Introduction
Chronic liver injury generally leads to hepatic
fibrosis and may subsequently progress to cirrhosis, a serious
liver disease that may eventually result in the development of
hepatocellular carcinoma (HCC) (1–3).
Numerous studies have demonstrated that the activation of hepatic
stellate cells (HSCs), which store fat in the liver, represent a
pivotal event in the initiation of hepatic fibrogenesis, during
which quiescent HSCs specialized for retinoid-storage are
transformed to contractile cells termed myofibroblast as a result
of liver damage (1). Activated
HSCs or myofibroblasts are characterized by positive expression of
α-smooth muscle actin (α-SMA) and by secretion of the extracellular
matrix (ECM) components such as collagen type I and collagen type
II, which are mainly responsible for the formation of scar tissue
and the development of cirrhosis (1). This phenotype shift of HSCs is also
accompanied by an increase in cell proliferation and contractility
of activated HSCs. Transforming growth factor-β (TGF-β) has been
shown to mediate myofibroblastic transformation of quiescent HSCs
and accumulation of ECM in response to liver injury (4–8). The
TGF-β family of cytokines possesses three isoforms, TGF-β1, TGF-β2,
and TGF-β3. These regulate various cellular processes including
cell proliferation, apoptosis, differentiation, and migration
through a number of signaling pathways (4,9).
However, the molecular mechanisms involved in the activation of
HSCs are not well understood.
Nitric oxide (NO) is an important cell signaling
molecule produced from oxidation of L-arginine, a step that is
enzymatically catalyzed by NO synthase (NOS). Studies have shown
that the activity of NOS is competitively inhibited by asymmetric
dimethylarginine (ADMA), a naturally occurring analogue of
L-arginine in humans. In humans, ADMA is catabolized by the enzyme
dimethylarginine dimethylaminohydrolase (DDAH); abnormal
down-regulation of DDAH or inhibition of its activity could result
in accumulation of ADMA in the plasma. DDAH has two isoforms:
DDAH1, a major isoform in the liver, and DDAH2 which is mainly
expressed in the endothelial cells (10). In a recent study, over-expression
of DDAH was shown to prevent renal fibrosis via suppression of
ADMA, a well-known inhibitor of NOS, and therefore reduction of
ADMA would lead to an increase in the production of NO (11,12).
Elevated plasma levels of ADMA were also found to be strongly
associated with the degree of liver damage and hepatic fibrosis
(13–15). Interestingly, rat primary HSCs
exposed to excessive ADMA displayed strong induction of α-SMA and
collagen type I, which suggests that the ADMA/DDAH pathway could be
involved in hepatic fibrogenesis in response to liver injury
(16–18).
In the present study, we investigated the role of
the ADMA/DDAH pathway in the TGF-β1-induced activation of freshly
isolated rat primary HSCs. We furher examined if the MAPK pathway
could participient in the TGF-β1-associated effects on DDAH/ADMA by
using specific inhibiors for three main subgroups in the MAPK
superfamily [p38 kinases (p38), extracellular signal regulated
kinases (ERK), and c-Jun N-terminal kinases (JNK)]. Our findings
may help understand the molecular mechanisms involved in activation
of HSCs, and help identify target molecules for the development of
preventive and therapeutic approaches to liver fibrosis.
Materials and methods
Reagents and materials
Rat anti-α-SMA monoclonal antibody and the enzyme
linked immunosorbent assay (ELISA) kit for collagen type I were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany); DMEM
cell culture medium was from Gibco (Cambridge, UK);
Mitogen-activated protein kinase (MAPK) specific inhibitors
SB203580, PD98059, and SP600125 were purchased from Beyotime
Biotechnology (Shanghai, China); Anti-DDAH1 and anti-TGF-β1
antibodies were purchased from Abcam (Grand Island, NY, USA).
Preparation, culture, and verification
of the rat primary HSCs
Male Sprague-Dawley (SD) rats (body weight: 350 to
400 g) were obtained from the Shanghai SLAC Laboratory Animal Inc.
(Shanghai, China). Fresh liver tissues of the SD rats were used for
isolation of primary HSCs. Prior to the experiments, all SD rats
were maintained in the Animal Center of Hunan Province and handled
according to the protocols that were approved by the Animal Care
and Ethics Committee at the Animal Center of Hunan Province [no.
SCXK (Xing) 2011-0003]. Rat primary HSCs were prepared and cultured
as described elsewhere (19–21).
Briefly, primary HSCs were isolated by digestion of the freshly
harvested rat liver tissues with in situ perfusion of
pronase and collagenase, followed by a single-step density Nycodenz
gradient centrifugation. During the centrifugation, HSCs cells were
separated from other hepatic cells due to the high lipid content of
HSCs.
Trypan Blue staining was used to assess the
viability of the freshly prepared HSCs. In brief, 100 µl of the
HSCs suspension were mixed with the equal value of 0.4% typan blue
solution, and then 10 µl of the resulting suspension were dropped
on a slide with a cell counting chamber, which was followed by
calculating the numbers of viable and nonviable HSCs under an
optical microscope (Olympus Corporation, Tokyo, Japan). Desmin
immunocytochemistry was utilized to examine the purity of the
isolated HSCs. Briefly, HSCs were fixed, permeabilized, and
incubated overnight in primary antibodies. Following three washes
with PBS, bound primary antibodies were detected with secondary
antibody. HSCs were visualized and characterized by microscopy
using a microscope (Olympus Corporation). Next, Image-Pro Plus
software 6.0 (Media Cybernetics, Silver Spring, MD, USA) was used
for analysis of the images, and the average optical density (AOD)
value was calculated to represent the expression levels of desmin.
Freshly prepared HSCs were grown in DMEM cell culture medium
supplemented with 20% fetal bovine serum (FBS) for 24 h, and the
culture medium was replaced with DMEM containing reduced
concentration FBS (0.5%), cultured for another 24 h, and followed
by exposure to different treatments.
