Introduction
The branchio-otic (BO) syndrome is characterized by
branchial arch and otic anomalies. It presents heterogeneously,
both clinically and genetically, and manifests with reduced
penetrance and variable expressivity (1). BO syndrome is a rare
autosomal-dominant disorder with a birth prevalence of about
1:40,000 (1). The first identified
causative gene was the human homologue of the Drosophila eyes
absent gene, EYA1 (2).
Vincent et al (3)
demonstrated that BOR (branchio-oto-renal syndrome 1, BOR1; OMIM
#113650) and BOS (branchio-otic syndrome 1, BOS1; OMIM #602588) are
allelic disorders. Subsequently, mutations in the two human
homologues of the Drosophila sine oculis homeobox 1 and 5
genes (SIX1, SIX5) have been detected (4,5). To
date, defects of SIX5 have been exclusively found in
patients who additionally presented with congenital renal
anomalies, whereas SIX1 mutations have been found in
patients with the classic BO phenotype. Evidence for further
genetic heterogeneity of BO syndrome was provided by Kumar et
al, who linked an additional form (BOS2; OMIM %120502) to a
region on chromosome 1q31 (6).
Esophageal atresia (EA) with or without
tracheo-esophageal fistula (TEF) are the most common malformations
of the upper digestive tract. EA/TEF comprises five anatomical
subtypes and these are classified on the basis of the location and
the type of anastomosis that exists between the trachea and the
esophagus (7). The birth
prevalence of EA/TEF has been reported with 1 in 3,000 live births
(8). Approximately 50% of affected
individuals show an isolated phenotype, while the remaining
patients present EA/TEF in combination with other congenital
malformations, e.g., cardiac or renal anomalies (9). Furthermore, EA/TEF have been observed
in over 50 distinct genetic syndromes, associations and sequences
(9). The likely causes of EA/TEF
are heterogeneous and, to date, remain poorly understood. However,
previous study has implicated several developmental genes with
emphasis on effectors of the Sonic Hedgehog (SHH) signaling pathway
[SHH, GLI family zinc finger 1 (GLI1), GLI2, GLI3] in
mouse models (10). In this
context, Motoyama et al (10) found that in
Gli2−/− mice, a reduction of 50% in the gene
dosage of Gli3 in a Gli2−/− background
resulted in EA/TEF and a severe lung phenotype, suggestive of a
possible digenic inheritance model.
In the present study, we investigated a family with
three affected members who all presented with classic BO-associated
symptoms. Interestingly, the index patient also showed the most
common EA/TEF subtype type 3b according to Vogt (7).
Materials and methods
Subjects
Blood samples were collected from all family members
of the index patient and a further 18 patients with EA/TEF and BO
syndrome-associated anomalies, such as hearing loss or malformation
of the ears. Written informed consent was obtained from all
participants or from their proxies in the case of legal minors. The
study was approved by the ethics committee of the Medical Faculty
of the University of Bonn and was conducted in accordance with the
principles of the Declaration of Helsinki.
Whole exome sequencing (WES) and data
analysis
Blood samples were obtained from the family under
study and isolation of genomic DNA from blood was carried out using
a Chemagic Magnetic Separation Module I (Chemagen, Baesweiler,
Germany).
Mutation analysis was performed on our patient by
WES (enrichment kit: Nimble Gene SeqCap ES Human Exome Library 2.0)
with the Genome Analyzer II (Illumina). Read alignment and
detection of variants was done with genome analyzing software
(Varbank; www.varbank.ccg.uni-koeln.de/). In particular, we
filtered for high quality (coverage of more than six reads, a
minimum quality score of 10, VQSLOD greater than −8) and rare
(allele frequency <0.5%) autosomal variants in EAY1,
SIX1 and EAY1-SIX1 pathway-related genes. In order to
exclude pipeline specific artifacts, we also filtered against an
in-house epilepsy cohort (n=511, AF <2%) of variations, which
were created with the same analysis pipeline. The filter conditions
were set to be more sensitive following manual inspections of
aligned reads.
