Introduction
Chronic myeloid leukemia (CML) is a rare hematologic
disease with low incidence but increasing prevalence (1). This progressive, hematopoietic
neoplasm is characterized by the presence of the BCR-ABL1
hybrid gene that is localized on the-so called Philadelphia (Ph+)
chromosome [t(9;22) (q34;q11)] (2)-which leads to the constitutively
active tyrosine kinase (TK) BCR-ABL1 causing leukemic cell
transformation (3–5). As the oncogenic TK BCR-ABL1 is
responsible for initiating the disease process (6), selective TK inhibitors (TKI) such as
imatinib (IMA; Glivec®/Gleevec®: Novartis,
Basel, Switzerland) were developed. Since 2001, (7–12)
IMA has become the standard front-line therapy for the treatment of
CML in adults (13). For pediatric
patients with CML, IMA was approved in Germany in 2003. However,
due to the increasing resistance or intolerance of leukemic cells
to IMA therapy (14),
second-generation TKIs like nilotinib (NIL; Tasigna®;
Novartis, Basel, Switzerland) were developed. NIL, an
aminopyrimidine-derivative based on imatinib mesylate (15), has a 20- to 50-fold higher
inhibitory activity in IMA-sensitive cells and a 3 to 7 times
higher inhibitory activity in IMA-resistant cells due to its higher
potency and selectivity for the BCR-ABL1 TK (16). Based upon its efficacy, NIL was
approved for the treatment of adult patients with CML in chronic
and advanced phases after IMA failure or intolerance in 2008
(1).
However, both TKIs show off-target effects on
further TKs such as PDGFR and CSF1R (c-FMS), which are involved in
the bone remodeling cycle. Especially for IMA it is known that
under prolonged treatment, adult CML patients revealed
hypophosphatemia and an increased bone mineralization whereas
pediatric CML patients develop growth retardation in up to 72.9% of
the cases (17–22).
Reports of growth retardation due to a long-term
application of IMA and related TKIs are increasing (13,17,21,23,24)
and are even more prominent in those patients, who started IMA
therapy at a prepubertal age. Additionally, pediatric patients
display reduced serum levels of 25-hydroxy-vitamin D3
(25-OH-VD3; calcidiol) and 1.25-dihydroxyvitamin
D3 (1.25-(OH)2-VD3; calcitriol) (25) under IMA treatment. At least, the
effects for NIL are expected to have a similar potential for
skeletal effects compared to IMA.
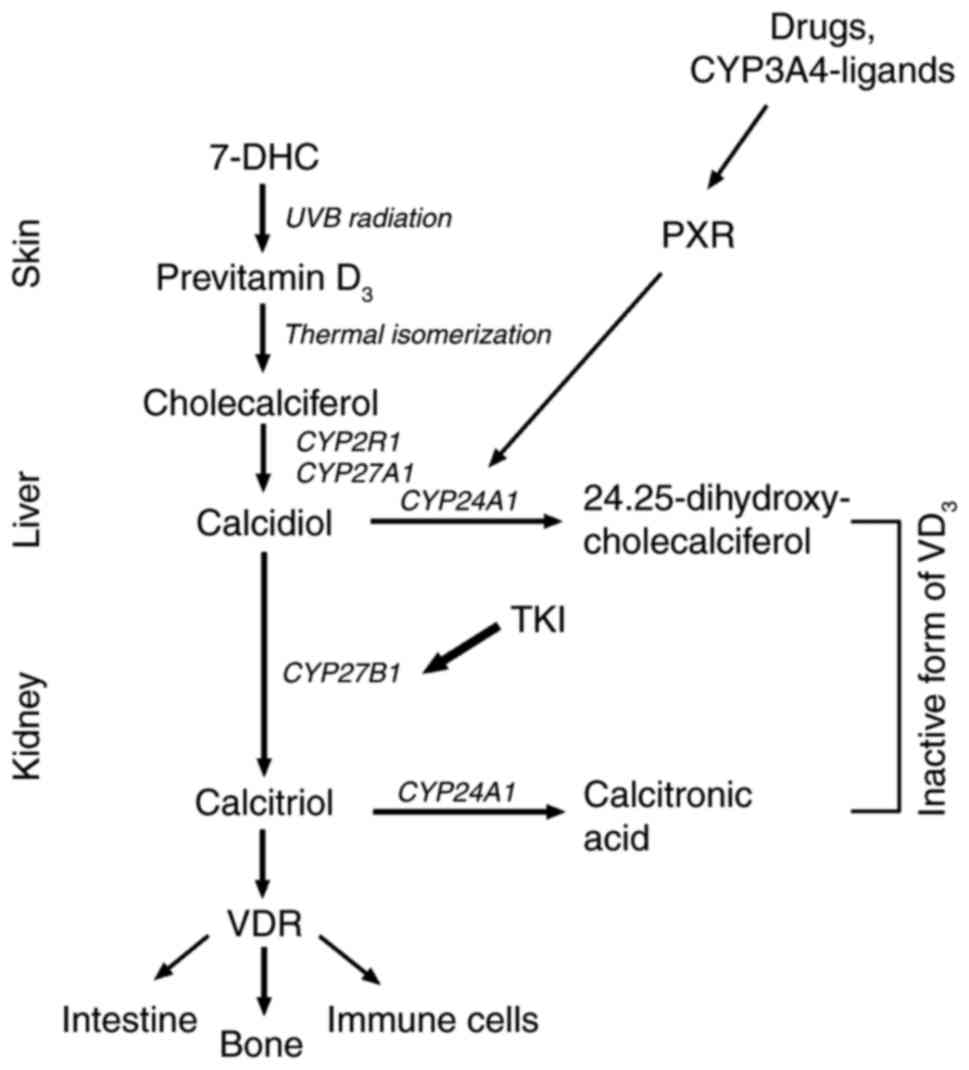
Vitamin D3 (VD3) synthesis is
initiated by UVB-induced photolysis of 7-dehydrocholesterol (7-DHC)
into previtamin D3 (26) that is then enzymatically
hydroxylated to calcidiol by CYP2R1 and/or CYP27A1 (27) in the liver which is further
metabolized to hormonally active calcitriol by CYP27B1 (28–30)
in the kidney (Fig. 1).