Treatments of the rat primary
HSCs
The HSCs were subsequently divided into different
groups as per the experimental protocol. To determine the effect of
TGF-β1 on DDAH/ADMA, HSCs were treated with different
concentrations of TGF-β1 (0, 1, 2.5, and 5 ng/ml) for 48 h. HSCs
were harvested for subsequent analysis of the protein expression of
DDAH1 and ADMA, and the activity of DDAH. In the ADMA treatment
group, HSCs were incubated with ADMA (0, 1.0, 2.5, 5.0 µM) for 48
h, cell culture medium and HSCs were collected for subsequent
measurement. In some experiments, we included specific inhibitors
of the mitogen-activated protein kinase (MAPK) superfamily: p38
kinases (p38) extracellular signal regulated kinases (ERK), and
c-Jun N-terminal kinases (JNK). Three inhibitors specific for p38
(SB203580), ERK (PD98059), and JNK (SP600125) were selected, and
HSC cells were pretreated with 2 µM of SB203580, PD98059, and
SP600125 for 30 min, prior to exposure to 5 ng/ml of TGF-β1. 48 h
post treatment, HSCs and cell culture were harvested for subsequent
analyses.
Immunohistochemical examination for
α-SMA positive HSCs
HSCs with positive expression of α-SMA were assessed
in the different experimental groups after immunohistochemical
staining using anti-α-SMA monoclonal antibodies. α-SMA-positive
HSCs were defined as HSCs with light or dark brown particles
localized in the cell membrane and/or cytoplasm. The staining
intensity was assessed using Image-ProPlus image processing
software and quantified by calculating the AOD.
ELISA for collagen type I
expression
ELISA for type I collagen was performed according to
the manufacturer's instructions (Sigma-Aldrich; Merck KGaA). In
brief, the HSCs culture medium was collected from the experimental
groups, diluted with PBS, and subsequently incubated with rabbit
anti-rat anti-type I collagen antiserum (1:2,000) and goat
anti-rabbit IgG HRP (1:1,000). 2 ml/l of H2SO4 was used to
terminate the reaction. Fresh serum-free medium was used as
negative control. The concentrations of type I collagen in the cell
culture medium were calculated by the absorbance obtained at
wavelength of 490 nm on a spectrophotometer.
Measurement of ADMA in the cell
culture medium and DDAH activity assay
Levels of ADMA in the cell culture medium were
measured with high performance liquid chromatography (HPLC), as
described elsewhere (22). The
DDAH activity assay was carried out as reported previously
(22). In brief, ADMA was added to
HSCs lysates at a final concentration of 500 µM, followed by
incubation at 37°C for 2 h. 30% of sulfurosalicylic acid, which is
able to inactivate DDAH, was used to terminate the reaction. The
remaining ADMA was measured by HPLC, and the reduction of ADMA
amount reflected the DDAH activity. DDAH activity in the various
experimental groups was expressed relative to that of the normal
control group.
Western blotting for protein
expression of DDAH1 and TGF-β1
Western blot analysis was performed to examine the
protein levels of DDAH1 and TGF-β1. In brief, total proteins
extracted from the HSCs in various experimental groups were
separated on 8% sodium dodecyl sulphate-polyacrylamide gel
electrophoresis (SDS-PAGE). The proteins were subsequently
transferred onto ImmunBlot PVDF membranes and incubated with
anti-DDAH1 (1:2,000) and anti-TGF-β1 (1:5,000) primary antibodies,
respectively, at room temperature for 2 h. The resulting bands were
visualized using ECL and analyzed on an Image J analyzer imaging
system. The intensity of DDAH1 and TGF-β1 bands was normalized to
that of β-actin as an internal control.
Cell proliferation assay
MTT assay was used to assess cell proliferation in
the control group, and three experimental groups: TGF-β1, ADMA, and
SB203580. The HSCs were exposed to 5.0 ng/ml TGF-β1, 5.0 µM ADMA,
or 3.0 µM SB203580 for different durations of time (0, 12, 24, 48,
72, 96 h). 10 µl of MTT working solution (5 mg/ml) was added to
each well, incubated at 37°C for 4 h, and mixed with 100 µl of
DMSO. Absorbance (A) was obtained on a microplate reader at a
wavelength of 490 nm.
Statistical analysis
All experiments in this study were performed for a
minimum of three times and all data were presented as mean values
with standard error (SE) (mean ± SE). Statistical analysis was
performed with IBM SPSS Statistics version 17.0 from SPSS, Inc.
(Chicago, IL, USA). Multi-factor analysis of variance (ANOVA) was
used to detect significant differences. P<0.05 was considered to
indicate a statistically significant difference.
Results
Viability and purity of the freshly
prepared rat primary HSCs
We first isolated primary HSCs from the liver
tissues of SD rats. The yield was approximately 7×107
cells per rat. The viability of freshly prepared HSCs was
determined by Trypan Blue staining and was found to be more than
92% (data not shown). The purity of the isolated HSCs was examined
using immunohistochemical staining for Desmin. Results were
analyzed under phase-contract microscopy, in combination with
analysis by ultraviolet-excited fluorescence microscopy. The purity
was greater than 90% (data not shown). The high viability and
purity of the extracted rat primary rat HSCs met the requirement
for the proposed experiments in this study.
