Introduction
The sympathetic nervous system, catecholamines and
adrenergic receptors function in cellular growth, differentiation
and the regulation of tumor growth and cancer progression (1–6).
Although the expression of β-adrenergic receptors in breast cancer
cells has been well-documented and extensively investigated
(7–13), the regulatory roles of these
receptors in breast cancer cell replication and proliferation are
not fully understood at present.
One of the key regulators of cellular growth and
differentiation, and tumor progression and invasion, is
extracellular signal-regulated kinase (ERK)1/2.
β2-adrenergic receptor stimulation regulates the
activity of ERK1/2 and, depending on the cell type, activates
ERK1/2 (14,15). On the other hand, in certain cancer
cell lines, including MDA-MB-231 breast cancer cells,
β2-adrenergic receptor stimulation inhibits ERK1/2
phosphorylation, causing its inactivation (16,17).
There are two key processes that determine the phosphorylation and
activity status of ERK1/2: Its phosphorylation rate by kinases and
its dephosphorylation rate by phosphatases (18). Inhibition of the activity of
kinases, which phosphorylate ERK1/2, and/or activation of
phosphatases, which dephosphorylate pERK1/2, may be induced by
β2-adrenergic receptor stimulation, which will reduce
phosphorylated (p)ERK1/2 levels in MDA-MB-231 cancer cell
lines.
Carie and Sebti (16) reported that inhibition of ERK1/2
phosphorylation induced by β2-adrenergic receptor
stimulation is mediated by inactivation of Raf-1
proto-oncogene/mitogen-activated protein kinase (MAPK)1 kinases by
a cyclic AMP-dependent pathway in MDA-MB-231 cells. This indicates
that inhibition of kinases is implicated in
β2-adrenergic receptor-mediated pERK1/2
dephosphorylation. However, certain reports have indicated that
β-adrenergic receptor stimulation may also influence the activity
of various phosphatases. β-adrenergic receptor stimulation
increases the expression of mitogen-activated dual-specificity
phosphatase (DUSP/MKP)1, which may mediate the rapid
dephosphorylation of ERK1/2 in the rat pineal gland (19). Another study that investigated the
phosphorylation status of several signaling proteins in mouse
embryonic fibroblast cells demonstrated that β-adrenergic receptor
stimulation causes the dephosphorylation of protein phosphatase
(PP)1 at tyrosine 320, leading to its activation (20). However, to the best of our
knowledge, no associations between the activity of these
phosphatases and β2-adrenergic receptor-mediated pERK1/2
dephosphorylation in cancer cell lines have been previously
reported. Considering the important roles of
β2-adrenergic receptor signaling and the activity status
of phosphatases in the regulation of cancer cell lines, we
hypothesize that determining the association between the
β2-adrenergic receptor and these phosphatases may
contribute to an improved understanding of breast cancer cell
regulation.
Therefore, the present study focused on
investigating the role of phosphatases in β2-adrenergic
receptor-mediated dephosphorylation of ERK1/2 in breast cancer
cells. MDA-MB-231 and MDA-MB-468 triple negative breast cancer cell
lines, which are negative for the estrogen receptor (ER-),
progesterone receptor (PR-) and human epidermal growth factor
receptor 2 (HER2-), were employed in the present study as these
cells express high levels of the β2-adrenergic receptor
(12) and exhibit a high activity
of ERK1/2, as high levels of pERK1/2 are present (21). In addition, as other breast cancer
cell lines differ in terms of the expression of HER2, ER and PR and
may lead to variability. Therefore, to reduce variability, the
present study performed the experiments in two triple negative
breast cancer cell lines. One important group of pERK1/2
phosphatases is DUSPs. There are nine different DUSPs, of which
DUSP6/MKP3 is primarily cytosolic. DUSP6 is able to interact with
ERK1/2 and cause dephosphorylation, which ultimately inactivates it
(22,23). DUSP1 is a phosphatase that is
localized in the nucleus and is also involved in the regulation of
ERK1/2 activity (22,24). The other protein serine/threonine
phosphatases, PP1 and PP2, also have regulatory roles in the
dephosphorylation of ERK1/2 (25).
The present study investigated the roles of DUSP1/6 and PP1/2 in
β2-adrenergic receptor-mediated ERK1/2 dephosphorylation
in MDA-MB-231 and MDA-MB-468 cells by using a DUSP1/6 inhibitor,
(E)-2-benzylidene-3-(cyclohexylamino) −2,3-dihydro-1H-inden-1-one
(BCI), and a PP1/2 inhibitor, calyculin A, and by determining the
expression level of these phosphatases and reducing their
expression level by transfection of small interfering (si)RNA.
Materials and methods
Materials
The β2 ligands terbutaline, clenbuterol,
formoterol, epinephrine, isoproterenol and ICI118,551 hydrochloride
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) were dissolved in
saline. The DUSP1/6 inhibitor, BCI (Merck KGaA), and the PP1/2
inhibitor, calyculin A (Cell Signaling Technology, Inc., Danvers,
MA, USA), were dissolved in dimethyl sulfoxide (DMSO;
Sigma-Aldrich; Merck KGaA) and diluted in Dulbecco's modified
Eagle's medium/F12 (DMEM/F12, Capricorn Scientific, Ebsdorfergrund,
Germany). The final concentration of DMSO did not exceed 0.2%. In
all control experiments, cells were incubated with the
corresponding dilution of solvent used for the ligand. The
antibodies used were as follows: Total (t)PP1 catalytic subunit α
(PP1A; 1:1,000; Sc-271762; Santa Cruz Biotechnology, Inc., TX,
USA), tPP2 (PP2A; 1:1,000; ab33537; Abcam, Cambridge, UK), pPP1A
(T320; 1:1,000; ab6234; Abcam), pPP2A (Y307; 1:1,000; Sc-12615,
Santa Cruz Biotechnology, Inc.), pERK1/2 (1:5,000; Sc-16982; Santa
Cruz Biotechnology, Inc.), tERK1/2 (1:5,000; Sc-154; Santa Cruz
Biotechnology, Inc.), MKP1/DUSP1 (1:1,000; Sc-1102; Santa Cruz
Biotechnology, Inc.), MKP3/DUSP6 (1:1,000; Sc-377070; Santa Cruz
Biotechnology, Inc.), GAPDH (1:5,000; Sc-166545; Santa Cruz
Biotechnology, Inc.), goat anti-rabbit (1:10,000; Sc-31460; Santa
Cruz Biotechnology, Inc.), rabbit anti-goat (1:10,000; Sc-2768;
Santa Cruz Biotechnology, Inc.) and donkey anti-mouse (1:10,000;
Sc-2314; Santa Cruz Biotechnology, Inc.).
Cell culture and stimulation
The cell lines were obtained from the American Type
Culture Collection (Manassas, VA, USA). MDA-MB-231 and MDA-MB-468
cells were cultured in 75 cm2 non-treated cell culture
flasks in DMEM/F12 (Capricorn Scientific) enriched with 10% fetal
bovine serum (Capricorn Scientific) and 1% penicillin (10,000
I.U/ml; Biochrom GmbH, Berlin, Germany) and 1% streptomycin (10,000
µg/ml; Biochrom GmbH) at 5% CO2, 37°C with 90–95%
humidity. For each experiment, cells (2.5×105
cells/well) were plated in a 6-well plate at 37°C with treatments
performed on the second day following overnight serum starvation.
The following treatments were included in the present study:
Terbutaline, 1 µM for 2, 5, 10 or 30 min; clenbuterol, 1 µM for 10
min; formoterol, 0.1 µM for 10 min; isoproterenol, 1 µM for 2, 5,
10 or 30 min; and epinephrine, 10 µM for 10 min. In certain
experiments, prior to β2 adrenergic stimulation, cells
were pretreated for 30 min at 37°C with one of the following
inhibitors: ICI118,551 hydrochloride (0.1 µM), BCI (10 µM) and
calyculin A (10 nM).
RNA interference
DUSP1 siRNA (cat. no. Sc-35937), PP1 siRNA (cat. no.
Sc-36299), negative control siRNA (cat. no. Sc-3707), transfection
reagent (cat. no. Sc-29528) and medium (cat. no. Sc-36868) were all
purchased from Santa Cruz Biotechnology, Inc. The cells
(2.5×105) were cultured in a 6-well-plate and 24 h
later, when the cells reached 70% confluency, DUSP1 siRNA (1 µM),
PP1 siRNA (1 µM) or negative control siRNA (1 µM) were transfected
according to the manufacturer's protocol. After transfection for 24
h at 37°C, the medium was replaced and the cells were cultured for
an additional 24 h. The transfected cells were stimulated with 0.1
or 1 mM (data not shown) terbutaline (5–10 min) or saline. pERK1/2,
tERK, DUSP1, PP1 and GAPDH levels were subsequently determined by
western blot analysis.
Protein isolation and western
blotting
Following treatments, cells were immediately placed
on ice, washed with ice-cold PBS and homogenized in 100 µl lysis
buffer (Roche Diagnostics GmbH, Mannheim, Germany) containing 1%
Nonidet P40, 0.02 M sodium orthovanadate and protease inhibitors.
Following homogenization, cells were incubated for 15 min and
centrifuged at 5,000 × g for 5 min at 4°C. The supernatant was
collected, protein concentration was determined using the Bradford
protein assay and stored at −80°C. Electrophoresis (20–30 µg
protein/per lane) was performed on newly-cast 8–10% sodium dodecyl
sulphate (SDS)-polyacrylamide gels followed by transfer onto
polyvinylidene difluoride membranes. The membranes were blocked for
2 h at 22°C in PBS with 20 mM
NaH2PO4-Na2HPO4 (pH
7.6) containing 154 mM NaCl, 5% nonfat dry milk and 0.1% Tween-20.
