF-R1 may induce the
release of inflammatory factors from LPS-induced macrophages
directly. In the present study, inhibition of CRF-R1 decreased the
number of macrophages infiltrating the colon in the DSS-induced
mice, which further illustrated that macrophages are a potential
target for CRF-R1 to exert its pro-inflammatory effect (17,20).
NF-κB is the crucial regulator of immune and
inflammatory responses. Activation of the NF-κB signalling pathway
in macrophages is crucial in the mechanism of UC, and the NF-κB
expression level was increased in the colon of experimental animal
models of colitis and patients with UC (29,30).
In the present study, p65-NF-κB phosphorylation was increased in
the colons of the DSS-treated mice compared with that in the
controls. Activation of CRF-R1 by CRF increased p65-NF-κB
phosphorylation, and inhibition of CRF-R1 by CP154526 decreased its
phosphorylation compared with that of the controls, which parallels
the extent of colitis and expression levels of TNF-α and IL-6.
Activation of NF-κB by CRF and associated peptides induces the
release of inflammatory cytokines, such as the chemoattractants
IL-8 and monocyte chemoattractant protein 1 in different cell
types, including macrophages (31,32).
Thus, NF-κB activation is involved in the effects of the CRF
receptor on the immune reaction. Hence, the current study
hypothesized that the pro-inflammatory effect of CRF-R1 on
DSS-induced colitis correlated with the activation of p65
NF-κB.
Based on the results, CRF-R1 activation increased
the expression levels of TNF-α and IL-6 in the colon samples of the
DSS-treated mice. By contrast, inhibition of CRF-R1 by CP154526
decreased the expression levels of TNF-α and IL-6 in the colon
tissues from the DSS-treated mice compared with that from the
controls, accompanied by a decrease in NF-κB phosphorylation and
macrophage infiltration in the colon. As indicated by previous
studies, activation of the CRF receptor increases the LPS-induced
synthesis and release of TNF-α and IL-6 from macrophages (9,17).
Furthermore, this role is associated with the regulation of TLR-2
and TLR-4, which activate macrophages to release pro-inflammatory
cytokines by phosphorylating NF-κB (20,12,33).
Here, the current study verified that CRF-R1 exerted a
pro-inflammatory role by activating NF-κB, which in turn increased
the expression levels of the pro-inflammatory cytokines, TNF-α and
IL-6 in vivo. These findings may be associated with
macrophage-mediated inhibition of CRF-R1, which may decrease the
number of macrophages infiltrating the colon in the present study.
Macrophage polarization balances immune reactions by releasing many
inflammation-associated factors. M1 polarization is stimulated by
interferon-γ and TLRs, and these cells produce pro-inflammatory
cytokines, such as IL-1β, IL-6 and TNF-α, to aggravate inflammation
(34). By contrast, M2
polarization is stimulated by IL-4, and these cells produce
anti-inflammatory cytokines to reverse or alleviate inflammation
(34,35). Together with previous studies on
CRF-mediated activation of macrophages to induce inflammatory
cytokine release in vitro, the current study hypothesizes
that the pro-inflammatory role of CRF-R1 in DSS-induced colitis may
be to regulate macrophage accumulation and activation. This action
may contribute to M1 polarization, thus activating NF-κB and
releasing TNF-α and IL-6.
In conclusion, CRF-R1 expression levels were
proportional to the severity of DSS-induced colitis. Activation of
CRF-R1 aggravated inflammation, and inhibition of CRF-R1
ameliorates inflammation evaluated by the DAI and histological
scores in the colon samples of the DSS-treated mice. These effects
are reliant, at least in part, on the number of macrophages
infiltrating the colon and trend to M1 polarization, exhibited as
NF-κB activation, and TNF-α and IL-6 release. These results provide
evidence for the pro-inflammatory role of CRF-R1 in a DSS-induced
UC model and the possible underlying mechanism. Further studies are
required to investigate markers of activation of M1 or M2 and the
involved signal transduction pathway in CRF-R1-aggravated
inflammation. Further investigation is required to understand
whether antagonists that target CRF-R1 serve as a potential
therapeutic approach for the treatment of UC.
Acknowledgements
The present study was supported by the Fundamental
Research Funds for the Central Universities (grant no. 2015gjhz22)
and the Shaanxi Science and Technology Research and Development
Project (grant no. 2014K11-03-03-05).