Effects of excessive TGF-β1 on the
expression and activity of DDAH, and levels of ADMA
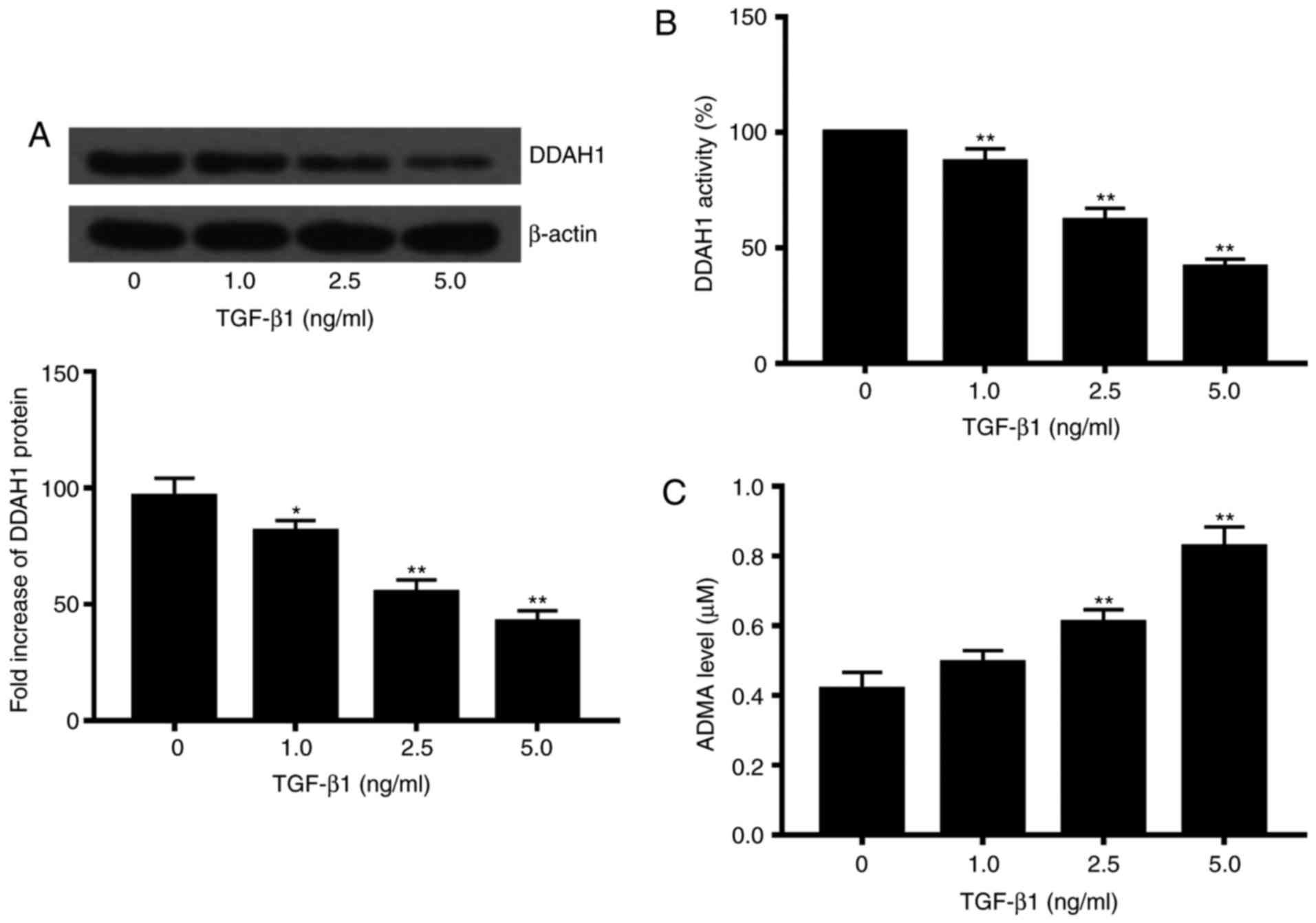
With the freshly isolated rat primary HSCs, we first
determined effects of TGF-β1 on the protein expression and activity
of DDAH in the HSCs, as well as levels of ADMA in the culture
medium. In this study, the isoform DDAH1 rather than DDAH2 was
selected. On exposure to various concentrations of TGF-β1 (1, 2.5
and 5 ng/ml), both DDAH1 protein expression and activity of the HSC
cells were significantly reduced as compared to that in controls
(P<0.05). Moreover, the effect was highly dose-dependent
(Fig. 1A and B). TGF-β1
concentrations of 2.5 and 5 ng/ml caused a significant increase in
ADMA in the cell culture medium as compared to that in controls
(P<0.05; Fig. 1C).
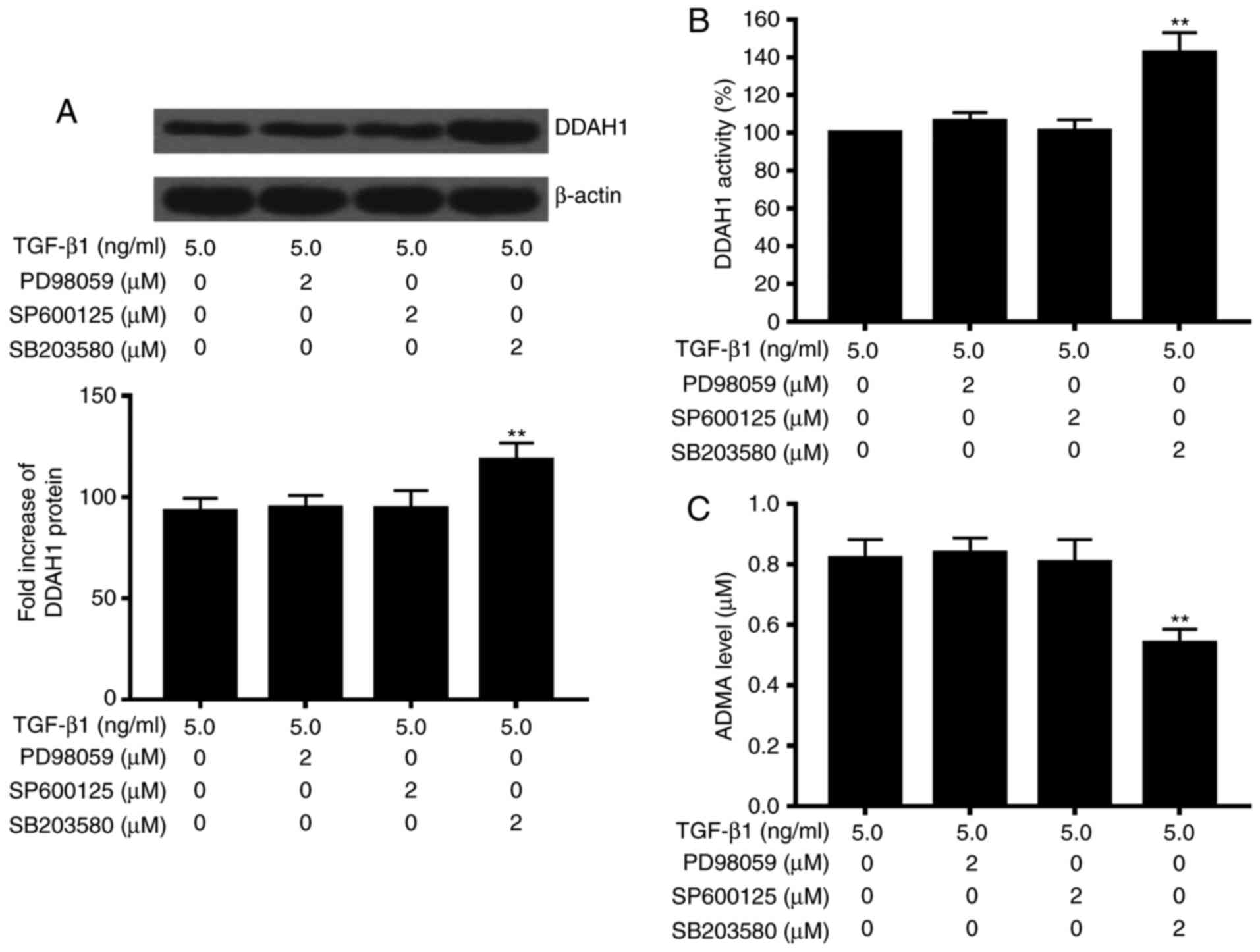
Role of p38 MAPK pathway in the
TGF-β1-associated effects on DDAH/ADMA
We next explored the potential involvement of the
MAPK molecular pathway in the TGF-β1-associated effects on
DDAH/ADMA, and included three inhibitors specific for p38