As calcitriol is essential in regulating the blood
levels of calcium and phosphorus (32), it plays a key role during bone
mineralization (33–35). Clinical studies revealed an
impaired growth especially during puberty and prepuberty under IMA
treatment (36). Furthermore an
association of VD3 deficiency was shown indicated by low
calcidiol/calcitriol blood levels, under IMA treatment as well as
impaired longitudinal growth (25).
In a previous study, the effect of IMA on
VD3 synthesis was investigated in HaCaT cells and
revealed significantly reduced calcitriol levels up to ~50%,
compared to untreated controls (37). However, as the mechanism is poorly
understood, the aim of the present study was to investigate the
effects of NIL in comparison to IMA and to elucidate the causative
mechanisms for this effect by means of the immortalized cell line
HaCaT and human keratinocytes expanded in culture from hair
follicles collected from pediatric CML patients under IMA
treatment.
Materials and methods
Cell culture protocol and cell
isolation
The human keratinocyte cell line HaCaT was purchased
from Leibniz Institute DSMZ-German Collection of Microorganisms and
Cell Cultures (Braunschweig, Germany). Cells were seeded at a
density of 1×105 cells/cm2 and grown in
Dulbecco's modified Eagle's medium (DMEM, Gibco, Eggenstein,
Germany) supplemented with 10% fetal bovine calf serum (FCS; Gibco,
Eggenstein, Germany) at 95% relative humidity, 5% CO2
and 37°C for 48 h. Subsequently, the medium was replaced for 18 h
by serum-free DMEM to induce synchronization of the cell cycle.
Afterwards cells were grown in fetal bovine serum-supplemented DMEM
for 8 h until they were almost confluent. To investigate vitamin
D3 metabolism, cells were seeded at a density of
5×104 cells/cm2 in culture dishes (Ø 30
mm).
ORS-KCs were prepared from human scalp hair
follicles of IMA-treated children and their healthy siblings
hailing from different regions all over Germany. Because of the
disease rareness, 16 IMA-treated children and adolescents between
10 and 22 years (ø16±4 years old; 6 male and 12 female), and 15
healthy subjects between 2 and 33 years old (ø15±11 years old; 7
male and 8 female) take part of this study. An ethic statement of
the University Hospital Carl Gustav Carus (EK28212200) and an
International Clinical Trials Identifier (NCT00445822) was
approved. Hair follicles were plucked by using a pair of tweezers
and the bulk of the hair shaft was cropped while the hair follicle
was immersed in DMEM buffered with 1 M HEPES (Gibco) and
supplemented with 1% PenStrep (Gibco) for 24 h. Afterwards, hair
follicles were applied on a feeder layer of 3T3 fibroblasts,
previously treated with 0.004 µg/ml mitomycin C (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany), and cultivated in a complex medium
containing 3 parts DMEM and 1 part HAMS F12 supplemented with 10%
FCS, 0.135 mM adenine (Sigma-Aldrich; Merck KGaA), 0.1 nM cholera
toxine (Sigma-Aldrich; Merck KGaA), 2 nM triiodothyronine
(Sigma-Aldrich; Merck KGaA), 1 pack epithelial cell growth medium
supplements (containing epidermal growth factor, hydrocortisone,
insulin and transferrine (Promocell, Heidelberg, Germany) 1%
PenStrep, 1% sodium pyruvate (100 mM; Gibco) and incubated at 95%
relative humidity, 5% CO2 and 37°C. Medium was changed 3
times a week. After 2–3 weeks in primary culture, 3T3 cells were
removed by trypsination and ORS-KCs were replated at a density of
1×105 cells/cm2 and grown in DermaLife K
complete medium (Cellsystems, Troisdorf, Germany).
Vitamin D3 assay
For investigation of vitamin D3
metabolism, HaCaT cells were incubated with 25 µM 7-DHC (dissolved
in 100% ethanol; Sigma-Aldrich; Merck KGaA) as substrate and
exposed to UVB (300 nm; application rate: 75 mJ/cm2).
Irradiation was carried out by using a tuneable high intensity
monochromator (FWHM, 5 nm; Dermolum Um, Müller Optik-Elektronik,
Moosinning, Germany). During irradiation, IMA or NIL (provided by
Novartis, Basel, Switzerland) were added to the cell culture medium
at a concentration of 1 µM (dissolved in 100% DMSO; Sigma-Aldrich;
Merck KGaA), respectively. After UVB irradiation and incubation for
24, 48 or 72 h, the medium and detached cells were collected and
extracted in a methanol:chloroform (1:1) (Sigma-Aldrich; Merck
KGaA) solution. Chloroform phase was used for quantitative
determination of calcidiol and calcitriol levels by using
commercially available enzyme assays (IDS, Frankfurt, Germany).
Results were normalized to 1×106 cells.
To analyze if the VD3 processing enzymes
CYP2R1, CYP27A1 and CYP27B1 are inhibited by IMA or NIL, specific
inhibitors of cytochrome P450 enzyme family (VID400, ketoconazol,
both Sigma-Aldrich; Merck KGaA) were investigated. Experiments were
carried out without irradiation. Cells were incubated for 0, 2, and
4 h with either 5 µM cholecalciferol or 5 µM calcidiol (both
Sigma-Aldrich; Merck KGaA and both dissolved in 100% ethanol) as
substrate. Before substrate incubation, cells were treated for 1 h
either with 200 nM VID400 or 10 µM ketoconazole (both dissolved in
100% ethanol) alone or in combination with 1 µM IMA or NIL,
respectively. All experiments were repeated at least 4 times.