The membranes were incubated with the appropriate primary
antibodies overnight at 4°C and washed three times for 10 min with
TBS-0.2% Tween-20 prior to incubation for 1 h at 22°C with
horseradish peroxidase conjugated anti-rabbit, anti-mouse or
anti-goat secondary antibody. Following washing, the membranes were
soaked in Clarity Western ECL Substrate (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) and imaged using a ChemiDoc MP Imaging
System (Bio-Rad Laboratories, Inc.). Band intensities were
quantified using Image Lab software (version 5; Bio-Rad
Laboratories, Inc.). Two separate bands were observed for pERK1/2
(42 and 44 kDa) and the sum of the intensities of these two bands
were determined. When required, membranes were stripped with 2% SDS
and 0.7% mercaptoethanol in 10 ml PBS prior to incubation with
another antibody overnight. Band intensities were presented
relative to tERK or GAPDH expression.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. n represents the number of independent experiments for
each indicated condition. Statistical analysis was performed using
SPSS 17.0 for Windows software (SPSS, Inc., Chicago, IL, USA).
Statistical analysis of data and comparisons between multiple
groups were performed by one-way analysis of variance followed by
Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
β2-adrenergic receptor
stimulation and pERK1/2 dephosphorylation in MDA-MB-231 and
MDA-MB-468 cells
As expected, two separate bands for pERK1/2 (42 and
44 kDa) were generally observed. However, for certain measurements,
due to the high level of pERK1/2 in MDA-MB-231 or MDA-MB-468, one
large, combined band was obtained located in the range of 42–44
kDa.
The results in Fig.
1 confirmed that β2-adrenergic receptor stimulation
led to the dephosphorylation of pERK1/2. β2-adrenergic
receptor-selective agonists, terbutaline (1 µM), formoterol (0.1
µM) and clenbuterol (1 µM), and nonselective β-adrenergic receptor
agonists, epinephrine (10 µM) and isoproterenol (1 µM), all
dephosphorylated pERK1/2 in MDA-MB-231 (Fig. 1A) and MDA-MB-468 (Fig. 1B) cells. Furthermore, pERK1/2
dephosphorylation induced by terbutaline was blocked by the
β2-adrenergic receptor antagonist ICI118,551
hydrochloride (0.1 µM), indicating that this response is primarily
mediated by the β2-adrenergic receptor (Fig. 1). pERK1/2 dephosphorylation was
observed following treatment with 10 nM-1 µM terbutaline (data not
shown).
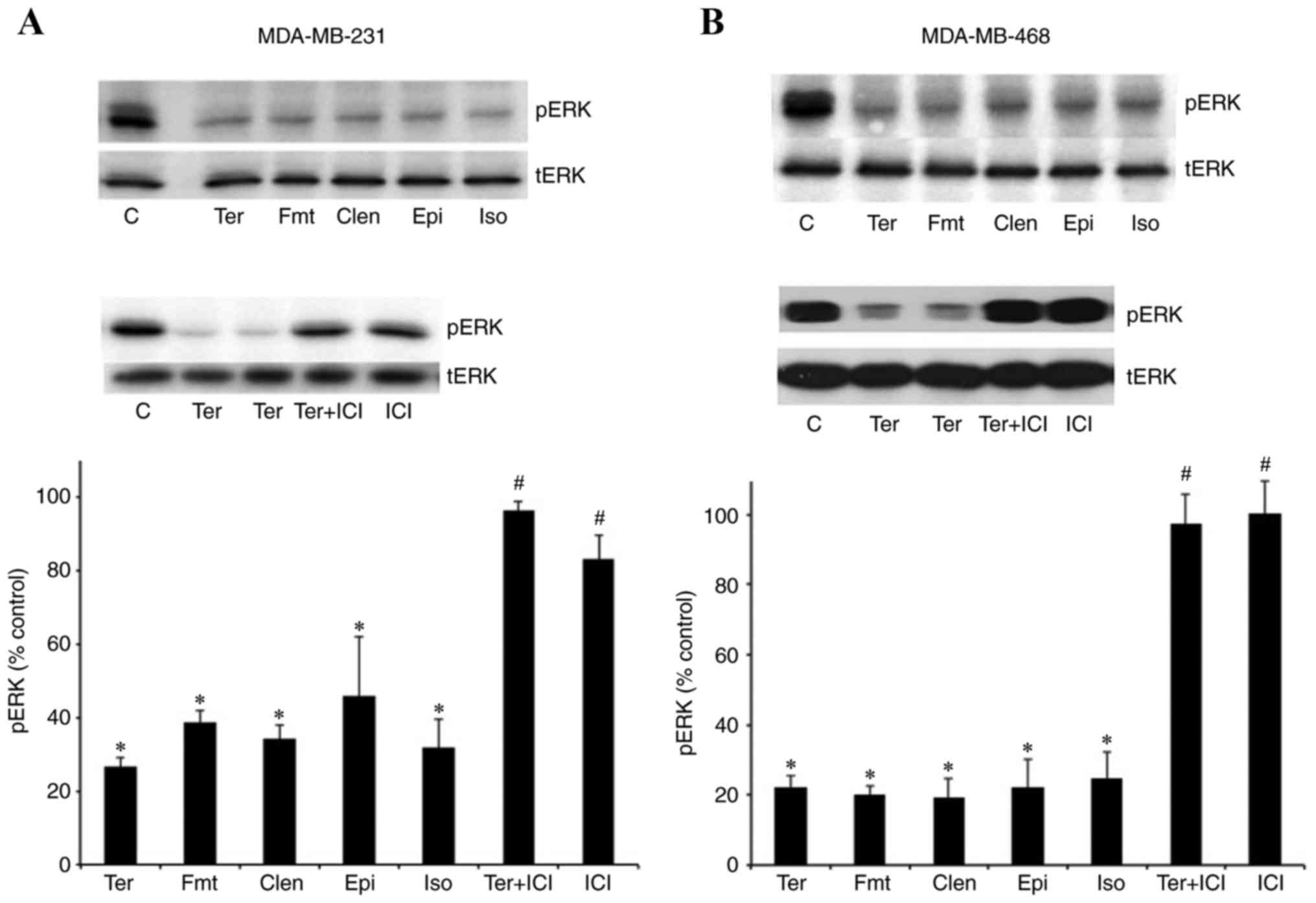 | Figure 1.β2-adrenergic receptor
agonists led to the dephosphorylation of pERK1/2 in MDA-MB-231 and
MDA-MB-468 cells. Treatment with the β2-adrenergic
receptor-selective agonists terbutaline (1 µM), clenbuterol (1 µM)
and formoterol (0.1 µM), and nonselective β-adrenergic receptor
agonists isoproterenol (1 µM) and epinephrine (10 µM), for 10 min
significantly inhibited the basal level of pERK1/2 in (A)
MDA-MB-231 and (B) MDA-MB468 cells. Pretreatment of cells with the
β2-adrenergic receptor antagonist, ICI118,551
hydrochloride (0.1 µM) for 30 min, completely antagonized
terbutaline-stimulated pERK1/2 dephosphorylation in both cell
lines. Representative western blot bands for pERK and tERK are
presented. Two replicate bands are presented for certain treatment
groups. tERK bands were used as a reference, and pERK band
intensities were normalized to tERK and presented as a percentage
of saline-treated control cells. Data are presented as the mean ±
standard error of the mean, n=4-5. *P<0.05 vs. control,
#P<0.05 vs. Ter group. ERK, extracellular
signal-regulated kinase; pERK, phosphorylated ERK; tERK, total ERK;
C, control; Ter, terbutaline; Fmt, formoterol; Clen, clenbuterol;
Epi, epinephrine; Iso, isoproterenol; ICI, ICI118,551
hydrochloride. |
The DUSP1 inhibitor, BCI, antagonizes
β2-adrenergic receptor-mediated pERK1/2
dephosphorylation
The activation of DUSP1/6 is reported to lead to the
dephosphorylation of pERK1/2 and negatively regulate its activity
(22–24). MDA-MB-231 and MDA-MB-468 cells were
treated with the DUSP1/6 inhibitor, BCI (10 µM), for 30 min prior
to terbutaline (1 µM) stimulation. BCI treatment of the cells
completely antagonized β2-adrenergic receptor-mediated
inhibition of ERK1/2 phosphorylation in MDA-MB-231 (Fig. 2A) and MDA-MB-468 (Fig. 2B) cells.
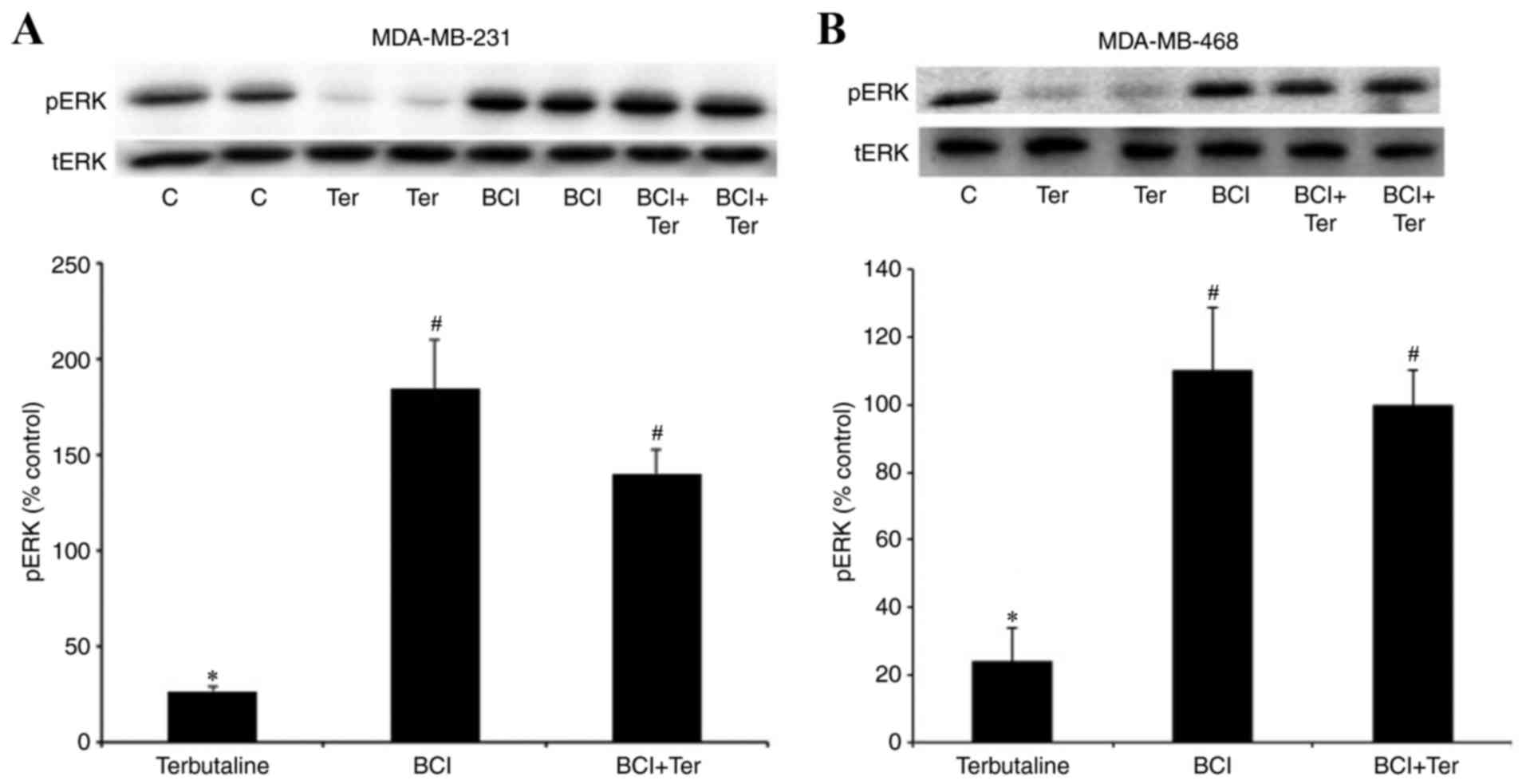 | Figure 2.The DUSP1 inhibitor, BCI, antagonized
β2-adrenergic receptor-mediated pERK1/2
dephosphorylation. Levels of pERK following treatment with the
β2-adrenergic receptor-selective agonist terbutaline (1
µM) for 10 min with or without pretreatment with the DUSP1/6
inhibitor BCI (10 µM) for 30 min in (A) MDA-MB-231 and (B)
MDA-MB-468 cells. BCI antagonized β2-adrenergic
receptor-mediated pERK1/2 dephosphorylation in both cell lines.