(SB203580), ERK (PD98059), and JNK (SP600125) in the subsequent
experiments. Freshly prepared rat primary HSCs were treated with
the p38 MAPK specific inhibitor SB203580 or vehicle only as
control. We also included an ERK inhibitor (PD98059) and JNK
inhibitor (SP600125) to determine if p38 MAPK could be specific to
the TGF-β1-induced alteration on DDAH/ADMA, and also to exclude the
potential contribution of ERK and JNK in the observed effects.
p38MAPK specific inhibitor inhibited the TGF-β1-induced effects on
DDAH/ADMA, while protein expression of DDAH1 and activity of DDAH
increased, and protein levels of ADMA in the cell culture medium
decreased (P<0.05 vs. TGF-β1 alone). In contrast, neither ERK
inhibitor PD98059 nor JNK inhibitor SP600125 displayed any effect
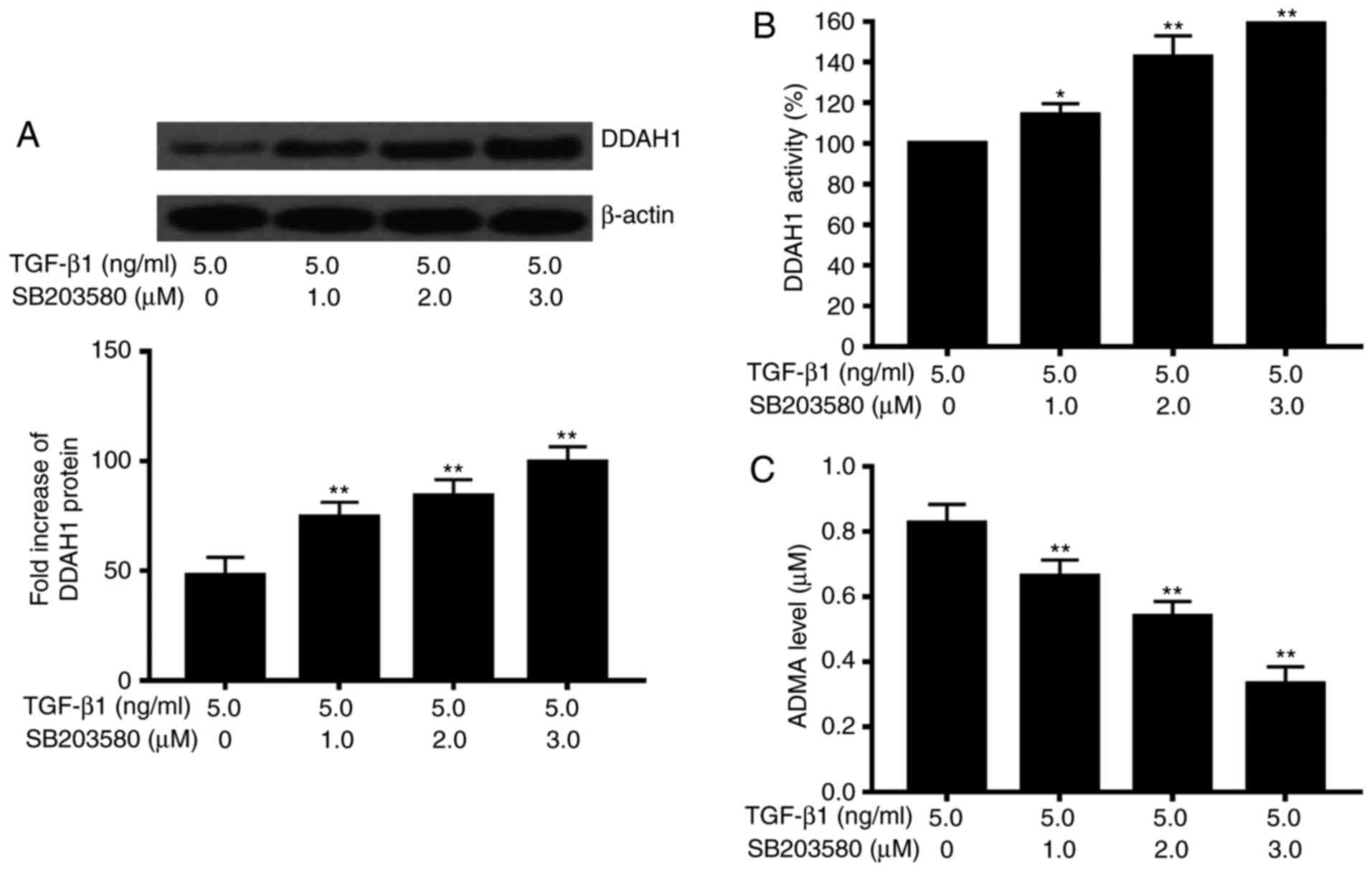
on the TGF-β1-associated effects on DDAH/ADMA (Fig. 2). We then examined the effect of
different doses of the p38MAPK specific inhibitor (0, 1, 2 and 3
µM) on TGF-β1-induced effects on DDAH/ADMA. The results showed that
the effects on the protein levels of DDAH1 and ADMA, and on the
activity of DDAH were dose-dependent (Fig. 3).