Statistical analysis
Statistical analysis at defined time points of
incubation was performed using one-way analysis of variance with
Bonferroni adjustment of P-values to evaluate the effects of IMA or
NIL-treated samples compared with untreated controls, using
GraphPad Prism 6.0 software (GraphPad Software, Inc., La Jolla, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Inhibitory effect of TKI on calcitriol
synthesis in HaCaT and ORS-KCs
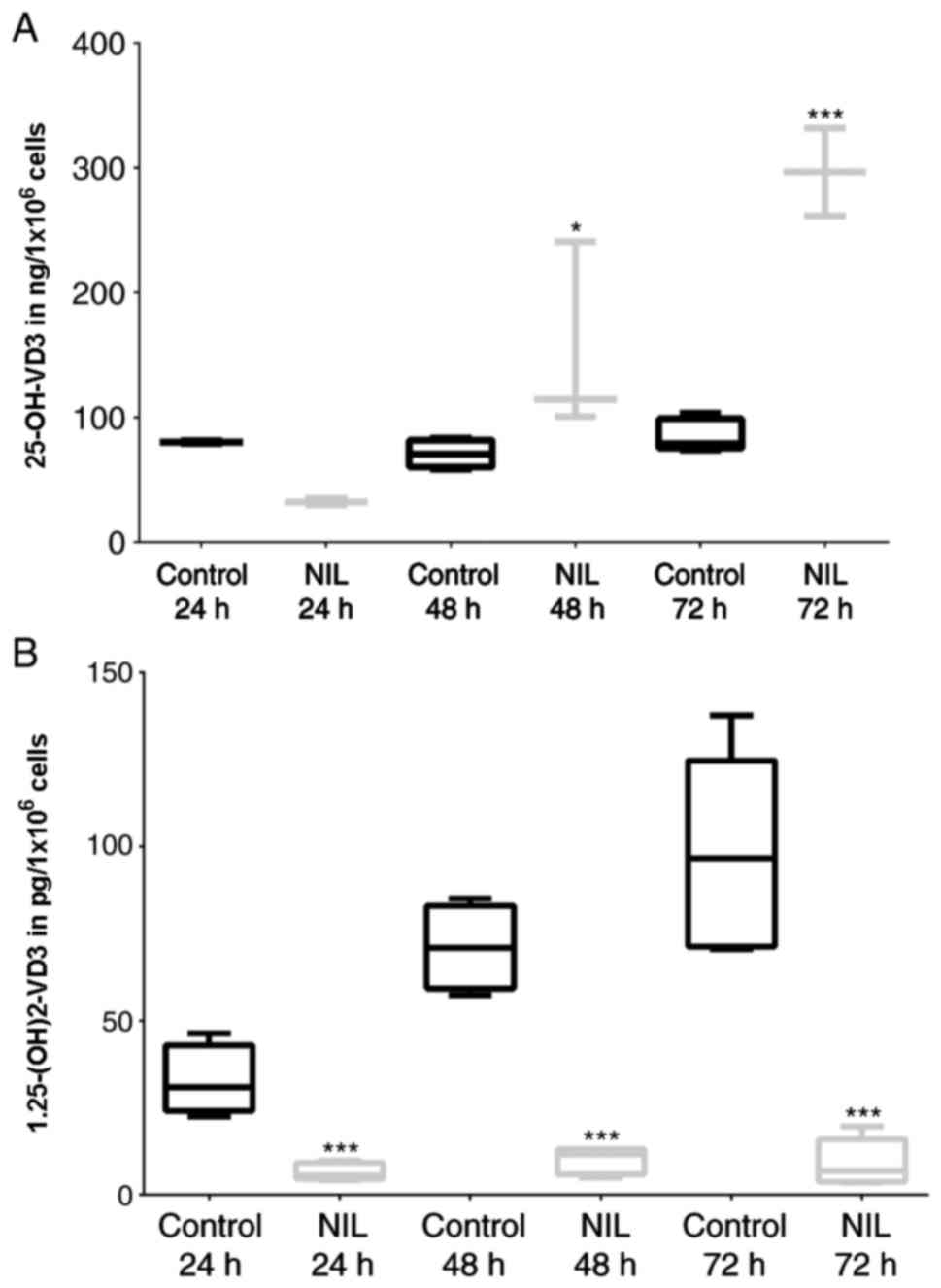
To determine the effect of IMA and NIL treatment on
VD3 metabolism, we cultured confluent HaCaT cells for a
maximum of 72 h with TKI (clinically effective concentration: 1 µM)
and measured calcidiol and calcitriol levels. Using 7-DHC as
substrate, NIL significantly increased calcidiol levels to 300% in
comparison to untreated controls (Fig.
2A) and significantly reduced calcitriol levels to 10%
(Fig. 2B). These data were
verified by repeating the experiments without irradiation and using
cholecalciferol as synthesis starting substrate.
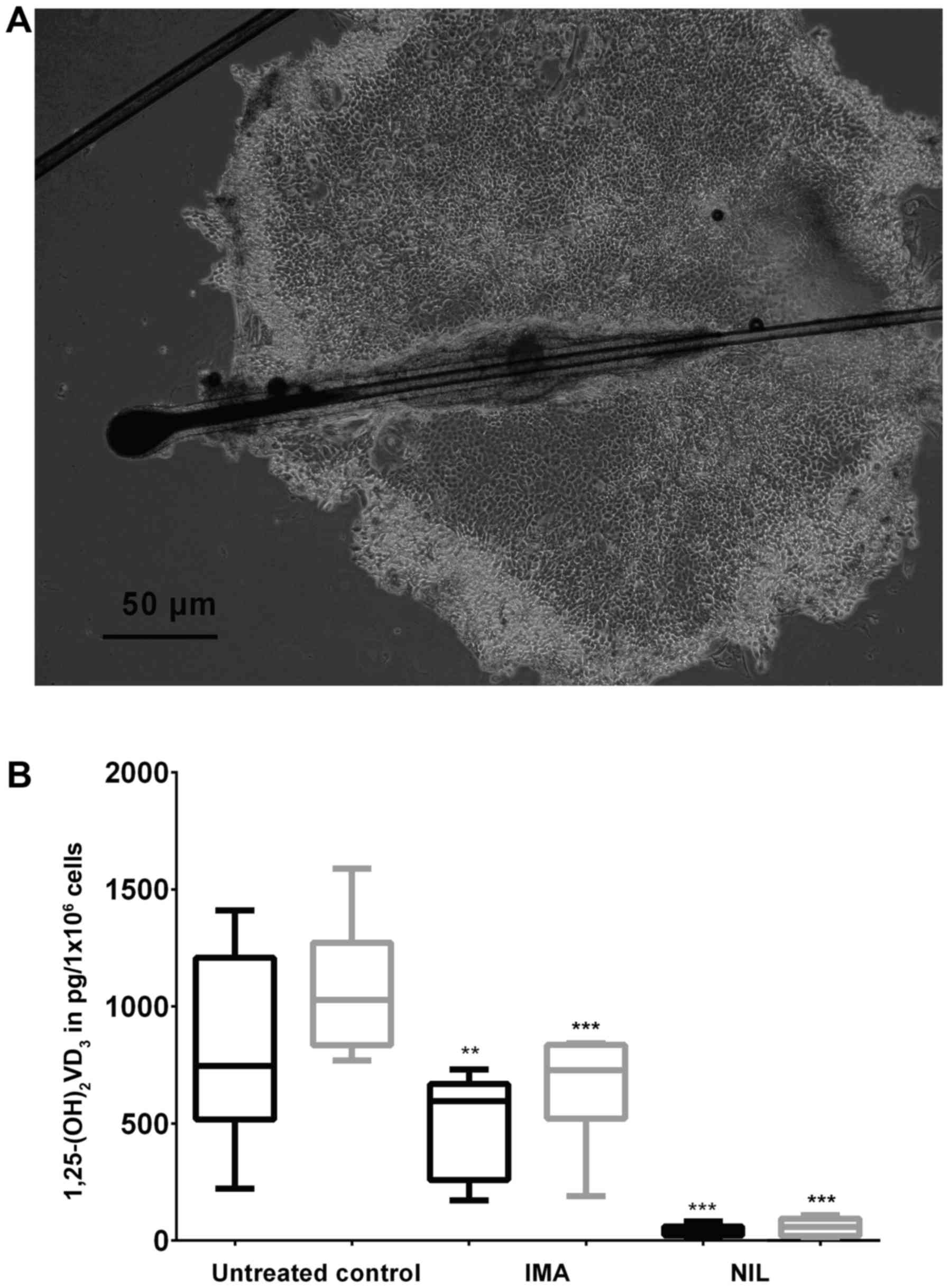
The same effect was found by repeating the described