References
|
1
|
Mawdsley JE and Rampton DS: Psychological
stress in IBD: New insights into pathogenic and therapeutic
implications. Gut. 54:1481–1491. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Taché Y, Kiank C and Stengel A: A role for
corticotropin-releasing factor in functional gastrointestinal
disorders. Curr Gastroenterol Rep. 11:270–277. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Taché Y and Bonaz B:
Corticotropin-releasing factor receptors and stress-related
alterations of gut motor function. J Clin Invest. 117:33–40. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Overman EL, Rivier JE and Moeser AJ: CRF
induces intestinal epithelial barrier injury via the release of
mast cell proteases and TNF-α. PLoS One. 7:e399352012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Buckinx R, Adriaensen D, Nassauw LV and
Timmermans JP: Corticotrophin-releasing factor, related peptides,
and receptors in the normal and inflamed gastrointestinal tract.
Front Neurosci. 5:542011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baker C, Richards LJ, Dayan CM and Jessop
DS: Corticotropin-releasing hormone immunoreactivity in human T and
B cells and macrophages: Colocalization with arginine vasopressin.
J Neuroendocrinol. 15:1070–1074. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kawahito Y, Sano H, Kawata M, Yuri K,
Mukai S, Yamamura Y, Kato H, Chrousos GP, Wilder RL and Kondo M:
Local secretion of corticotropin-releasing hormone by
enterochromaffin cells in human colon. Gastroenterology.
106:859–865. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yuan PQ, Wu SV, Elliott J, Anton PA,
Chatzaki E, Million M and Taché Y: Expression of corticotropin
releasing factor receptor type 1 (CRF1) in the human
gastrointestinal tract and upregulation in the colonic mucosa in
patients with ulcerative colitis. Peptides. 38:62–69. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Agelaki S, Tsatsanis C, Gravanis A and
Margioris AN: Corticotropin-releasing hormone augments
proinflammatory cytokine production from macrophages in vitro and
in lipopolysaccharide-induced endotoxin shock in mice. Infect
Immun. 70:6068–6074. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Y, Fang X, Yuan J, Sun Z, Li C, Li R,
Li L, Zhu C, Wan R, Guo R, et al: The role of
corticotropin-releasing hormone receptor 1 in the development of
colitis-associated cancer in mouse model. Endocr Relat Cancer.
21:639–651. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang J, Adams MR, Clifton MS, Liao M,
Brooks JH, Hasdemir B and Bhargava A: Urocortin 1 modulates
immunosignaling in a rat model of colitis via
corticotropin-releasing factor receptor 2. Am J Physiol
Gastrointest Liver Physiol. 300:G884–G894. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chaniotou Z, Giannogonas P, Theoharis S,
Teli T, Gay J, Savidge T, Koutmani Y, Brugni J, Kokkotou E,
Pothoulakis C and Karalis KP: Corticotropin-releasing factor
regulates TLR4 expression in the colon and protects mice from
colitis. Gastroenterology. 139:2083–2092. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cantarella G, Lempereur L, Lombardo G,
Chiarenza A, Pafumi C, Zappla G and Bernadini R: Divergent effects
of corticotropin releasing hormone on endothelial cell nitric oxide
synthase are associated with different expression of CRH type 1 and
2 receptors. Br J Pharmacol. 134:837–844. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dieleman LA, Palmen MJ, Akol H, Bloemena
E, Peña AS, Meuwissen SG and Van Rees EP: Chronic experimental
colitis induced by dextran sulphate sodium (DSS) is characterized
by Th1 and Th2 cytokines. Clin Exp Immunol. 114:385–391. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mahida YR, Wu K and Jewell DP: Enhanced
production of interleukin 1-beta by mononuclear cells isolated from
mucosa with active ulcerative colitis of Crohn's disease. Gut.
30:835–838. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tsatsanis C, Androulidaki A, Dermitzaki E,
Charalampopoulos I, Spiess J, Gravanis A and Margioris AN:
Urocortin 1 and urocortin 2 induce macrophage apoptosis via CRFR 2.
FEBS Lett. 579:4259–4264. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsatsanis C, Androulidaki A, Dermitzaki E,
Gravanis A and Margioris AN: Corticotropin releasing factor
receptor 1 (CRF 1) and CRF 2 agonists exert an anti-inflammatory
effect during the early phase of inflammation suppressing
LPS-induced TNF-alpha release from macrophages via induction of
COX-2 and PGE2. J Cell Physiol. 210:774–783. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Neurath MF, Pettersson S, zum Büschenfelde
Meyer KH and Strober W: Local administration of antisense
phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B
abrogates established experimental colitis in mice. Nat med.
2:998–1004. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rogler G, Brand K, Vogl D, Page S,
Hofmeister R, Andus T, Knuechel R, Baeuerle PA, Schölmerich J and
Gross V: Nuclear factor kappaB is activated in macrophages and
epithelial cells of inflamed intestinal mucosa. Gastroenterology.