Effects of excessive ADMA on levels of
α-SMA and collagen type I
We next asked if increased levels of ADMA induced by
TGF-β1 could alter the expressions of fibrotic markers α-SMA and
collagen type I. We found that the cell number of HSCs positive for
α-SMA increased significantly in response to excessive ADMA
treatment at different concentrations (0, 1, 2.5 and 5 µM) as
compared to that in the controls, and that the effect was
dose-dependent (P<0.05; Fig.
4A-C). Rat primary HSCs exposed to ADMA (2.5 and 5 µM) showed
significant enhancement in the production of collagen type I vs.
control cells without ADMA treatment (P<0.05; Fig. 4D). Moreover, excessive ADMA at the
indicated concentrations (1, 2.5 and 5 µM) significantly increased
the protein expression of collagen type I in comparison with
control HSCs without exposure to ADMA (P<0.05; Fig. 4E and F).
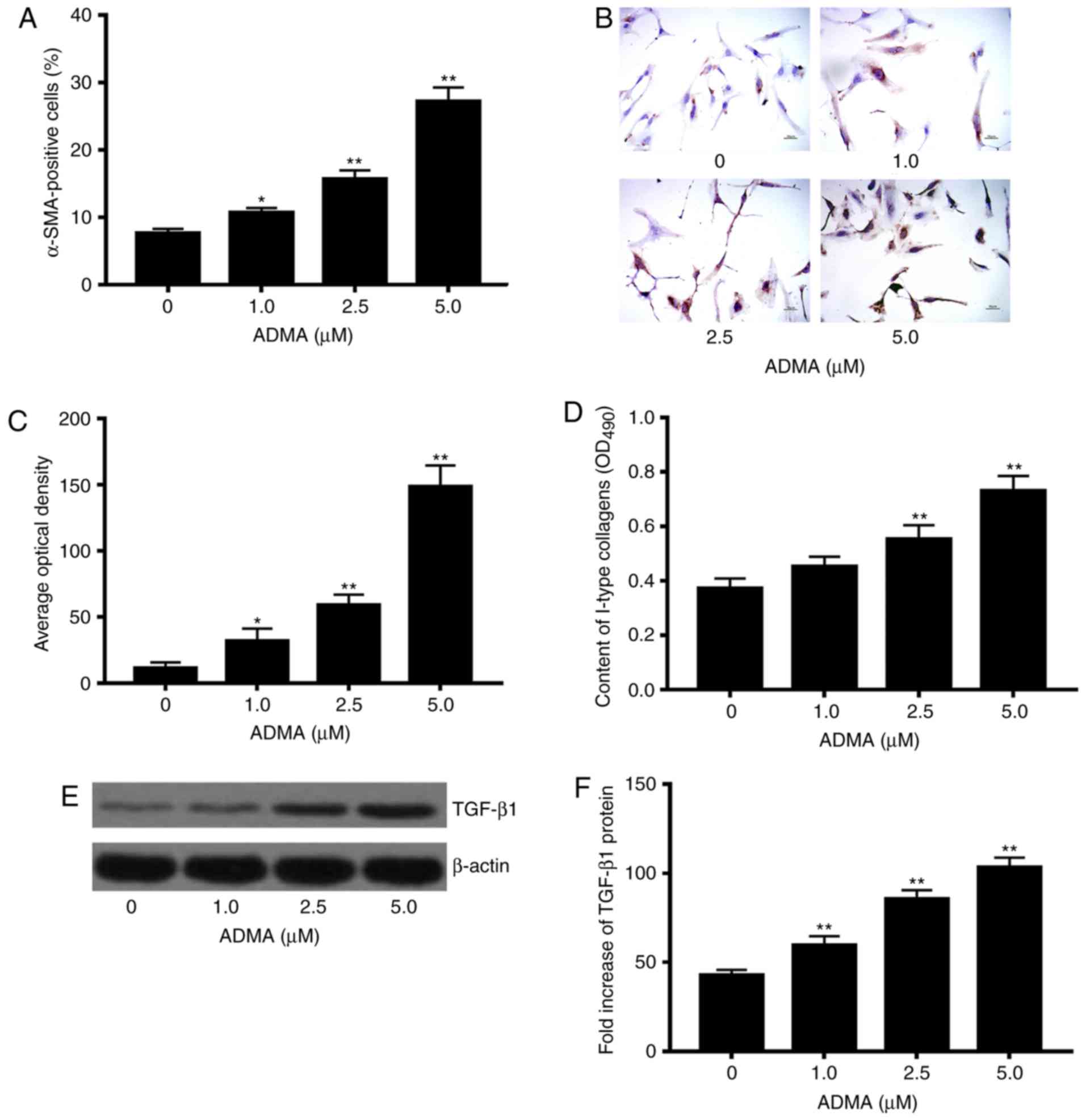 | Figure 4.Effects of asymmetric dimethylarginine
(ADMA) treatment on α-SMA-positive hepatic stellate cells (HSCs),
intensity of α-SMA, and type I collagen in rat primary HSCs.
Freshly prepared rat primary HSCs were treated with different
concentrations of ADMA (0, 1, 2.5 and 5 µM) for 48 h.