experiments using ORS-KCs from IMA-treated children with CML and
their healthy siblings as controls. The experiments with ORS-KCs
were performed with IMA and NIL, respectively. Data of IMA-treated
children and healthy subjects were summarized and shown as one bar,
respectively (Fig. 3). However,
compared to KCs of healthy subjects, KCs of children with CML
revealed no differences in their capability to synthesize
calcitriol under identical physiological conditions.
Effects of TKI in presence of specific
inhibitors on the vitamin D3 cascade
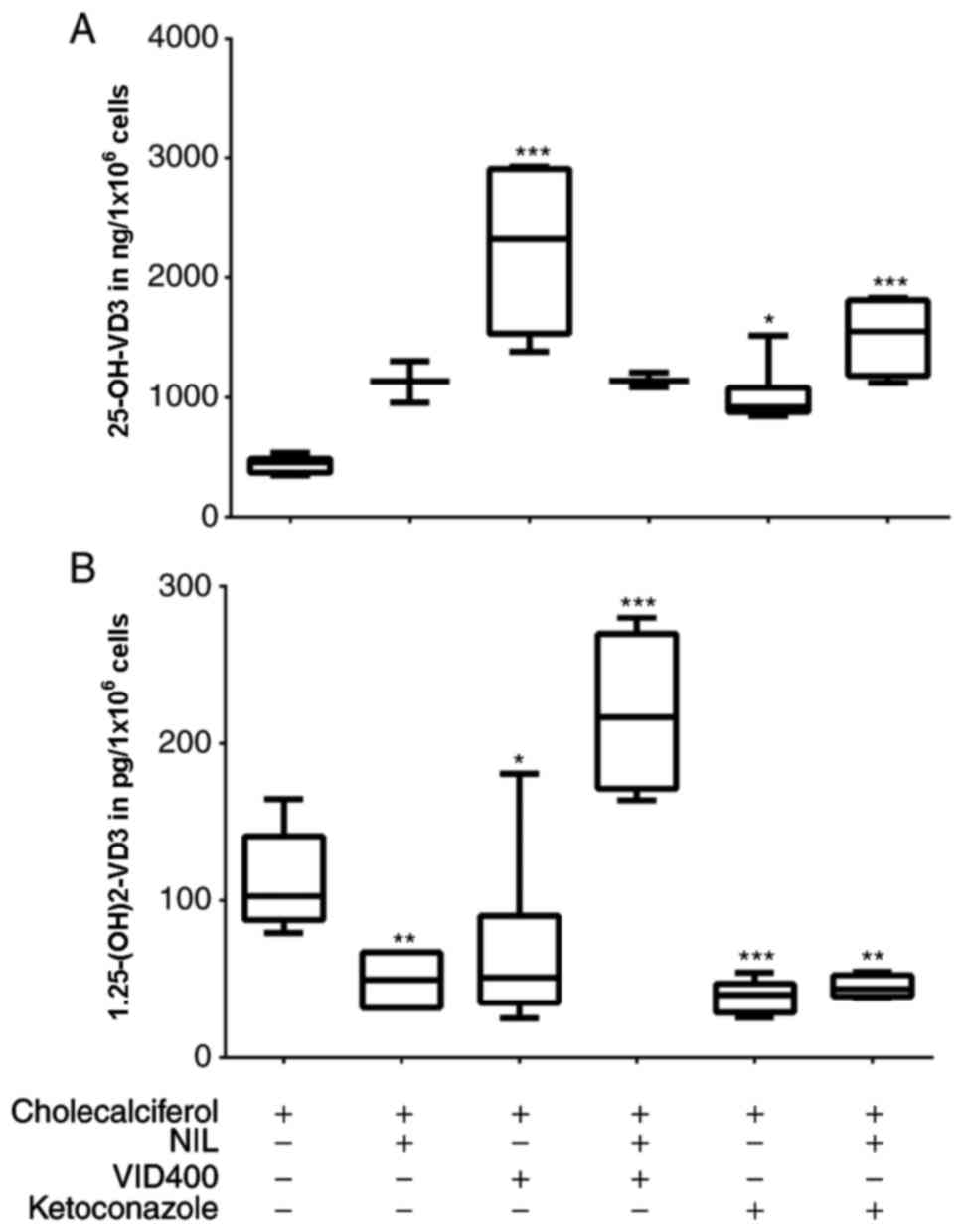
For identification of the potential target of TKI
within the VD3 cascade, we examined confluent HaCaT
cells under exposure to selective cytochrome P450 inhibitors such
as VID400 and ketoconazole. While ketoconazole is known to be a
general inhibitor of P450 enzymes, VID400 only blocks CYP24A1 at a
specific concentration. Experiments were carried out in combination
with and without TKI by using cholecalciferol as synthesis-starting
substrate, so that no irradiation of cells was necessary.