Representative western blot bands for pERK and tERK are presented.
Two replicate bands are presented for certain treatment groups.
pERK band intensities were normalized to tERK and presented as a
percentage of saline + 0.1% DMSO-treated control cells. DMSO (0.1%)
was the final dilution of the solvent of BCI and was used as the
control for pretreatment with BCI. Data are presented as the mean ±
standard error of the mean, n=4-5. *P<0.05 vs. control,
#P<0.05 vs. Ter. DUSP, dual-specificity phosphatase;
BCI,
(E)-2-benzylidene-3-(cyclohexylamino)-2,3-dihydro-1H-inden-1-one;
ERK, extracellular signal-regulated kinase; pERK, phosphorylated
ERK; tERK, total ERK; DMSO, dimethyl sulfoxide; C, control; Ter,
terbutaline. |
β2-adrenergic receptor
stimulation induces the expression of DUSP1
As DUSP1 is an inducible protein (19,22,26,27),
the present study also investigated DUSP1 expression levels by
western blot analysis with or without terbutaline (1 µM) or
isoproterenol (1 µM) treatment. Terbutaline and isoproterenol
treatment (5, 10 and 30 min) increased the protein expression of
DUSP1 in MDA-MB-231 cells, compared with control treatment
(Fig. 3A). In addition, DUSP1
protein expression was also increased in MDA-MB-468 cells following
10 min terbutaline treatment (Fig.
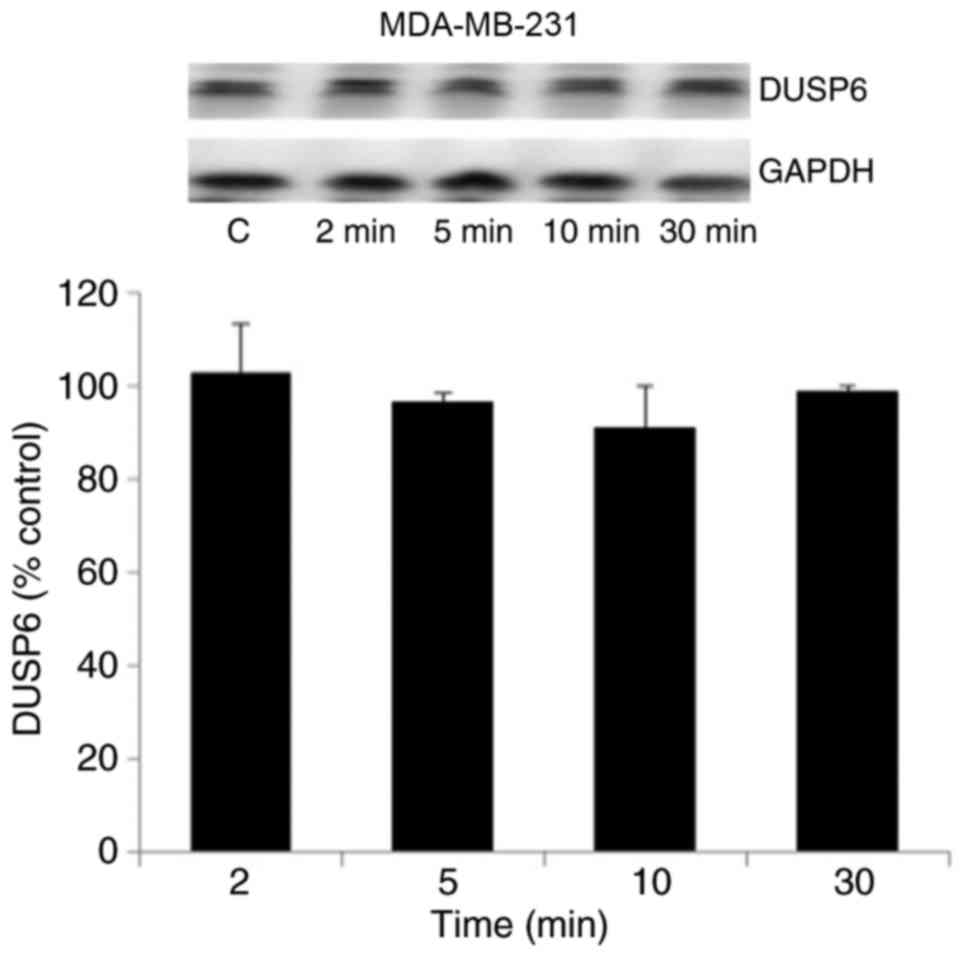
3B). However, the results in Fig.
4 indicate that DUSP6 levels in MDA-MB-231 cells were not
altered by terbutaline treatment for 2–30 min.
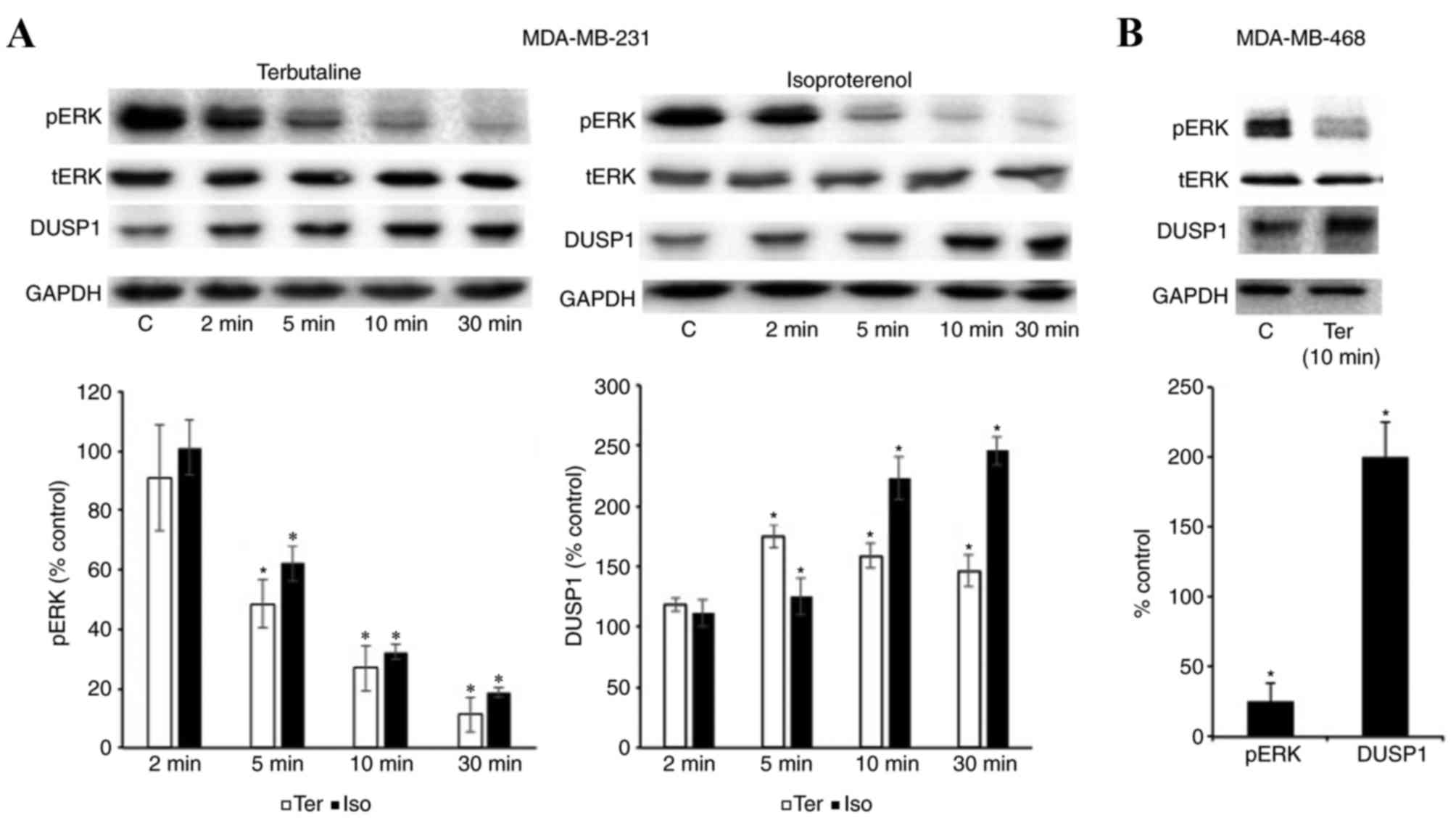 | Figure 3.β2-adrenergic receptor
stimulation induced the expression of DUSP1. (A) The protein
expression of pERK, tERK, DUSP1 and GAPDH was investigated
following treatment with terbutaline (1 µM) or isoproterenol (1 µM)
treatment for 2–30 min in MDA-MB-231 cells. (B) The expression of
pERK, tERK, DUSP1 and GAPDH following treatment with terbutaline (1
µM) for 10 min in MDA-MB-468 cells. Representative western blot
bands for pERK, tERK, DUSP1 and GAPDH are presented. In both cell
lines, β2-adrenergic receptor stimulation increased the
expression level of DUSP1. Band intensities were normalized to
GAPDH for DUSP1 and tERK for pERK, and presented as a percentage of
saline-treated control cells. Data are presented as the mean ±
standard error of the mean, n=4-5. *P<0.05 vs. control cells.