α-SMA-positive HSCs were assayed by IHC. α-SMA-positive HSCs were
defined as those with light or dark brown particles visualized in
the cell membrane and/or cytoplasm. The intensity of the
α-SMA-positive HSCs was quantified with Image-ProPlus 6 digital
medical image system. (A) Effect of ADMA on the number of the
α-SMA-positive HSCs; (B) Representative images of IHC staining for
α-SMA protein in the rat HSCs treated with increasing
concentrations of ADMA; (C) effect of ADMA on α-SMA expression in
rat HSCs. IHC staining intensity of the images as illustrated in
(B) was quantified by calculating the average optical density (AOD)
using Image-Pro Plus software 6.0 (Media Cybernetics, Silver
Spring, MD, USA). ELISA for type I collagen was performed, and the
concentrations of type I collagen were calculated by absorbance at
490 nm wavelength on a spectrophotometer, while western blot
analysis was used to examine the protein levels of transforming
growth factor (TGF)-β1 and β-actin; (D) effect of ADMA on levels of
type I collagen in rat HSCs; (E) Effects of ADMA on protein
expression of TGF-β1 in rat HSCs; (F) the bar graphs were generated
from the above western blot analysis, showing the quantitative
results after the amounts of TGF-β1 protein were normalized to
those for β-actin, which did not vary with ADMA treatment.
*P<0.05, **P<0.01 vs. control. Magnification, ×200. |
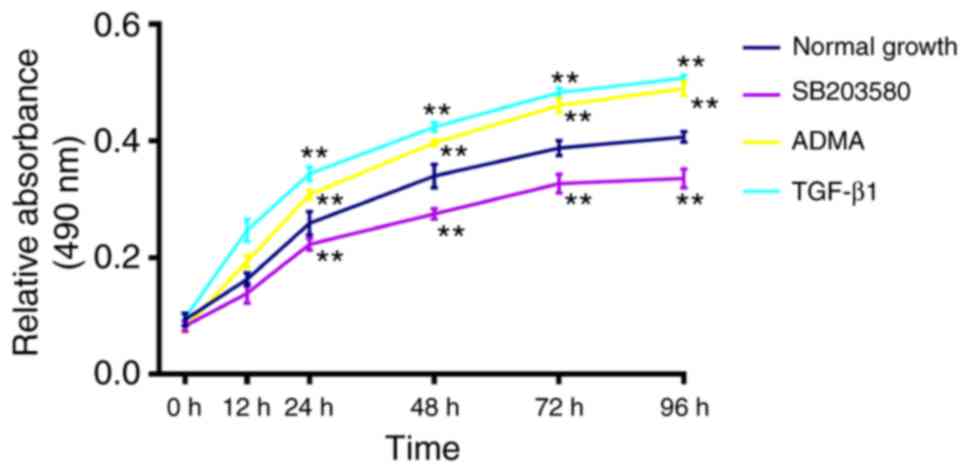
Effects of ADMA and the p38 MAPK
specific inhibitor on the proliferation of HSCs
Since cell proliferation is generally promoted in
the activated HSCs, we further evaluated the role for the DDAH/ADMA
pathway on the hepatic fibrogenesis and conducted comparative
studies to assess the effects of excessive ADMA, TGF-β1, and the
p38 MAPK specific inhibitor on the proliferation of HSCs. TGF-β1
showed the greatest increase in the proliferation of HSCs, followed
by the ADMA treatment in contrast to controls, whereas the p38 MAPK
displayed a significant inhibitory effect on the growth of HSCs as
compared to that of control HSCs (P<0.01; Fig. 5).
Discussion
The molecular mechanisms whereby TGF-β induces the
activation of HSCs are not well understood. The key novel findings
from this study are as follows: i) TGF-β1 significantly suppressed
the DDAH protein expression and activity, and increased levels of
ADMA in rat primary HSCs (Fig. 1);
ii) the TGF-β1-mediated effects on DDAH/ADMA were significantly
abrogated by the p38 MAPK specific inhibitor SB203580, but not the
ERK- and JNK-specific inhibitors (Figs. 2 and 3); and iii) Enhancement of ADMA
significantly induced expression of α-SMA and type I collagen in
the rat primary HSCs (Fig. 4), and
promoted cell proliferation of HSCs as compared to that in controls
(Fig. 5).
It has been well-documented that the TGF-β family
members including TGF-β1, TGF-β2, and TGF-β3 in mammals are major
mediators of fibrosis in response to tissue injury of the liver and
kidney through the TGF-β-mediated signaling pathways (5,9). In
the present study, we demonstrated that TGF-β1 suppressed the
protein expression and activity of DDAH, and in turn led to an
accumulation of ADMA in the culture medium of rat primary HSCs, and
that the p38 MAPK pathway participated in the TGF-β1-associated
effects on DDAH/ADMA. The findings, to our knowledge, have not been
reported previously and may represent an important advance in our
understanding of the roles of TGF-β1 and ADMA in the pathogenesis
of hepatic and renal fibrosis. It has been reported that the
regulation of DDAH activity by TGF-β1 comprises of both inhibition
of translation activity and that of translation level protein
expression (5,9). In this study, it is likely that
reduced protein expression of DDAH1 by TGF-β1 led to a decrease in
its activity. Of note, the above TGF-β1-mediated effects on
DDAH/ADMA were abrogated by the presence of p38 MAPK specific
inhibitor, but not by the inhibitors of JNK and ERK, which
indicates an involvement of p38 MAPK pathway rather than JNK-MAPK
and ERK-MAPK pathways in this effect.
The activation of HSCs is well recognized as a
central event in hepatic fibrosis, which involves a range of
signaling pathways. A number of studies have shown that ADMA, an
endogenous molecule (23), is
associated with the severity and progression of liver damage
(15,24–27).