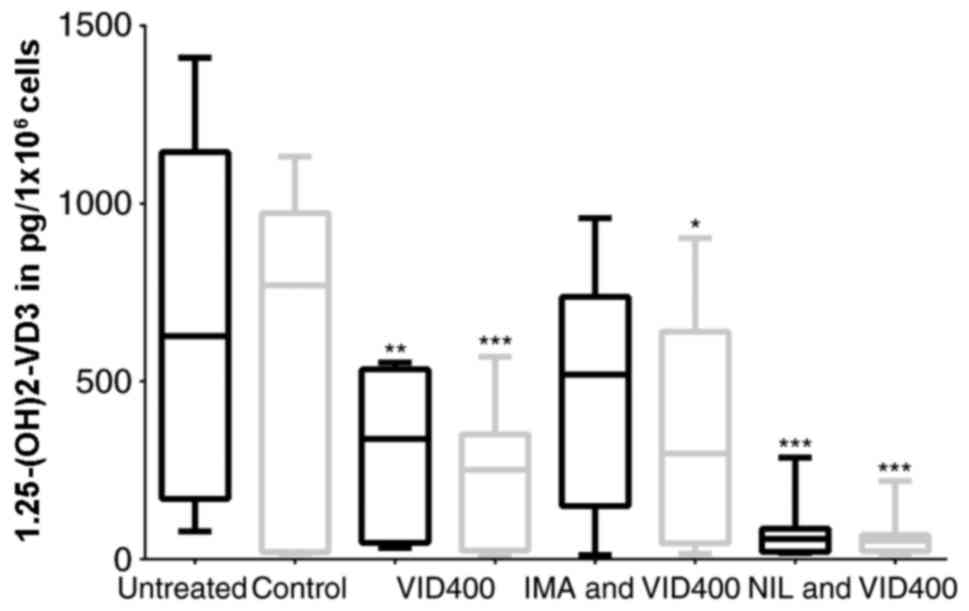
The results were comparable to those described
before. Cells treated with NIL alone revealed an increase of
calcidiol level to 250% whereas calcitriol levels were lowered down
to 50% in comparison to those without TKI (Fig. 4). Treating cells with NIL and
VID400 revealed calcitriol levels at the same level as cells
treated with NIL alone, while calcidiol levels were significantly
increased to 400% (Fig. 4).
Treatment with NIL and ketoconazole had no remarkable effect in
HaCaT cell line. Experiments with ORS-KCs were repeated with TKI
and VID400 treatment only. Similar to the results for the HaCaT
cell line, ORS-KCs from IMA-treated children with CML and their
healthy siblings showed under TKI and VID400 treatment reduced
calcitriol levels whereby this effect was more pronounced with NIL
as IMA (Fig. 5). No difference was
detectable between the VD3 synthesis of ORS-KCs from
IMA-treated children with CML and their healthy siblings. Repeating
the experiments with calcidiol as substrate showed the same effect
confirming the observed data.
Discussion
VD3 plays a primary role in the human
body by maintaining the extracellular calcium level, acts as an
important immune modulator, potentiates apoptosis or inhibits
angiogenesis (38). Especially in
children, VD3 is necessary during bone mineralization
and in this context for growth but also for prevention of rickets
(39).
The presented study describes an off-target effect
of the TKIs IMA and NIL on human VD3 metabolism, which
might play a central role in the complexity of longitudinal growth
retardation during CML therapy with TKI treatment. Under prolonged
IMA therapy, growth retardation is increasingly reported as a main
side effect in children (18–20,40–50).
Additionally, VD3 deficiency is often described in
children who have been treated for different kinds of cancer
(38,51) may due to lack of sun exposure
and/or poor nutrition and/or drug interactions (51). Concerning pediatric CML patients,
Jaeger et al (25)
investigated for the first time serum bone markers in 17 pediatric
patients with CML (age: 4–17 years) under ongoing IMA therapy and
reported VD3 insufficiency or deficiency in addition to
impaired bone metabolism (25). As
it is now speculative if VD3 insufficiency or deficiency
is caused by the disease itself, the impaired bone metabolism or
due to a direct effect of TKI on VD3 metabolism, we
investigated the inhibitory effect of IMA and NIL on VD3
metabolism in human keratinocyte cell line HaCaT and ORS-KCs of
IMA-treated children.
In the skin synthesized VD3 undergoes
25-hydroxylation in the liver followed by 1α-hydroxylation in the
kidney to build the biologically active hormone. For catalysing the
25-hydroxylation step in the liver, at least six cytochrome P450
enzymes (CYPs) are involved whereby CYP27A1 and CYP2R1 (52) are the most viable ones. In the
kidney, CYP27B1 is responsible for 1α-hydroxylation of
VD3 to hormonally active calcitriol (Fig. 1). These enzymes are also found in
various extra renal tissues including epidermal keratinocytes.
Keratinocytes are able to synthesize and catabolize calcitriol as
well as harbouring the vitamin D receptor (VDR) (53). As described for the TKI IMA before
(37), IMA inhibits CYP27B1
leading to a decrease of calcitriol in combination with an increase
of calcidiol in HaCaT and ORS-KC cells. However, here we could show
that NIL, according to its 20-fold stronger inhibition properties
to BCR-ABL1 (16), demonstrated
more pronounced inhibition of calcitriol synthesis up to 95% in
comparison to untreated controls. While IMA needs to be metabolized
by CYP3A4 and CYP3A5 to an active metabolite (54–56),
NIL itself is an orally active drug (15). This probably leads to an even more
rapid effect in comparison to IMA and agrees with our results.
Interestingly, independent of starting substrate,
TKI treatment, or application of CYP450 inhibitors differences
between OTC-KCs of IMA-treated patients and healthy siblings and
their ability to synthesize calcidiol or calcitriol were not
detected. This could be explained by the extensive cultivation
period of the primary culture where the majority of OTC-KCs from
IMA-treated children seem to be TKI naïve and thus a possible
effect of long-term application of TKI on the cells would be lost.
Gender and age of the IMA-treated children, adolescents and healthy
subjects had no influence on the outcomes. Therefore, concerning
their physiological VD3 metabolism, OTC-KCs of IMA-treated children
are comparable to cells of healthy siblings.