DUSP, dual-specificity phosphatase; ERK, extracellular
signal-regulated kinase; pERK, phosphorylated ERK; tERK, total ERK;
C, control; Ter, terbutaline; Iso, isoproterenol. |
Downregulation of DUSP1 reduces
β2-adrenergic receptor-mediated pERK1/2
dephosphorylation
Western blotting results in Fig. 5 indicate that DUSP1 siRNA
transfection successfully reduced the expression of DUSP1 compared
with the control siRNA transfection group in MDA-MB-231 and
MDA-MB-468 cells. Terbutaline-mediated dephosphorylation of pERK1/2
significantly declined following downregulation of DUSP1, compared
with the control siRNA transfection group. While 0.1 µM terbutaline
led to 75±3 and 70±5% dephosphorylation of ERK1/2 in MDA-MB-231 and
MDA-MB-468 cells, respectively, dephosphorylation was 43±7.9 and
47±6%, respectively, following downregulation of DUSP1 (Fig. 5). In the present study, 0.1 and 1
µM terbutaline were generally employed. There was no significant
difference in the 0.1 or 1 µM terbutaline-induced pERK1/2
dephosphorylation (data not shown). Terbutaline treatment following
downregulation of DUSP1 was performed with 0.1 and 1 (data not
shown) µM terbutaline, and results demonstrated that the inhibition
of dephosphorylation of pERK1/2 was pronounced when 0.1 µM
terbutaline was employed. Therefore, results for 0.1 µM terbutaline
treatment are presented in Fig. 5.
We hypothesized that signal strength is lower when a lower
concentration of terbutaline (0.1 µM) was employed and, therefore,
a lower concentration of terbutaline-mediated ERK1/2
dephosphorylation was more sensitive to the downregulation of DUSP1
levels. Therefore, terbutaline-mediated pERK1/2 dephosphorylation
may depend on the expression level of DUSP1.
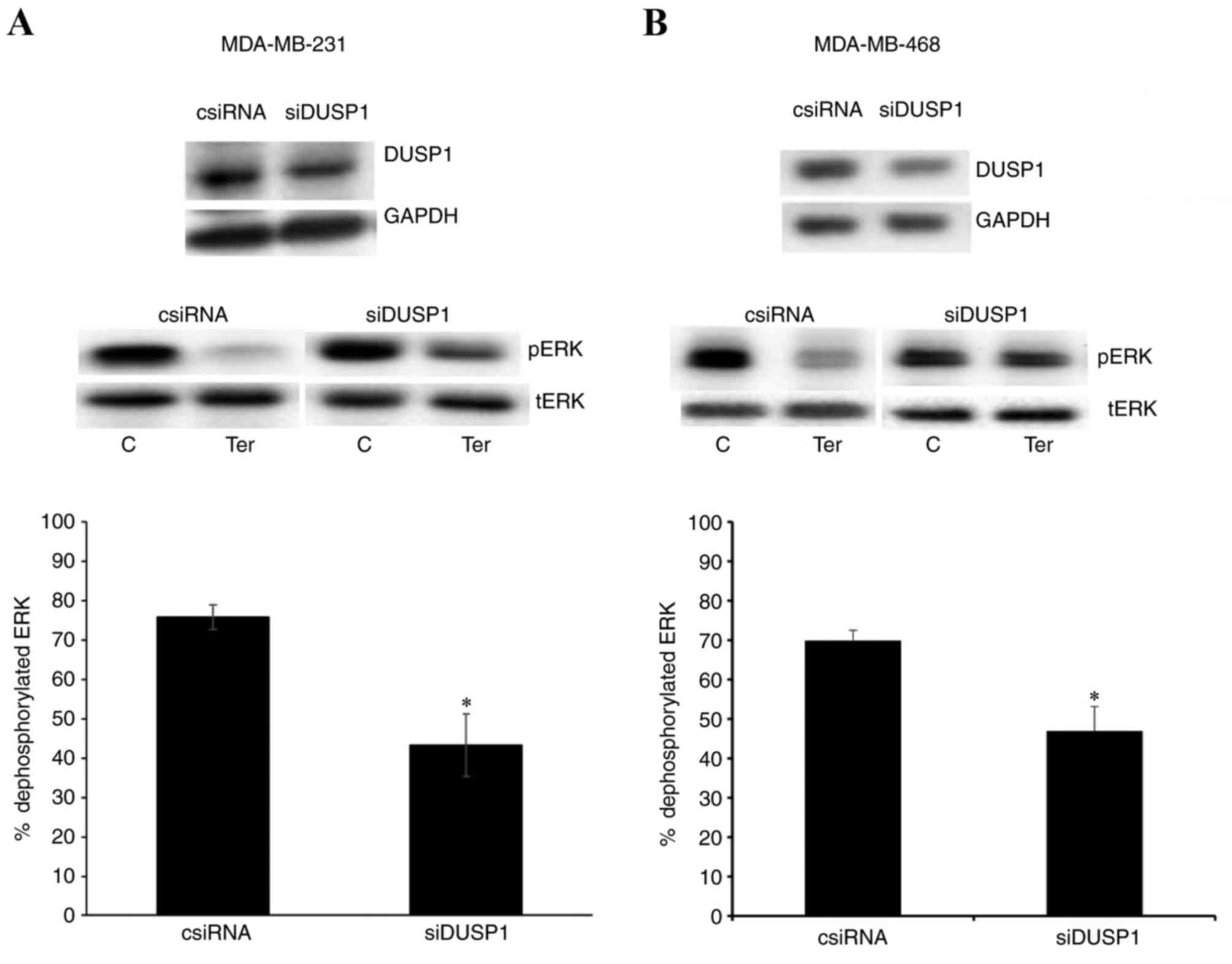 | Figure 5.Downregulation of DUSP1 reduced
β2-adrenergic receptor-mediated pERK1/2
dephosphorylation. Results demonstrating the expression levels of
DUSP1 following transfection with control (1 µM) or DUSP1 (1 µM)
siRNA, and pERK expression in control and DUSP1 siRNA-transfected
cells following treatment with terbutaline (0.1 µM) or saline, are
presented for (A) MDA-MB-231 and (B) MDA-MB-468 cells.
Representative western blot bands for pERK, tERK, DUSP1 and GAPDH
are presented. Band intensities were normalized to GAPDH for DUSP1
and tERK for pERK, and presented as a percentage of saline-treated
control cells. Bar graphs indicate the percentage of
dephosphorylated ERK1/2 induced by terbutaline treatment in control
or DUSP1 siRNA-transfected cells. Data are presented as the mean ±
standard error of the mean, n=4. *P<0.05 vs. control cells.
DUSP, dual-specificity phosphatase; ERK, extracellular
signal-regulated kinase; pERK, phosphorylated ERK; siRNA, small
interfering RNA; tERK, total ERK; csiRNA, control siRNA; siDUSP1,
siRNA targeting DUSP1; C, control; Ter, terbutaline. |
The PP1 inhibitor, calyculin A,
antagonizes β2-adrenergic receptor-mediated pERK1/2
dephosphorylation
Additional phosphatases that regulate ERK1/2
phosphorylation are the serine/threonine phosphatases PP1 and PP2.
Therefore, the present study also investigated the potential
involvement of PP1 and PP2 in β2-adrenergic
receptor-mediated dephosphorylation of pERK1/2. To investigate
their roles, calyculin A, a PP1/2 inhibitor, was employed. The
results demonstrated that terbutaline-mediated ERK1/2
dephosphorylation was reversed by 30 min pretreatment of MDA-MB-231
and MDA-MB-468 cells with 10 nM calyculin A (Fig. 6).