In a study by Li et al (28), it was important to note that
excessive ADMA significantly induced mRNA expression of α-SMA,
increased α-SMA-positive cells ratio, and synthesis of type I
collagen in dose- and time-dependent manners in HSCs through, at
least in part, the ROS-NF-κB molecular pathway, which suggests that
ADMA is involved in the activation of HSCs as a potentially novel
mediator of transformation of HSCs (28). Besides its potential role in liver
fibrosis, ADMA has also been implicated in renal fibrosis (29,30)
and in tubuleinterstitial ischaemia on the early stage in diabetic
nephropathy (DN) (31). With a rat
model of DN, Shibata et al (31) found that reduction of AMDA by
over-expression of DDAH diminished levels of TGF-beta 1, also
well-documented a critical mediator in renal fibrogenesis, and that
the tubuleinterstitial ischaemia was subsequently improved. Lluch
et al (32) examined plasma
concentrations of ADMA in patients with compensated alcohol
cirrhosis and with advance cirrhosis in comparison with healthy
controls, and found that the cirrhotic subjects had higher plasma
levels of ADMA than the healthy subjects (ADMA ranged from 0.3 to
0.5 µmol/l). In this study, we initially performed a dose-dependent
in vitro experiment by including lower concentrations of
ADMA, and only at higher concentrations caused significant effects
on the tested liver fibrotic markers, collagen type I (ADMA at
concentrations of 2.5 and 5 µM) and α-SMA (ADMA at concentrations
1, 2.5, 5 µM). Based on this result, higher concentrations of ADMA
were selected in this study, and we demonstrated that both α-SMA
and type I collagen were markedly up-regulated in a dose-dependent
fashion after exposure of freshly isolated HSCs to increasing
concentrations of ADMA. These results were in agreement with the
previous reports of the role of ADMA/DDAH in tissue fibrosis. It
was worthy of more attention in our study that the ADMA-mediated
action appeared to be achieved through the p38MAPK pathway as it
was disrupted by the p38 MAPK specific inhibitor SB253080 but not
by the inhibitors for the JNK- and ERK-MAPKs. In addition, the
results obtained in this study supported the potentially novel
mediator for the ADMA in the activation of HSCs and hepatic
fibrosis. Studies have revealed a role of cytoskeleton actin in the
ADMA-mediated activation of NF-κB and up-regulation of TGF-β in
human renal glomerular endothelial cells (HRGECs) (29). Indeed, ADMA treatment stimulated
assembly of stress fibers, induced DNA binding of NF-κB, and
induced an increase in TGF-β1 expression in HRGECs. After
cytoskeleton actin was disrupted, the activation of NF-κB was
suppressed (29). ADMA has also
been reported to increase TGF-β expression (33) and to induce kidney fibrosis
(29) via activation of the NF-κB
signaling pathway. However, the underlying molecular mechanisms
were not elucidated.
Our study may have a number of limitations. First,
the effective concentrations of TGF-β1 used in this in vitro
study were similar to those reported previously, but much higher
than the plasma levels of TGF-β1 observed in patients with liver
fibrosis. Due to the short duration of exposure, lower
concentration of TGF-β1 may fail to exert any effect on DDAH/ADMA,
while long-term treatment in an experiment with freshly isolated
HSCs seems to be challenging. Thus, a further study with long-term
exposure to cytokine TGF-β1 in an in vivo animal model is
required to clarify the effects of TGF-β1 on DDAH/ADMA and ADMA on
the activation of HSCs. Second, the present study demonstrated that
TGF-β1 decreases protein levels of DDAH, which may involve the
p38MAPK signaling pathway, as disturbance of the p38 abrogates
significantly the TGF-β1-mediated effects on DDAH/ADMA, while we
could not exclude the possibility that the effect of TGF-β1 on
DDAH/ADMA could be indirect. These need to be clarified by further
investigations in future. Further studies are underway in our
laboratory in this direction.
In summary, our findings on the effects of TGF-β on
DDAH/ADMA, along with those reported in previous studies, suggest
an importance of the interaction of TGF-β1 and DDAH/ADMA pathways
in inducing HSCs activation, and that suppression of the ADMA/DDAH1
pathway could be an alterative approach to combating liver
fibrosis.
Acknowledgements
This study was financially supported by the Special
National International Technology Cooperation of China (no.
2015DFA31490); National Natural Sciences Foundation of China (no.
81272253); National Major Sciences research Program of China (973
Program) (no. 2013CB910502); General plan of Hunan Science and
Technology Department (no. 2009WK3056).
References
|
1
|
Koyama Y, Xu J, Liu X and Brenner DA: New
developments on the treatment of liver fibrosis. Dig Dis.
34:589–596. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee YA, Wallace MC and Friedman SL:
Pathobiology of liver fibrosis: A translational success story. Gut.
64:830–841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou WC, Zhang QB and Qiao L: Pathogenesis
of liver cirrhosis. World J Gastroenterol. 20:7312–7324. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li J, Wang W and Shen JL: The role of
TGFbeta1 and IL-13 in cellular signal transduction of hepatic
fibrosis of schistosomiasi. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng
Chong Bing Za Zhi. 27:357–360. 2009.(In Chinese). PubMed/NCBI
|
|
5
|
Fabregat I, Moreno-Càceres J, Sánchez A,
Dooley S, Dewidar B, Giannelli G and Ten Dijke P; IT-LIVER
Consortium, : TGF-β signalling and liver disease. FEBS J.
283:2219–2232. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dooley S and ten Dijke P: TGF-β in
progression of liver disease. Cell Tissue Res. 347:245–256. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sakai K, Jawaid S, Sasaki T, Bou-Gharios G
and Sakai T: Transforming growth factor-β-independent role of
connective tissue growth factor in the development of liver
fibrosis. Am J Pathol. 184:2611–2617. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hayashi H and Sakai T: Biological
significance of local TGF-β activation in liver diseases. Front
Physiol. 3:122012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Massague J: TGFβ signalling in context.
Nat Rev Mol Cell Biol. 13:616–630. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dayoub H, Achan V, Adimoolam S, Jacobi J,
Stuehlinger MC, Wang BY, Tsao PS, Kimoto M, Vallance P, Patterson
AJ and Cooke JP: Dimethylarginine dimethylaminohydrolase regulates
nitric oxide synthesis: Genetic and physiological evidence.