For inhibition of specific enzymes involved in the
VD3 cascade (CYP24A1, CYP27A1, CYP27B1), we used VID400
and ketoconazole. VID400 acts dose-dependently with complete
inhibition of CYP24A1 activity and partial inhibition of 30% of
CYP27B1 (57).
Here we could demonstrate that VID400 treatment
alone stabilized the levels of endogenously produced calcitriol in
HaCaT. In general, it is described that under VID400 treatment the
expression of the CYP24A1 enzyme is strongly amplified and
prolonged (58,59). CYP24A1 catalyses the metabolization
of calcidiol and calcitriol (Fig.
1) and is thereby regulated by a negative feedback loop of
calcitriol concentration. For cancer cells, especially for prostate
cancer cells, it has been suggested, that a rapid breakdown of the
calcitriol levels are caused by an overactive CYP24A1 (60).
VID400 in combination with TKI increased calcidiol
levels whereby the effect was more pronounced for NIL treatment in
comparison to IMA. However, this result indicates that beside an
inhibition of CYP24A1 by VID400, CYP27B1 might be affected by IMA
(37) and NIL resulting in an
accumulation of calcidiol. This may be due to the binding affinity
of IMA and NIL to microsomal 25-hydroxylases. IMA and NIL are both
metabolized by cytochrome P450 isoenzymes like CYP3A4 and CYP3A5 in
the liver (54,61). Like CYP3A4, CYP27B1 in VD3 cascade
is known to be a human microsomal vitamin D 25-hydroxylase as well
(62).
The antifungal agent ketoconazole is a known general
CYP inhibitor (63) including
vitamin D hydroxylating enzymes such as CYP24A1, CYP27A1 and
CYP27B1 (64). Here we could
demonstrate that a treatment with ketoconazole led to increased
calcidiol and decreased calcitriol levels. The same effect was
shown with an application of ketoconazole and NIL.
We conclude that NIL interferes with the binding of
ketoconazole and might compete for binding sites on one or more
CYPs. In regard to the described interaction with CYP27B1 (37) this is also displaying the reason
for the interference of TKI with the vitamin D3
metabolism.
To summarize, our results indicate a competitive
inhibition of CYP27B1 by IMA and NIL, but being more pronounced by
NIL. Because CYPs in general act dose-dependently to redress a
balance of metabolites, increasing calcidiol levels resulted in
decreasing calcitriol levels. Keeping in mind the stronger
properties of NIL in comparison to IMA possibly such distinctive
effects in another context e.g., calcitriol synthesis are
supposable.
In addition to the inhibition of CYP27B1 and as
described for different drugs, an additional impairment of CYP24A1
is imaginable, leading to elevated calcidiol levels. However, the
detailed mechanism remains weakly understood and additional
investigations are needed. Knowing that pediatric oncology patients
would have a- at least transiently-higher prevalence of
VD3 hypovitaminosis (25,38),
further investigations are needed to identify the reasons for
VD3 deficiency in children with CML exhibiting growth
delay.
Acknowledgements
The authors of the present study would like to thank
Mr. Peter Knuschke for the introduction to solar radiation and for
scientific discussion. They are also grateful to Novartis Pharma AG
(grant no. HTAS-079; Basel, Switzerland) for the supply of TKIs and
financial support of this study.
Glossary
Abbreviations
Abbreviations:
|
7-DHC
|
7-dehydrocholesterol
|
|
CYP2R1
|
cytochrome P450 family 2, subfamily R,
polypeptide 1 (vitamin D 25-hydroxylase)
|
|
CYP24A1
|
cytochrome P450, family 22, subfamily
a, polypeptide1 (1.25-dihydroxyvitamin D3
24-hydroxylase)
|
|
CYP27A1
|
cytochrome P450, family 27, subfamily
A, polypeptide 1 (vitamin D 25-hydroxylase)
|
|
CYP27B1
|
cytochrome P450, family 27, subfamily
B, polypeptide 1 (1α-Hydroxylase)
|
|
PXR
|
pregnan × receptor
|
|
VDR
|
vitamin D receptor
|
|
VD3
|
vitamin D3
|
References
|
1
|
Jabbour E, El AS, Cortes J and Kantarjian
H: Nilotinib: A novel Bcr-Abl tyrosine kinase inhibitor for the
treatment of leukemias. Expert Opin Investig Drugs. 17:1127–1136.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tipping AJ, Mahon FX, Zafirides G, Lagarde
V, Goldman JM and Melo JV: Drug responses of imatinib
mesylate-resistant cells: Synergism of imatinib with other
chemotherapeutic drugs. Leukemia. 16:2349–2357. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Capdeville R, Silberman S and Dimitrijevic
S: Imatinib: The first 3 years. Eur J Cancer. 38 Suppl 5:S77–S82.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Daley GQ, Van Etten RA and Baltimore D:
Induction of chronic myelogenous leukemia in mice by the
P210bcr/abl gene of the Philadelphia chromosome. Science.
247:824–830. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cohen MH, Williams G, Johnson JR, Duan J,
Gobburu J, Rahman A, Benson K, Leighton J, Kim SK, Wood R, et al:
Approval summary for imatinib mesylate capsules in the treatment of
chronic myelogenous leukemia. Clin Cancer Res. 8:935–942.
2002.PubMed/NCBI
|
|
6
|
Pasternak G, Hochhaus A, Schultheis B and
Hehlmann R: Chronic myelogenous leukemia: Molecular and cellular
aspects. J Cancer Res Clin Oncol. 124:643–660. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Champagne MA, Capdeville R, Krailo M, Qu
W, Peng B, Rosamilia M, Therrien M, Zoellner U, Blaney SM and
Bernstein M: Children's Oncology Group phase 1 study: Imatinib
mesylate (STI571) for treatment of children with Philadelphia
chromosome-positive leukemia: Results from a children's oncology
group phase 1 study. Blood. 104:2655–2660. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Druker BJ, Tamura S, Buchdunger E, Ohno S,
Segal GM, Fanning S, Zimmermann J and Lydon NB: Effects of a
selective inhibitor of the Abl tyrosine kinase on the growth of
Bcr-Abl positive cells. Nat Med. 2:561–566. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Druker BJ, Talpaz M, Resta DJ, Peng B,
Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R,
Ohno-Jones S and Sawyers CL: Efficacy and safety of a specific
inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid
leukemia. N Engl J Med. 344:1031–1037. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grigg A and Hughes T: Role of allogeneic
stem cell transplantation for adult chronic myeloid leukemia in the
imatinib era. Biol Blood Marrow Transplant. 12:795–807. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Millot F, Guilhot J, Nelken B, Leblanc T,
De Bont ES, Békassy AN, Gadner H, Sufliarska S, Stary J,
Gschaidmeier H, et al: Imatinib mesylate is effective in children
with chronic myelogenous leukemia in late chronic and advanced
phase and in relapse after stem cell transplantation. Leukemia.