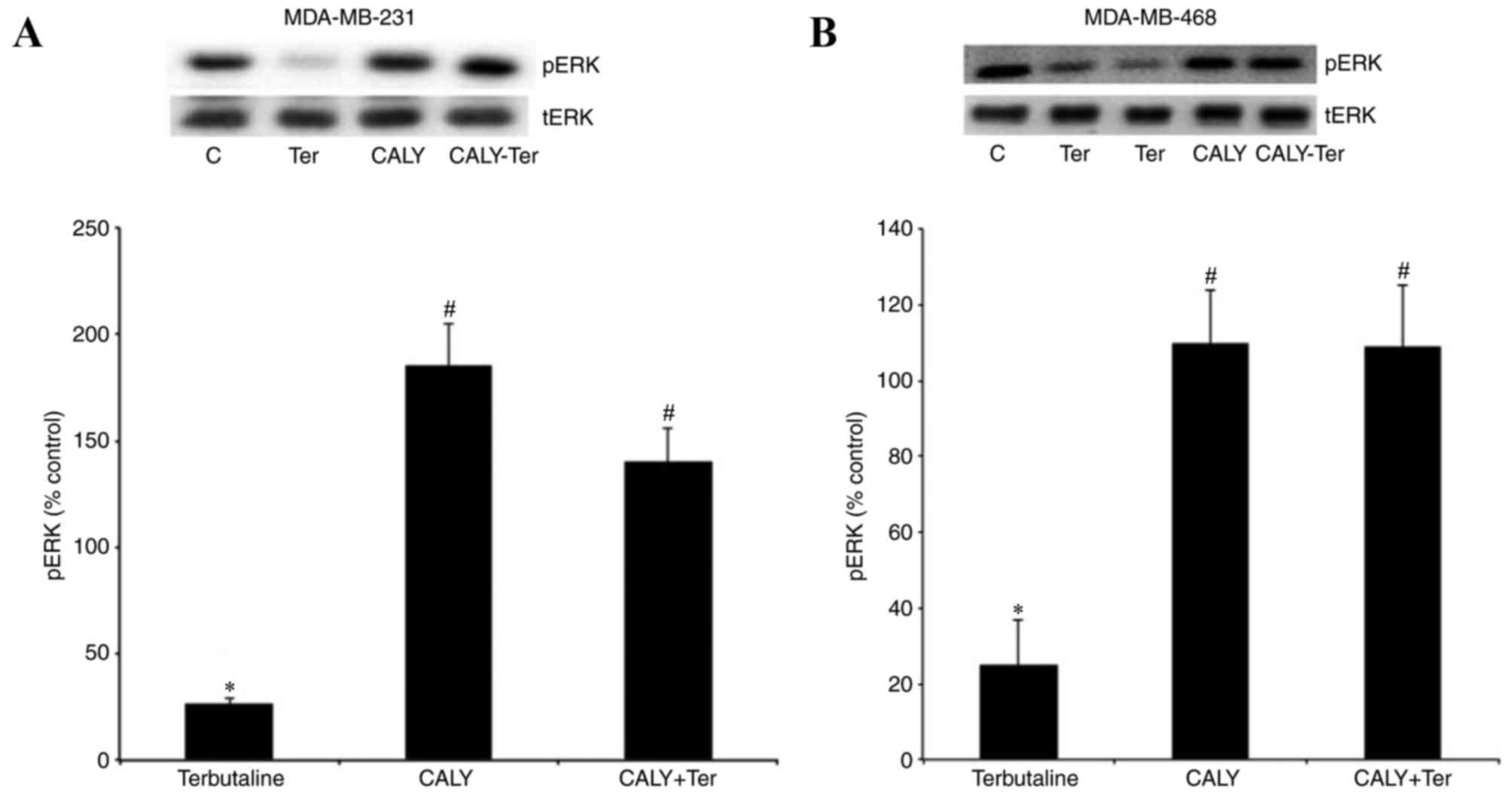 | Figure 6.The PP1 inhibitor, calyculin A,
antagonized β2-adrenergic receptor-mediated pERK1/2
dephosphorylation. pERK levels following treatment with terbutaline
(1 µM) for 10 min with or without pretreatment with the PP1
inhibitor calyculin A (10 nM) for 30 min are presented for (A)
MDA-MB-231 and (B) MDA-MB-468 cells. Calyculin A inhibited
β2-adrenergic receptor-mediated pERK1/2
dephosphorylation in both cell lines. Representative Western blot
bands for pERK and tERK are presented. Two replicate bands are
presented for certain treatment groups. pERK band intensities were
normalized to tERK and presented as a percentage of saline + 0.1%
DMSO-treated control cells. DMSO (0.1%) was the final dilution of
the solvent of calyculin A and was used as the control for
pretreatment with calyculin A. Data are presented as the mean ±
standard error of the mean, n=4-5. *P<0.05 vs. control,
#P<0.05 vs. Ter. PP, protein phosphatase; ERK,
extracellular signal-regulated kinase; pERK, phosphorylated ERK;
tERK, total ERK; DMSO, dimethyl sulfoxide; C, control; Ter,
terbutaline; CALY, calyculin A. |
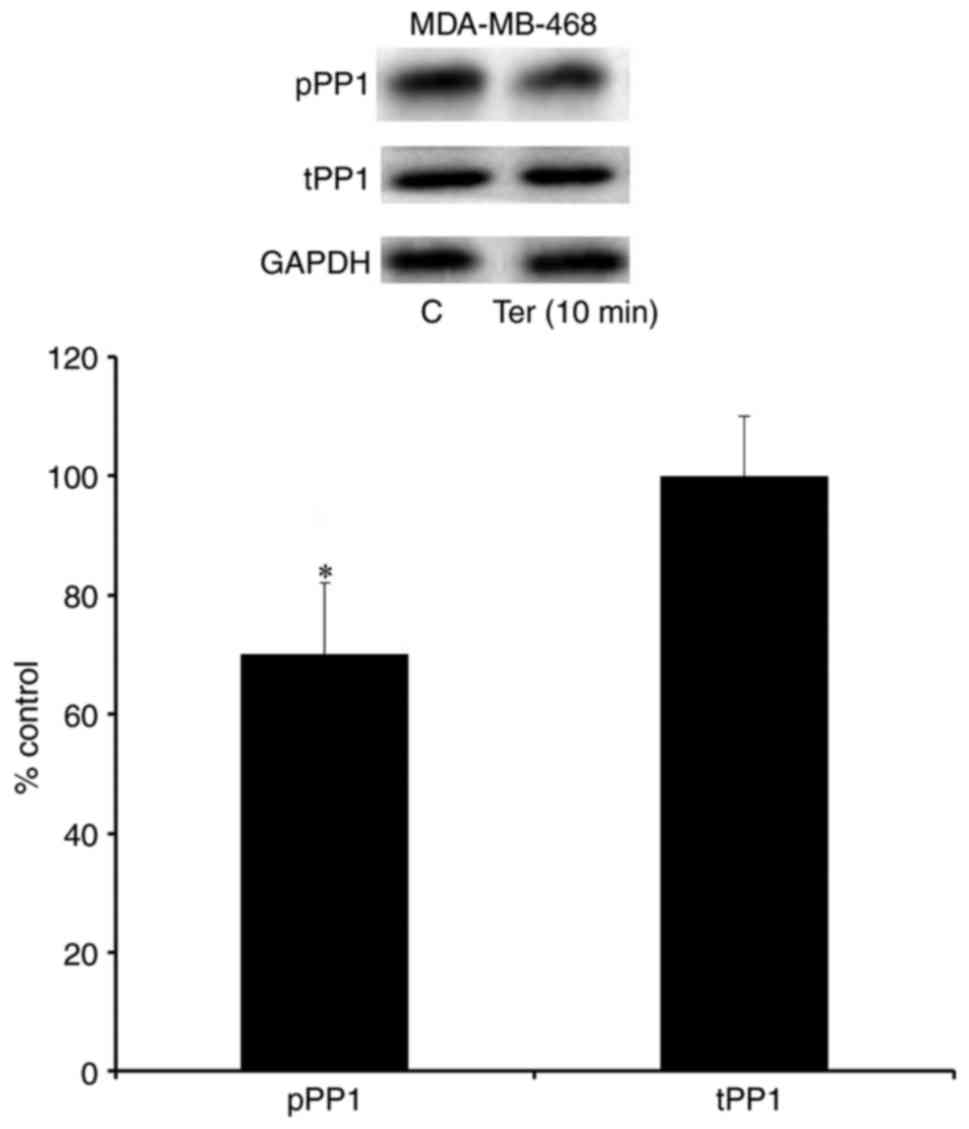
β2-adrenergic receptor
stimulation enhances the expression of the active form of PP1
Phosphorylation of PP1 at tyrosine 320 and PP2 at
tyrosine 307 represent inhibited forms of these phosphatases,
whereas dephosphorylation of PP1 (tyrosine 320) and PP2 (tyrosine
307) are associated with enhanced activity of these phosphatases
(28,29). The present study investigated the
expression levels of the phosphorylated forms of PP1 and PP2
following stimulation with terbutaline (1 µM). The results
demonstrated that the phosphorylation of PP1 was inhibited
following 2–10 min treatment with terbutaline in MDA-MB-231 cells
(Fig. 7). Furthermore, a decrease
in the level of pPP1 was observed following terbutaline treatment,
while pPP2 levels were not significantly altered, in MDA-MB-231
cells (Fig. 7). However,
terbutaline did not alter the expression levels of tPP1 and tPP2 in
MDA-MB-231 cells (Fig. 7). In
MDA-MB-468 cells, 1 µM terbutaline for 10 min also reduced pPP1
levels without affecting tPP1 levels (Fig. 8). These results indicate that by
enhancing the expression of the active form of PP1, terbutaline
stimulation may contribute to ERK1/2 dephosphorylation.