Circulation. 108:3042–3047. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Arrigoni F, Ahmetaj B and Leiper J: The
biology and therapeutic potential of the DDAH/ADMA pathway. Curr
Pharm Des. 16:4089–4102. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Matsumoto Y, Ueda S, Yamagishi S,
Matsuguma K, Shibata R, Fukami K, Matsuoka H, Imaizumi T and Okuda
S: Dimethylarginine dimethylaminohydrolase prevents progression of
renal dysfunction by inhibiting loss of peritubular capillaries and
tubulointerstitial fibrosis in a rat model of chronic kidney
disease. J Am Soc Nephrol. 18:1525–1533. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ferrigno A, Rizzo V, Bianchi A, Di Pasqua
LG, Berardo C, Richelmi P and Vairetti M: Changes in ADMA/DDAH
pathway after hepatic ischemia/reperfusion injury in rats: the role
of bile. Biomed Res Int. 2014:6274342014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baranyi A, Meinitzer A, Putz-Bankuti C,
Stauber R, Kapfhammer HP and Rothenhäusler HB: Asymmetric
dimethylarginine responses during interferon-α-induced depression
in patients with chronic hepatitis C infection. Psychosom Med.
76:197–207. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mookerjee RP, Malaki M, Davies NA, Hodges
SJ, Dalton RN, Turner C, Sen S, Williams R, Leiper J, Vallance P
and Jalan R: Increasing dimethylarginine levels are associated with
adverse clinical outcome in severe alcoholic hepatitis. Hepatology.
45:62–71. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sharma JN, Al-Omran A and Parvathy SS:
Role of nitric oxide in inflammatory diseases.
Inflammopharmacology. 15:252–259. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kelly LK, Wedgwood S, Steinhorn RH and
Black SM: Nitric oxide decreases endothelin-1 secretion through the
activation of soluble guanylate cyclase. Am J Physiol Lung Cell Mol
Physiol. 286:L984–L991. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Failli P, DeFRANCO RM, Caligiuri A,
Gentilini A, Romanelli RG, Marra F, Batignani G, Guerra CT, Laffi
G, Gentilini P and Pinzani M: Nitrovasodilators inhibit
platelet-derived growth factor-induced proliferation and migration
of activated human hepatic stellate cells. Gastroenterology.
119:479–492. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Knook DL, Seffelaar AM and de Leeuw AM:
Fat-storing cells of the rat liver. Their isolation and
purification. Exp Cell Res. 139:468–471. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shu JC, Zhao JR, Yang DH, Shen Y and Zhong
CC: An improved method for the isolation of rat hepatic stellate
cells. Zhonghua Gan Zang Bing Za Zhi. 12:353–355. 2004.(In
Chinese). PubMed/NCBI
|
|
21
|
Chang W, Yang M, Song L, Shen K, Wang H,
Gao X, Li M, Niu W and Qin X: Isolation and culture of hepatic
stellate cells from mouse liver. Acta Biochim Biophys Sin
(Shanghai). 46:291–298. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang DJ, Jiang JL, Tan GS, Du YH, Xu KP
and Li YJ: Protective effects of daviditin A against endothelial
damage induced by lysophosphatidylcholine. Naunyn Schmiedebergs
Arch Pharmacol. 367:600–606. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kasumov T, Edmison JM, Dasarathy S,
Bennett C, Lopez R and Kalhan SC: Plasma levels of asymmetric
dimethylarginine in patients with biopsy-proven nonalcoholic fatty
liver disease. Metabolism. 60:776–781. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ferrigno A, Di Pasqua LG, Berardo C,
Richelmi P and Vairetti M: Liver plays a central role in asymmetric
dimethylarginine-mediated organ injury. World J Gastroenterol.
21:5131–5137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lluch P, Segarra G and Medina P:
Asymmetric dimethylarginine as a mediator of vascular dysfunction
in cirrhosis. World J Gastroenterol. 21:9466–9475. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang W, Zhao C, Zhou J, Zhen Z, Wang Y and
Shen C: Simvastatin ameliorates liver fibrosis via mediating nitric
oxide synthase in rats with non-alcoholic steatohepatitis-related
liver fibrosis. PLoS One. 8:e765382013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sharma V, Ten Have GA, Ytrebo L, Sen S,
Rose CF, Dalton RN, Turner C, Revhaug A, van-Eijk HM, Deutz NE, et
al: Nitric oxide and L-arginine metabolism in a devascularized
porcine model of acute liver failure. Am J Physiol Gastrointest
Liver Physiol. 303:G435–G441. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li JC, Chang L, Lu D, Jiang DJ and Tan DM:
Effect of asymmetric dimethylarginine on the activation of hepatic
stellate cells and its mechanism. Zhong Nan Da Xue Xue Bao Yi Xue
Ban. 32:427–432. 2007.(In Chinese). PubMed/NCBI
|
|
29
|
Wang L, Zhang D, Zheng J, Feng Y, Zhang Y
and Liu W: Actin cytoskeleton-dependent pathways for ADMA-induced
NF-κB activation and TGF-β high expression in human renal
glomerular endothelial cells. Acta Biochim Biophys Sin (Shanghai).
44:918–923. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mihout F, Shweke N, Bigé N, Jouanneau C,
Dussaule JC, Ronco P, Chatziantoniou C and Boffa JJ: Asymmetric
dimethylarginine (ADMA) induces chronic kidney disease through a
mechanism involving collagen and TGF-β1 synthesis. J Pathol.
223:37–45. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shibata R, Ueda S, Yamagishi S, Kaida Y,
Matsumoto Y, Fukami K, Hayashida A, Matsuoka H, Kato S, Kimoto M
and Okuda S: Involvement of asymmetric dimethylarginine (ADMA) in
tubulointerstitial ischaemia in the early phase of diabetic
nephropathy. Nephrol Dial Transplant. 24:1162–1169. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lluch P, Torondel B, Medina P, Segarra G,
Del Olmo JA, Serra MA and Rodrigo JM: Plasma concentrations of
nitric oxide and asymmetric dimethylarginine in human alcoholic
cirrhosis. J Hepatol. 41:55–59. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Feng Y, Zhang D, Zhang Y, Zhang Q and Liu
W: The mechanism of long-term low-dose asymmetric dimethylarginine
inducing transforming growth factor-β expression in endothelial
cells. Int J Mol Med. 31:67–74. 2013. View Article : Google Scholar : PubMed/NCBI
|