20:187–192. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Roy L, Guilhot J, Krahnke T,
Guerci-Bresler A, Druker BJ, Larson RA, O'Brien S, So C, Massimini
G and Guilhot F: Survival advantage from imatinib compared with the
combination interferon-alpha plus cytarabine in chronic-phase
chronic myelogenous leukemia: Historical comparison between two
phase 3 trials. Blood. 108:1478–1484. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hobernicht SL, Schweiger B, Zeitler P,
Wang M and Hunger SP: Acquired growth hormone deficiency in a girl
with chronic myelogenous leukemia treated with tyrosine kinase
inhibitor therapy. Pediatr Blood Cancer. 56:671–673. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deguchi Y, Kimura S, Ashihara E, Niwa T,
Hodohara K, Fujiyama Y and Maekawa T: Comparison of imatinib,
dasatinib, nilotinib and INNO-406 in imatinib-resistant cell lines.
Leuk Res. 32:980–983. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim TD, le Coutre P, Schwarz M, Grille P,
Levitin M, Fateh-Moghadam S, Giles FJ, Dörken B, Haverkamp W and
Köhncke C: Clinical cardiac safety profile of nilotinib.
Haematologica. 97:883–889. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kantarjian H, Giles F, Wunderle L, Bhalla
K, O'Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W,
et al: Nilotinib in imatinib-resistant CML and Philadelphia
chromosome-positive ALL. N Engl J Med. 354:2542–2551. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shima H, Tokuyama M, Tanizawa A, Tono C,
Hamamoto K, Muramatsu H, Watanabe A, Hotta N, Ito M, Kurosawa H, et
al: Distinct impact of imatinib on growth at prepubertal and
pubertal ages of children with chronic myeloid leukemia. J Pediatr.
159:676–681. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Berman E, Nicolaides M, Maki RG, Fleisher
M, Chanel S, Scheu K, Wilson BA, Heller G and Sauter NP: Altered
bone and mineral metabolism in patients receiving imatinib
mesylate. N Engl J Med. 354:2006–2013. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fierro F, Illmer T, Jing D, Schleyer E,
Ehninger G, Boxberger S and Bornhäuser M: Inhibition of
platelet-derived growth factor receptorbeta by imatinib mesylate
suppresses proliferation and alters differentiation of human
mesenchymal stem cells in vitro. Cell Prolif. 40:355–366. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fitter S, Dewar AL, Kostakis P, To LB,
Hughes TP, Roberts MM, Lynch K, Vernon-Roberts B and Zannettino AC:
Long-term imatinib therapy promotes bone formation in CML patients.
Blood. 111:2538–2547. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schmid H, Jaeger BA, Lohse J and Suttorp
M: Longitudinal growth retardation in a prepuberal girl with
chronic myeloid leukemia on long-term treatment with imatinib.
Haematologica. 94:1177–1179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hijiya N, Schultz KR, Metzler M, Millot F
and Suttorp M: Pediatric chronic myeloid leukemia is a unique
disease that requires a different approach. Blood. 127:392–399.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kimoto T, Inoue M and Kawa K: Growth
deceleration in a girl treated with imatinib. Int J Hematol.
89:251–252. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mariani S, Giona F, Basciani S, Brama M
and Gnessi L: Low bone density and decreased inhibin-B/FSH ratio in
a boy treated with imatinib during puberty. Lancet. 372:111–112.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jaeger BA, Tauer JT, Ulmer A, Kuhlisch E,
Roth HJ and Suttorp M: Changes in bone metabolic parameters in
children with chronic myeloid leukemia on imatinib treatment. Med
Sci Monit. 18:CR721–CR728. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lehmann B, Sauter W, Knuschke P, Dressler
S and Meurer M: Demonstration of UVB-induced synthesis of 1
alpha,25-dihydroxyvitamin D3 (calcitriol) in human skin by
microdialysis. Arch Dermatol Res. 295:24–28. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lehmann B and Meurer M: Vitamin D
metabolism. Dermatol Ther. 23:2–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Holick MF: Vitamin D deficiency. N Engl J
Med. 357:266–281. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Holick MF: Resurrection of vitamin D
deficiency and rickets. J Clin Invest. 116:2062–2072. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
DeLuca HF: Overview of general physiologic
features and functions of vitamin D. Am J Clin Nutr. 80 6
Suppl:1689S–1696S. 2004.PubMed/NCBI
|
|
31
|
Schuster I, Egger H, Herzig G, Reddy GS,
Schmid JA, Schüssler M and Vorisek G: Selective inhibitors of
vitamin D metabolism-new concepts and perspectives. Anticancer Res.
26:2653–2568. 2006.PubMed/NCBI
|
|
32
|
Bogh MK, Schmedes AV, Philipsen PA,
Thieden E and Wulf HC: Interdependence between body surface area
and ultraviolet B dose in vitamin D production: A randomized
controlled trial. Br J Dermatol. 164:163–169. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kremer R, Campbell PP, Reinhardt T and
Gilsanz V: Vitamin D status and its relationship to body fat, final
height, and peak bone mass in young women. J Clin Endocrinol Metab.
94:67–73. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Davis CD and Dwyer JT: The ‘sunshine
vitamin’: Benefits beyond bone? J Natl Cancer Inst. 99:1563–1565.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mathieu C and Badenhoop K: Vitamin D and
type 1 diabetes mellitus: State of the art. Trends Endocrinol
Metab. 16:261–266. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pettifor JM: Rickets and vitamin D
deficiency in children and adolescents. Endocrinol Metab Clin North
Am. 34(537–553): vii2005.
|
|
37
|
Mehlig LM, Garve C, Tauer JT, Suttorp M
and Bauer A: Inhibitory effects of imatinib on vitamin
D3 synthesis in human keratinocytes. Mol Med Rep.