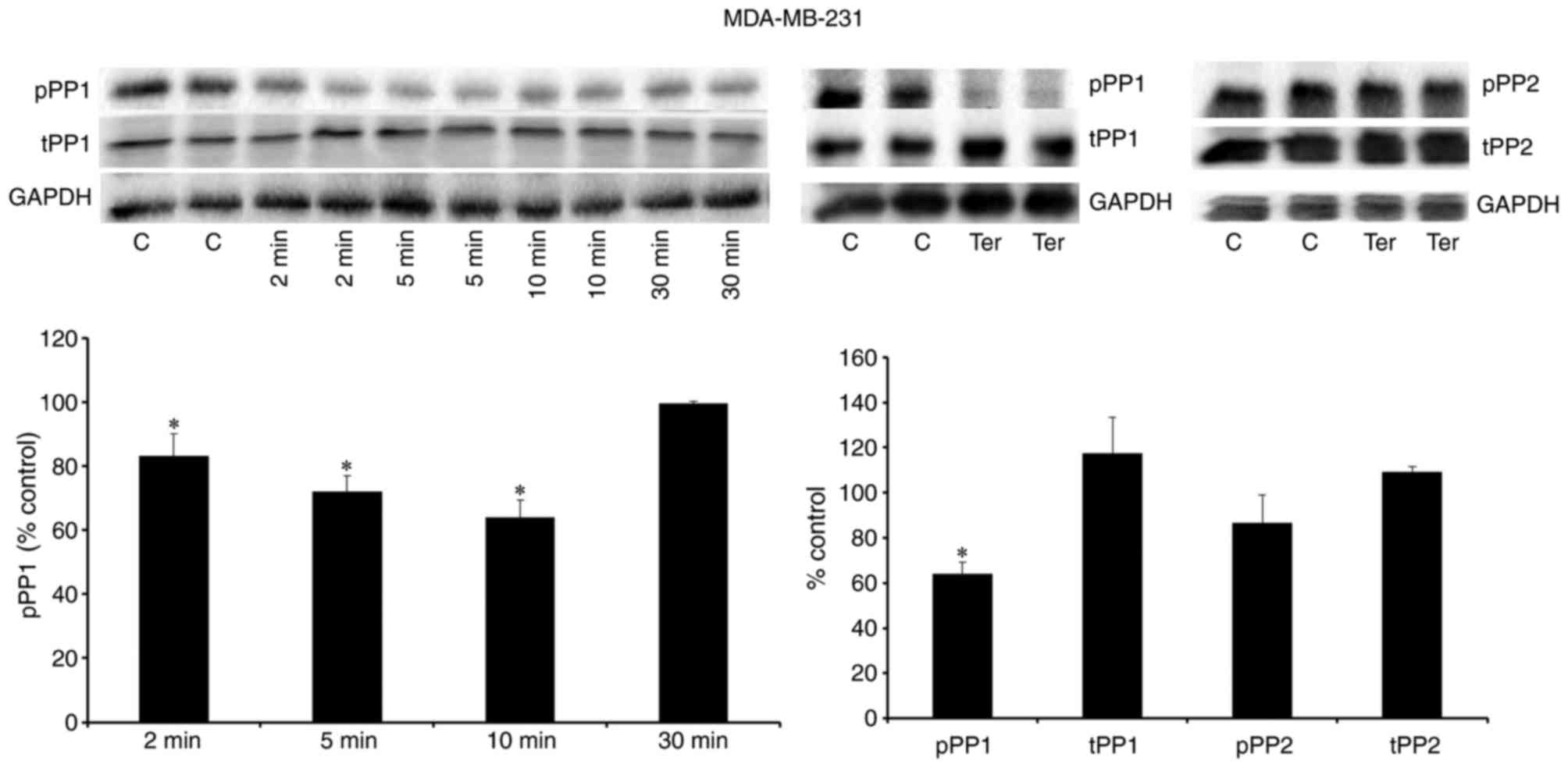 | Figure 7.β2-adrenergic receptor
stimulation reduced the level of the inactive form of PP1 in
MDA-MB-231 cells. Representative western blot bands are presented
for pPP1, tPP1 and GAPDH following treatment of MDA-MB 231 cells
with terbutaline (1 µM) for 2–30 min, and for pPP1, tPP1, pPP, tPP2
and GAPDH following treatment of MDA-MB 231 cells with terbutaline
(1 µM) for 10 min. Terbutaline treatment reduced the level of the
inactive form of PP1 (pPP1) and had no effect on the levels of
tPP1, pPP2 and tPP2. Two replicate bands are presented for
treatment groups. pPP1 and pPP2 band intensities were normalized to
tPP1 and tPP2, respectively, and tPP1 and tPP2 band intensities
were normalized to GAPDH and presented as a percentage of
saline-treated control cells. Data are presented as the mean ±
standard error of the mean, n=4-5. *P<0.05 vs. control cells.
PP, protein phosphatase; pPP, phosphorylated PP; tPP, total PP; C,
control; Ter, terbutaline. |
Downregulation of PP1 reduces
β2-adrenergic receptor-mediated pERK1/2
dephosphorylation
The present study also downregulated PP1 expression
by transfecting cells with PP1 siRNA. Western blotting results in
Fig. 9 demonstrated that PP1 was
successfully downregulated following transfection with PP1 siRNA,
compared with the control siRNA transfection group, in MDA-MB-231
and MDA-MB-468 cells. Terbutaline (0.1 µM)-mediated pERK1/2
dephosphorylation was significantly reduced following
downregulation of PP1 expression, compared with the control siRNA
transfection group (Fig. 9).
Similar to the results observed in the DUSP1 downregulation
experiments, while 0.1 µM terbutaline caused 76±4 and 70±5%
dephosphorylation of ERK1/2 in MDA-MB-231 and MDA-MB-468 cells,
respectively, these values reduced to 44±6 and 30±10%,
respectively, following downregulation of PP1. Experiments
involving the downregulation of PP1 employed 0.1 and 1 µM (data not
shown) terbutaline and, similar to the results for the
downregulation of DUSP1, a lower concentration of terbutaline (0.1
µM) was selected. ERK1/2 dephosphorylation was more sensitive to
the downregulation of PP1 levels at this lower concentration.
Therefore, terbutaline-mediated pERK1/2 dephosphorylation may
depend on the expression levels of both PP1 and DUSP1.
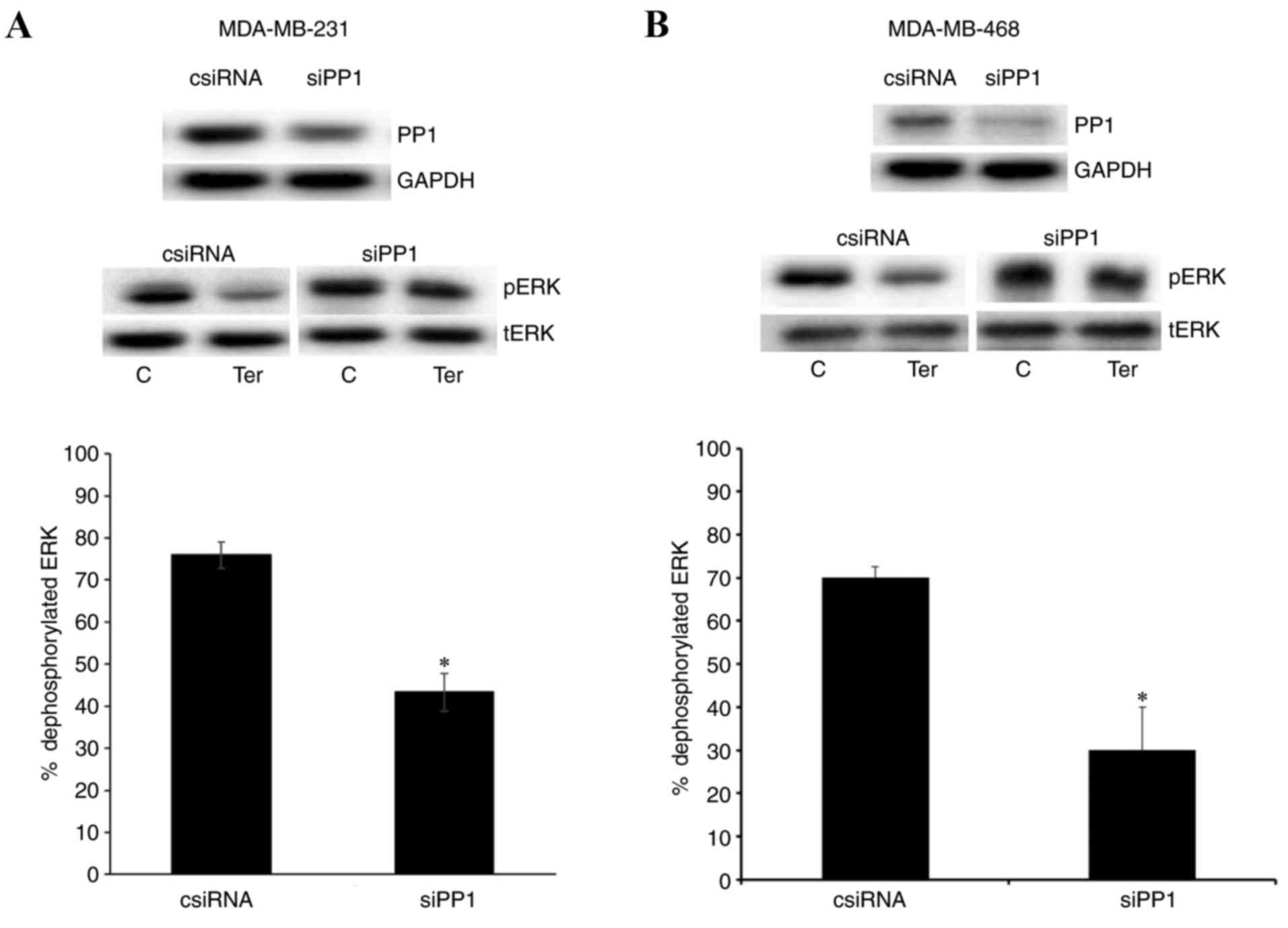 | Figure 9.Downregulation of PP1 reduced
β2-adrenergic receptor-mediated pERK1/2
dephosphorylation. (A) Expression levels of PP1, GAPDH, pERK and
tERK following transfection with control siRNA (1 µM) or PP1 siRNA
(1 µM), and levels of PP1, GAPDH, pERK and tERK following treatment
of control or PP1 siRNA-transfected cells with terbutaline (0.1 µM)
or saline for 5–10 min, for (A) MDA-MB-231 and (B) MDA-MB-468
cells. Transfection of cells with PP1 siRNA downregulated PP1 in
both cell. Downregulation of PP1 reduced terbutaline-mediated
dephosphorylation of pERK. Band intensities were normalized to
GAPDH for PP1 and tERK for pERK. Results in the bar graph indicate
the percentage of dephosphorylated ERK following terbutaline
treatment in control and PP1 siRNA-transfected MDA-MB-231 and
MDA-MB-468 cells, and are presented as a percentage of
saline-treated control cells. Data are presented as the mean ±
standard error of the mean, n=4. *P<0.05 vs. control cells. PP,
protein phosphatase; ERK, extracellular signal-regulated kinase;
pERK, phosphorylated ERK; tERK, total ERK; siRNA, small interfering
RNA; csiRNA, control siRNA; siPP1, siRNA targeting PP1; C, control;
Ter, terbutaline. |
Discussion
MDA-MB-231 and MDA-MB-468 triple negative breast
cancer cell lines express the β2-adrenergic receptor,
and the results of several previous studies indicate that the
β2-adrenergic receptor signaling pathway may be involved
in tumor development, metastasis and cancer progression (4,5,13).