115:357–369. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsatsanis C, Androulidaki A, Alissafi T,
Charalampopoulos I, Dermitzaki E, Roger T, Gravanis A and Margioris
AN: Corticotropin-releasing factor and the urocortins induce the
expression of TLR4 in macrophages via activation of the
transcription factors PU.1 and AP-1. J Immunol. 176:1869–1877.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Teitelbaum AA, Gareau MG, Jury J, Yang PC
and Perdue MH: Chronic peripheral administration of
corticotropin-releasing factor causes colonic barrier dysfunction
similar to psychological stress. Am J Physiol Gastrointest Liver
Physil. 295:G452–G459. 2008. View Article : Google Scholar
|
|
22
|
Saito-Nakaya K, Hasegawa R, Nagura Y, Ito
H and Fukudo S: Corticotropin-releasing hormone receptor 1
antagonist blocks colonic hypersensitivity induced by a combination
of inflammation and repetitive colorectal distension.
Neurogastroenterol Motil. 20:1147–1156. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Castagliuolo I, Lamont JT, Qiu B, Fleming
SM, Bhaskar KR, Nikulasson ST, Kornetsky C and Pothoulakis C: Acute
stress causes mucin release from rat colon: Role of corticotropin
releasing factor and mast cells. Am J Physiol. 271:G884–G892.
1996.PubMed/NCBI
|
|
24
|
Cooper HS, Murthy SN, Shah RS and
Sedergran DJ: Clinicopathologic study of dextran sulfate sodium
experimental murine colitis. Lab Invest. 69:238–249.
1993.PubMed/NCBI
|
|
25
|
Van der Sluis M, De Koning BA, De Bruijn
AC, Velcich A, Meijerink JP, Van Goudoever JB, Büller HA, Dekker J,
Van Seuningen I, Renes IB and Einerhand AW: Muc2-deficient mice
spontaneously develop colitis, indicating that Muc2 is critical for
colonic protection. Gastroenterology. 131:117–129. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wlk M, Wang CC, Venihaki M, Liu J, Zhao D,
Anton PM, Mykoniatis A, Pan A, Zacks J, Karalis K and Pothoulakis
C: Corticotropin-releasing hormone antagonists possess
anti-inflammatory effects in the mouse ileum. Gastroenterology.
123:505–515. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Saruta M, Takahashi K, Suzuki T, Torii A,
Kawakami M and Sasano H: Urocortin 1 in colonic mucosa in patients
with ulcerative colitis. J Clin Endocrinol Metab. 89:5352–5361.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Im E, Rhee SH, Park YS, Fiocchi C, Taché Y
and Pothoulakis C: The corticotropin releasing hormone family of
peptides regulates intestinal angiogenesis. Gastroenterology.
138:2457–2467. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pandurangan AK, Ismail S, Saadatdoust Z
and Esa NM: Allicin alleviates dextran sodium sulfate-(DSS-)
induced ulcerative colitis in BALB/c mice. Oxid Med Cell Longev.
2015:6052082015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pandurangan AK, Mohebali N, Mohd EN, Looi
CY, Ismail S and Saadatdoust Z: Gallic acid suppresses inflammation
in dextran sodium sulfate-induced colitis in mice: Possible
mechanisms. Int Immunopharmacol. 28:1034–1043. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kokkotou E, Torres D, Moss AC, O'Brien M,
Grigoriadis DE, Karalis K and Pothoulakis C:
Corticotropin-releasing hormone receptor 2-deficient mice have
reduced intestinal inflammatory responses. J Immunol.
177:3355–3361. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moss AC, Anton P, Savidge T, Newman P,
Cheifetz AS, Gay J, Paraschos S, Winter MW, Moyer MP, Karalis K, et
al: Urocortin II mediates pro-inflammatory effects in human
colonocytes via corticotropin-releasing hormone receptor 2 alpha.
Gut. 56:1210–1217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hermoso MA, Matsuguchi T, Smoak K and
Cidlowski JA: Glucocorticoids and tumor necrosis factor alpha
cooperatively regulate toll-like receptor 2 gene expression. Mol
Cell Biol. 24:4743–4756. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Murray PJ, Allen JE, Biswas SK, Fisher EA,
Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence
T, et al: Macrophage activation and polarization: Nomenclature and
experimental guidelines. Immunity. 41:14–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Van Welden S, De Vos M, Wielockx B,
Tavernier SJ, Dullaers M, Neyt S, Descamps B, Devisscher L,
Devriese S, Van den Bossche L, et al: Haematopoietic prolyl
hydroxylase-1 deficiency promotes M2 macrophage polarization and is
both necessary and sufficient to protect against experimental
colitis. J Pathol. 241:547–558. 2017. View Article : Google Scholar : PubMed/NCBI
|