11:3143–3147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Helou M, Ning Y, Yang S, Irvine P,
Bachmann LM, Godder K and Massey G: Vitamin D deficiency in
children with cancer. J Pediatr Hematol Oncol. 36:212–217. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lips P: Vitamin D status and nutrition in
Europe and Asia. J Steroid Biochem Mol Biol. 103:620–625. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tibullo D, Giallongo C, La Cava P,
Berretta S, Stagno F, Chiarenza A, Conticello C, Palumbo GA and Di
Raimondo F: Effects of imatinib mesylate in osteoblastogenesis. Exp
Hematol. 37:461–468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
O'Sullivan S, Naot D, Callon K, Porteous
F, Horne A, Wattie D, Watson M, Cornish J, Browett P and Grey A:
Imatinib promotes osteoblast differentiation by inhibiting PDGFR
signaling and inhibits osteoclastogenesis by both direct and
stromal cell-dependent mechanisms. J Bone Miner Res. 22:1679–1689.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dewar AL, Zannettino AC, Hughes TP and
Lyons AB: Inhibition of c-fms by imatinib: Expanding the spectrum
of treatment. Cell Cycle. 4:851–853. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dewar AL, Cambareri AC, Zannettino AC,
Miller BL, Doherty KV, Hughes TP and Lyons AB: Macrophage
colony-stimulating factor receptor c-fms is a novel target of
imatinib. Blood. 105:3127–3132. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dewar AL, Domaschenz RM, Doherty KV,
Hughes TP and Lyons AB: Imatinib inhibits the in vitro development
of the monocyte/macrophage lineage from normal human bone marrow
progenitors. Leukemia. 17:1713–1721. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Owen S, Hatfield A and Letvak L: Imatinib
and altered bone and mineral metabolism. N Engl J Med. 355:627–629.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
O'Sullivan S, Horne A, Wattie D, Porteous
F, Callon K, Gamble G, Ebeling P, Browett P and Grey A: Decreased
bone turnover despite persistent secondary hyperparathyroidism
during prolonged treatment with imatinib. J Clin Endocrinol Metab.
94:1131–1136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
El Hajj Dib I, Gallet M, Mentaverri R,
Sévenet N, Brazier M and Kamel S: Imatinib mesylate (Gleevec)
enhances mature osteoclast apoptosis and suppresses osteoclast bone
resorbing activity. Eur J Pharmacol. 551:27–33. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Grey A, O'Sullivan S, Reid IR and Browett
P: Imatinib mesylate, increased bone formation, and secondary
hyperparathyroidism. N Engl J Med. 355:2494–2495. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jönsson S, Olsson B, Ohlsson C, Lorentzon
M, Mellström D and Wadenvik H: Increased cortical bone
mineralization in imatinib treated patients with chronic
myelogenous leukemia. Haematologica. 93:1101–1103. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vandyke K, Fitter S, Dewar AL, Hughes TP
and Zannettino AC: Dysregulation of bone remodeling by imatinib
mesylate. Blood. 115:766–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Genc DB, Ozkan MA and Buyukgebiz A:
Vitamin D in childhood cancer: A promising anticancer agent?
Pediatr Endocrinol Rev. 10:485–493. 2013.PubMed/NCBI
|
|
52
|
Cheng JB, Levine MA, Bell NH, Mangelsdorf
DJ and Russell DW: Genetic evidence that the human CYP2R1 enzyme is
a key vitamin D 25-hydroxylase. Proc Natl Acad Sci USA.
101:7711–7715. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lehmann B, Rudolph T, Pietzsch J and
Meurer M: Conversion of vitamin D3 to 1alpha,25-dihydroxyvitamin D3
in human skin equivalents. Exp Dermatol. 9:97–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Peng B, Lloyd P and Schran H: Clinical
pharmacokinetics of imatinib. Clin Pharmacokinet. 44:879–894. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gschwind HP, Pfaar U, Waldmeier F,
Zollinger M, Sayer C, Zbinden P, Hayes M, Pokorny R, Seiberling M,
Ben-Am M, et al: Metabolism and disposition of imatinib mesylate in
healthy volunteers. Drug Metab Dispos. 33:1503–1512. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Rochat B: Role of cytochrome P450 activity
in the fate of anticancer agents and in drug resistance: Focus on
tamoxifen, paclitaxel and imatinib metabolism. Clin Pharmacokinet.
44:349–366. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xie Z, Munson SJ, Huang N, Portale AA,
Miller WL and Bikle DD: The mechanism of 1,25-dihydroxyvitamin D(3)
autoregulation in keratinocytes. J Biol Chem. 277:36987–36990.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Schuster I, Egger H, Reddy GS and Vorisek
G: Combination of vitamin D metabolites with selective inhibitors
of vitamin D metabolism. Recent Results Cancer Res. 164:169–188.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Schuster I, Egger H, Nussbaumer P and
Kroemer RT: Inhibitors of vitamin D hydroxylases:
Structure-activity relationships. J Cell Biochem. 88:372–380. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yee SW, Campbell MJ and Simons C:
Inhibition of Vitamin D3 metabolism enhances VDR signalling in
androgen-independent prostate cancer cells. J Steroid Biochem Mol
Biol. 98:228–235. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yin OQ, Gallagher N, Tanaka C, Fisher D,
Sethuraman V, Zhou W, Lin TH, Heuman D and Schran H: Effects of
hepatic impairment on the pharmacokinetics of nilotinib: An
open-label, single-dose, parallel-group study. Clin Ther.
31:2459–2469. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Gupta RP, Hollis BW, Patel SB, Patrick KS
and Bell NH: CYP3A4 is a human microsomal vitamin D 25-hydroxylase.
J Bone Miner Res. 19:680–688. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nguyen M, Boutignon H, Mallet E, Linglart
A, Guillozo H, Jehan F and Garabedian M: Infantile hypercalcemia
and hypercalciuria: New insights into a vitamin D-dependent
mechanism and response to ketoconazole treatment. J Pediatr.
157:296–302. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Segersten U, Björklund P, Hellman P,
Akerström G and Westin G: Potentiating effects of nonactive/active
vitamin D analogues and ketoconazole in parathyroid cells. Clin
Endocrinol (Oxf). 66:399–404. 2007. View Article : Google Scholar : PubMed/NCBI
|