DUSP1 and PP1 are also reported to be involved in the regulation of
cell proliferation, and cancer development and progression, and
they have been proposed as potential targets for cancer treatment
(30–34). As stimulation of the
β2-adrenergic receptor with endogenous catecholamines
may lead to the activation of DUSP1 and PP1, their targets,
including p38, c-Jun N-terminal kinase and ERK1/2, may subsequently
be affected. Therefore, as these affected proteins are key
components of cancer pathways, the results of the present study,
which demonstrated the activation of DUSP1 and PP1 phosphatases
following stimulation of β2-adrenergic receptors, are
important for cancer research. Triple negative breast cancer cells
overexpress epidermal growth factor receptor and mutations in KRAS
proto-oncogene, B-Raf proto-oncogene and phosphatase and tensin
homolog, which lead to high MAPK/ERK1/2 activity and subsequent
resistance to therapeutic agents (35). Although the molecular mechanisms
have not been completely clarified, previous research has
demonstrated that β2-adrenergic receptor stimulation
mediates pERK1/2 dephosphorylation and inactivation in MDA-MB-231
cells (16,17). Therefore, it is important to
investigate the activation of pathways that inactivate MAPK/ERK1/2
in β2-adrenergic receptor signaling in these cancer cell
lines, including DUSP1 and PP1.
Consistent with previous studies, the present study
demonstrated that stimulation of MDA-MB-231 and MDA-MB-468 cells
with β2-adrenergic receptor agonists resulted in the
dephosphorylation of basal pERK1/2 (9,16,17).
In addition, the results of the current study indicated that
β2-adrenergic receptor-mediated dephosphorylation of
ERK1/2 was associated with the activity of the protein phosphatases
DUSP1 and PP1; DUSP1 and PP1 inhibitors antagonized
β2-adrenergic receptor-mediated dephosphorylation of
ERK1/2. The treatment of MDA-MB-231 and MDA-MB-468 cells with
terbutaline increased the protein expression levels of DUSP1 and
enhanced the levels of the active form of PP1. Furthermore,
reducing the expression of DUSP1 or PP1 reduced
β2-adrenergic receptor-mediated dephosphorylation of
pERK1/2.
The present study investigated the activities of
enzymes that may cause pERK1/2 dephosphorylation during
β2-adrenergic receptor stimulation. Two different types
of enzymes, serine/threonine kinases and phosphatases, primarily
regulate ERK1/2 phosphorylation status. In particular, pERK1/2
dephosphorylation has been reported to be tightly regulated by
DUSP1/6 (22,23,36).
Therefore, the activation of DUSP1/6 following
β2-adrenergic receptor stimulation may trigger the
dephosphorylation of pERK1/2. Swingle et al (37) identified a small inhibitor molecule
of DUSP1/6, BCI, which directly binds to these phosphatases to
inhibit DUSP1 and DUSP6 with IC50 values of 11.5±2.8 and
12.3±4.0 µM, respectively. The results of the current study
demonstrated that 10 µM BCI completely reversed
terbutaline-mediated dephosphorylation of pERK1/2, therefore
indicating that the β2-adrenergic receptor may mediate
the activation of DUSP1/6. Several studies have reported that DUSP1
is a labile and inducible enzyme that is primarily localized in the
nucleus (22). Price et al
(19) demonstrated that
β-adrenergic receptor stimulation led to a rapid increase in DUSP1
mRNA and protein levels. Considering this, the present study
measured the levels of DUSP1 protein in MDA-MB-231 and MDA-MB-468
cells following stimulation with terbutaline and observed a rapid
increase in cellular DUSP1 protein levels within 5–30 min of
terbutaline stimulation. These results indicate that
β2-adrenergic receptor stimulation increases the level
of DUSP1 protein. Wu et al (36) reported an association between DUSP1
induction and ERK1/2 inhibition. The current study also observed a
clear association between the induction of DUSP1 expression and
ERK1/2 dephosphorylation following β2-adrenergic
receptor stimulation. Therefore, it was predicted that inhibiting
the expression level of DUSP1 may lead to a decline in
terbutaline-mediated pERK1/2 dephosphorylation. In the current
study, downregulation of DUSP1 expression by siRNA transfection led
to a decline in terbutaline-mediated pERK1/2 dephosphorylation.
These results also indicate an association between
β2-adrenergic receptor stimulation and the expression
and activation of DUSP1.
DUSP6, which is generally localized in the cytosol
as a phosphatase, is primarily responsible for ERK1/2
dephosphorylation (22,23). Although our experiments did not
demonstrate a significant alteration in the level of DUSP6 protein
following terbutaline stimulation, this does not eliminate a
potential role for DUSP6 protein in β2-adrenergic
receptor-mediated ERK1/2 dephosphorylation. The expression and
activity of DUSPs may be involved in terbutaline-mediated
dephosphorylation of ERK1/2. BCI inhibits both DUSP1 and DUSP6,
and, in the present study, it completely reversed the
dephosphorylation of ERK1/2. Further studies are therefore required
to clarify the influence of β2-adrenergic receptor
stimulation on DUSP6 activity.
It is established that ERK1/2 phosphorylation is
also regulated by the serine/threonine phosphatases PP1 and PP2
(25). Calyculin A is a PP1 and
PP2 inhibitor with IC50 values of 0.4 and 0.25 nM,
respectively (37). At
concentrations of 50–100 nM, calyculin A is cytotoxic, killing the
majority of human cell types, while at 10 nM it is suitable for
treating cells (37). Therefore,
if there is an association between PP1/2 activity and
β2-adrenergic receptor-mediated dephosphorylation of
pERK1/2, calyculin A should influence the response. In the present
study, treatment of cells with 10 nM calyculin A antagonized the
terbutaline-mediated dephosphorylation of pERK1/2. Following the
use of an enzyme inhibitor to block the response as a
pharmacological approach, the present study also determined the
activity of these enzymes in β2-adrenergic
receptor-mediated ERK1/2 dephosphorylation by measuring pPP1 and
pPP2 levels (inactivated forms of PP1 and PP2) using western blot
analysis. Stimulation of the cells with terbutaline (2, 5 and 10
min) significantly diminished pPP1 in the cells, while pPP2 levels
were not affected, indicating that the β2-adrenergic
receptor may mediate the activation of PP1. However, the same
response for pPP1 levels was not observed when the cells were
stimulated with terbutaline for 30 min. These results clearly
indicate the presence of a similar pattern between
terbutaline-mediated pERK1/2 and pPP1 dephosphorylation during the
30 min stimulation. As β2-adrenergic receptor
stimulation led to increased DUSP1 expression, rather than
phosphorylation status, tPP1/tPP2 expression levels were also
measured, and the results demonstrated that tPP1 and tPP2 levels
were not altered following terbutaline stimulation. This indicates
that the β2-adrenergic receptor-mediated decline in the
inactive form of PP1 was not due to altered tPP1 protein expression
level. If the activation of the β2-adrenergic receptor
activates PP1 then downregulation of PP1 expression should reduce
the β2-adrenergic receptor-mediated activation of PP1
and, consequently, the dephosphorylation of ERK1/2. To test this
hypothesis, in the present study, cells were transfected with PP1
siRNA to downregulate its cellular expression, which reduced
terbutaline-mediated pERK1/2 dephosphorylation. These results
indicate that PP1 activity may be important in
β2-adrenergic receptor-mediated ERK1/2
dephosphorylation. Consistent with the results of the current
study, Chruscinski et al (20) investigated the phosphorylation
status of several signaling proteins in mouse embryonic fibroblast
cells and showed that β-adrenergic receptor stimulation resulted in
the dephosphorylation and activation of PP1.
In the current study, β2-adrenergic
receptor-mediated dephosphorylation was investigated in MDA-MB-231
and MDA-MB-468 triple negative breast cancer cell lines. These
cells express high levels of the β2-adrenergic receptor
(12) and high ERK1/2 activity,
with high pERK1/2 levels (21).
Other breast cancer cell lines differ in terms of the expression of
HER2, ER and PR, and these cell lines possess different properties
compared with triple negative breast cancer cell lines. These
differences may result in variability with regards to the mechanism
of β2-adrenergic receptor-mediated dephosphorylation of
ERK1/2. Therefore, to reduce variability, the present study
performed experiments in only two different, triple negative breast
cancer cell lines. Further studies are required to investigate the
associations among the β2-adrenergic receptor, pERK1/2,
DUSP1 and PP1 in breast cancer cells other than the triple negative
type. In addition, further studies, such as directly measuring the
activity of DUSP1 and PP1 with β2-adrenergic receptor
stimulation in breast cancer cell lines, should be performed to
further confirm these results, as only western blot analysis was
performed in the present study to identify the association between
the β2-adrenergic receptor and DUSP1 and PP1.
In conclusion, the present study demonstrated that
β2-adrenergic receptor stimulation led to the
dephosphorylation of ERK1/2, which was inhibited by the DUSP1
inhibitor BCI and the PP1 inhibitor calyculin A, and increased
DUSP1 expression and PP1 activity. Furthermore, downregulation of
DUSP1 and PP1 expression reduced terbutaline-mediated pERK1/2
dephosphorylation. Therefore, the results of the present study
demonstrated that DUSP1 and PP1 may have important roles in
β2-adrenergic receptor-mediated ERK1/2 dephosphorylation
in MDA-MB-231 and MDA-MB-468 breast cancer cells.
β2-adrenergic receptor-mediated inactivation of ERK1/2
or activation of DUSP1 and PP1 can dephosphorylate and inactivate
some malignant signaling molecules and may effect cancer
progression. The consequences of this action of the
β2-adrenergic receptor should be examined in preclinical
and clinical studies, particularly when β2-adrenergic
blockers are used in patients with triple negative breast
cancer.
Acknowledgements
The authors thank Dr Bala Gur Dedeoğlu
(Biotechnology Institute, University of Ankara, Ankara, Turkey) and
Dr H. Ongun Onaran (Department of Medical Pharmacology, Faculty of
Medicine, University of Ankara) for critical reading of the
manuscript. The present study was supported by a research grant
from the Scientific and Technological Research Council of Turkey
(TÜBITAK; grant no. SBAG-113S396).
Glossary
Abbreviations
Abbreviations:
|
BCI
|
2-benzylidene-3-(cyclohexylamino)-2,3-dihydro-1H-inden-1-one
|
|
DUSP
|
dual-specificity phosphatase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
PP
|
protein phosphatase
|
References
|
1
|
Marchetti B, Spinola PG, Pelletier G and
Labrie F: A potential role for catecholamines in the development
and progression of carcinogen-induced mammary tumors: Hormonal
control of beta-adrenergic receptors and correlation with tumor
growth. J Steroid Biochem Mol Biol. 38:307–320. 1991. View Article : Google Scholar
|
|
2
|
Entschladen F, Drell TL IV, Lang K, Joseph
J and Zaenker KS: Tumour-cell migration, invasion, and metastasis:
Navigation by neurotransmitters. Lancet Oncol. 5:254–258. 2004.
View Article : Google Scholar
|
|
3
|
Antoni MH, Lutgendorf SK, Cole SW, Dhabhar
FS, Sephton SE, McDonald PG, Stefanek M and Sood AK: The influence
of bio-behavioural factors on tumour biology: Pathways and
mechanisms. Nat Rev Cancer. 6:240–248. 2006. View Article : Google Scholar :
|
|
4
|
Thaker PH, Lutgendorf SK and Sood AK: The
neuroendocrine impact of chronic stress on cancer. Cell Cycle.
6:430–433. 2007. View Article : Google Scholar
|
|
5
|
Lutgendorf SK, Sood AK and Antoni MH: Host
factors and cancer progression: Biobehavioral signaling pathways
and interventions. J Clin Oncol. 28:4094–4099. 2010. View Article : Google Scholar :
|
|
6
|
Cole SW, Nagaraja AS, Lutgendorf SK, Green
PA and Sood AK: Sympathetic nervous system regulation of the tumour
microenvironment. Nat Rev Cancer. 15:563–572. 2015. View Article : Google Scholar :
|
|
7
|
Vandewalle B, Revillion F and Lefebvre J:
Functional beta-adrenergic receptors in breast cancer cells. J
Cancer Res Clin Oncol. 116:303–306. 1990. View Article : Google Scholar
|
|
8
|
Draoui A, Vandewalle B, Hornez L,
Revillion F and Lefebvre J: Beta-adrenergic receptors in human
breast cancer: Identification, characterization and correlation
with progesterone and estradiol receptors. Anticancer Res.
11:677–680. 1991.
|
|
9
|
Slotkin TA, Zhang J, Dancel R, Garcia SJ,
Willis C and Seidler FJ: Beta-adrenoceptor signaling and its
control of cell replication in MDA-MB-231 human breast cancer
cells. Breast Cancer Res Treat. 60:153–166. 2000. View Article : Google Scholar
|
|
10
|
Powe DG, Voss MJ, Habashy HO, Zänker KS,
Green AR, Ellis IO and Entschladen F: Alpha- and beta-adrenergic
receptor (AR) protein expression is associated with poor clinical
outcome in breast cancer: An immunohistochemical study. Breast
Cancer Res Treat. 130:457–463. 2011. View Article : Google Scholar
|
|
11
|
Shi M, Liu D, Duan H, Qian L, Wang L, Niu
L, Zhang H, Yong Z, Gong Z, Song L, et al: The β2-adrenergic
receptor and Her2 comprise a positive feedback loop in human breast
cancer cells. Breast Cancer Res Treat. 125:351–362. 2011.
View Article : Google Scholar
|
|
12
|
Madden KS, Szpunar MJ and Brown EB:
β-Adrenergic receptors (β-AR) regulate VEGF and IL-6 production by
divergent pathways in high β-AR-expressing breast cancer cell
lines. Breast Cancer Res Treat. 130:747–758. 2011. View Article : Google Scholar :
|
|
13
|
Barron TI, Sharp L and Visvanathan K:
Beta-adrenergic blocking drugs in breast cancer: A perspective
review. Ther Adv Med Oncol. 4:113–125. 2012. View Article : Google Scholar :
|
|
14
|
Zhang P, He X, Tan J, Zhou X and Zou L:
β-arrestin2 mediates β-2 adrenergic receptor signaling inducing
prostate cancer cell progression. Oncol Rep. 26:1471–1477.
2011.
|
|
15
|
Ji Y, Chen S, Li K, Xiao X, Zheng S and Xu
T: The role of β-adrenergic receptor signaling in the proliferation
of hemangioma-derived endothelial cells. Cell Div. 8:12013.
View Article : Google Scholar :
|
|
16
|
Carie AE and Sebti SM: A chemical biology
approach identifies a beta-2 adrenergic receptor agonist that
causes human tumor regression by blocking the Raf-1/Mek-1/Erk1/2
pathway. Oncogene. 26:3777–3788. 2007. View Article : Google Scholar
|
|
17
|
Piñero Pérez C, Bruzzone A, Sarappa MG,
Castillo LF and Lüthy IA: Involvement of α2- and β2-adrenoceptors
on breast cancer cell proliferation and tumour growth regulation.
Br J Pharmacol. 166:721–736. 2012. View Article : Google Scholar :
|
|
18
|
Roskoski R Jr: ERK1/2 MAP kinases:
Structure, function, and regulation. Pharmacol Res. 66:105–143.
2012. View Article : Google Scholar
|
|
19
|
Price DM, Chik CL and Ho AK:
Norepinephrine induction of mitogen-activated protein kinase
phosphatase-1 expression in rat pinealocytes: Distinct roles of
alpha- and beta-adrenergic receptors. Endocrinology. 145:5723–5733.
2004. View Article : Google Scholar
|
|
20
|
Chruscinski AJ, Singh H, Chan SM and Utz
PJ: Broad-scale phosphoprotein profiling of beta adrenergic
receptor (β-AR) signaling reveals novel phosphorylation and
dephosphorylation events. PLoS One. 8:e821642013. View Article : Google Scholar :
|
|
21
|
Bartholomeusz C, Gonzalez-Angulo AM, Liu
P, Hayashi N, Lluch A, Ferrer-Lozano J and Hortobágyi GN: High ERK
protein expression levels correlate with shorter survival in
triple-negative breast cancer patients. Oncologist. 17:766–774.
2012. View Article : Google Scholar :
|
|
22
|
Boutros T, Chevet E and Metrakos P:
Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase
regulation: Roles in cell growth, death, and cancer. Pharmacol Rev.
60:261–310. 2008. View Article : Google Scholar
|
|
23
|
Bermudez O, Marchetti S, Pagès G and
Gimond C: Post-translational regulation of the ERK phosphatase
DUSP6/MKP3 by the mTOR pathway. Oncogene. 27:3685–3691. 2008.
View Article : Google Scholar
|
|
24
|
Wu JJ, Zhang L and Bennett AM: The
noncatalytic amino terminus of mitogen-activated protein kinase
phosphatase 1 directs nuclear targeting and serum response element
transcriptional regulation. Mol Cell Biol. 25:4792–4803. 2005.
View Article : Google Scholar :
|
|
25
|
Zhou B, Wang ZX, Zhao Y, Brautigan DL and
Zhang ZY: The specificity of extracellular signal-regulated kinase
2 dephosphorylation by protein phosphatases. J Biol Chem.
277:31818–31825. 2002. View Article : Google Scholar
|
|
26
|
Zhang Z, Kobayashi S, Borczuk AC, Leidner
RS, Laframboise T, Levine AD and Halmos B: Dual specificity
phosphatase 6 (DUSP6) is an ETS-regulated negative feedback
mediator of oncogenic ERK signaling in lung cancer cells.
Carcinogenesis. 31:577–586. 2010. View Article : Google Scholar :
|
|
27
|
Brondello J, Brunet A, Pouysségur J and
Mckenzie FR: The dual specificity mitogen-activated protein kinase
phosphatase-1 and −2 are induced by the p42/p44MAPK cascade. J Biol
Chem. 272:1368–1376. 1997. View Article : Google Scholar
|
|
28
|
Chen J, Martin BL and Brautigan DL:
Regulation of protein serine-threonine phosphatase type-2A by
tyrosine phosphorylation. Science. 257:1261–1264. 1992. View Article : Google Scholar
|
|
29
|
Guo CY, Brautigan DL and Larner JM:
Ionizing radiation activates nuclear protein phosphatase-1 by
ATM-dependent dephosphorylation. J Biol Chem. 277:41756–41761.
2002. View Article : Google Scholar
|
|
30
|
Candas D, Lu CL, Fan M, Chuang FY, Sweeney
C, Borowsky AD and Li JJ: Mitochondrial MKP1 is a target for
therapy-resistant HER2-positive breast cancer cells. Cancer Res.
74:7498–7509. 2014. View Article : Google Scholar :
|
|
31
|
Haagenson KK, Zhang JW, Xu Z, Shekhar MP
and Wu GS: Functional analysis of MKP-1 and MKP-2 in breast cancer
tamoxifen sensitivity. Oncotarget. 5:1101–1110. 2014. View Article : Google Scholar :
|
|
32
|
Nunes-Xavier C, Romá-Mateo C, Ríos P,
Tárrega C, Cejudo-Marín R, Tabernero L and Pulido R:
Dual-specificity MAP kinase phosphatases as targets of cancer
treatment. Anticancer Agents Med Chem. 11:109–132. 2011. View Article : Google Scholar
|
|
33
|
Hill TA, Stewart SG, Sauer B, Gilbert J,
Ackland SP, Sakoff JA and McCluskey A: Heterocyclic substituted
cantharidin and norcantharidin analogues-synthesis, protein
phosphatase (1 and 2A) inhibition, and anti-cancer activity. Bioorg
Med Chem Lett. 17:3392–3397. 2007. View Article : Google Scholar
|
|
34
|
Winter SL, Bosnoyan-Collins L, Pinnaduwage
D and Andrulis IL: The interaction of PP1 with BRCA1 and analysis
of their expression in breast tumors. BMC Cancer. 7:852007.
View Article : Google Scholar :
|
|
35
|
Brand TM, Iida M and Wheeler DL: Molecular
mechanisms of resistance to the EGFR monoclonal antibody cetuximab.
Cancer Biol Ther. 11:777–792. 2011. View Article : Google Scholar :
|
|
36
|
Wu W, Pew T, Zou M, Pang D and Conzen SD:
Glucocorticoid receptor-induced MAPK phosphatase-1 (MPK-1)
expression inhibits paclitaxel-associated MAPK activation and
contributes to breast cancer cell survival. J Biol Chem.
280:4117–4124. 2005. View Article : Google Scholar
|
|
37
|
Swingle M, Ni L and Honkanen RE:
Small-molecule inhibitors of ser/thr protein phosphatases:
Specificity, use and common forms of abuse. Methods Mol Biol.
365:23–38. 2007.
|