Variation analysis
Variations identified by WES were amplified from
genomic DNA by polymerase chain reaction (PCR) and automated
sequence analysis was carried out using standard procedures. In
brief, primers were directed to all observed variations and the
resultant PCR products were subjected to direct automated BigDye
terminator sequencing (3130XL Genetic Analyzer; Applied Biosystems;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). Both strands
from each amplicon were sequenced and segregation of the variations
in the family was investigated by sequencing the respective PCR
product in all members. Primer sequences for all gene variants
under investigation are available upon request.
Data from the observed allele frequencies harboring
the variants were obtained from the ExAc database (exac.broadinstitute.org). Interpretation of
identified missense variants was carried out with the following
prediction programs: MutPred (www.mutpred1.mutdb.org), Polyphen-2 (www.genetics.bwh.harvard.edu/pph2/),
HumVar (included in Polyphen-2), SIFT (sift.jcvi.org)
and PROVEAN (included in SIFT). The GLI3 splice variant was
analyzed according to Shapiro and Senapathy (11) and Human Splicing Finder 3.0
(12).
Results
Clinical observations
The investigated family has three affected members
who all presented with classic BO-associated symptoms (Table I and Fig. 1). Interestingly, the index patient
(II.3) presented with branchial anomalies (bilateral branchial
cleft fistulas and preauricular pits) and the most common EA/TEF
subtype type 3b according to Vogt (7). The sister (II.2) and the mother (I.2)
of the patient also presented with BO syndrome-associated symptoms
(hearing loss or impairment, ear and neck fistulas). His elder
brother (II.1) had a preauricular tag. The father showed no
anomalies (Fig. 1). To the best of
our knowledge, this is the first report on the concurrence of BO
syndrome and EA/TEF to date.
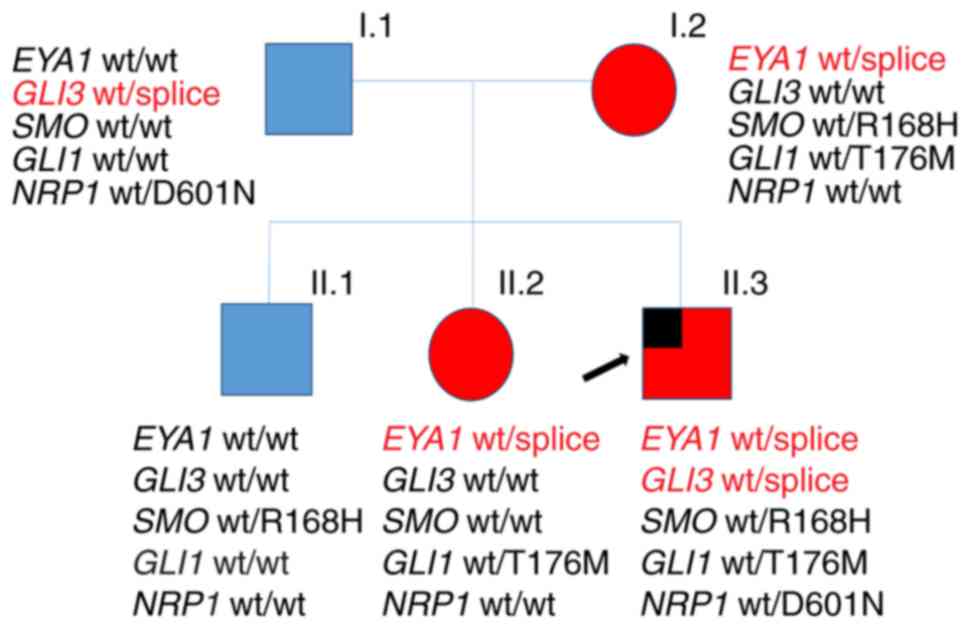 | Figure 1.Pedigree of the family with BO
syndrome. The index patient, also presenting with esophageal
atresia, is marked by an arrow. The presence/absence of gene
variants detected by whole-exome sequencing is indicated. Members
affected with BO are shown in red, while unaffected members are
shown in blue; males and females are indicated by squares and
circles, respectively. BO, branchio-otic; wt, wild type; EYA1,
Drosophila eyes absent; GLI, GLI family zinc finger; SMO,
smoothened, frizzled class receptor; NRP1, neuropilin 1. |
 | Table I.Phenotypic features identified in
patients with the Drosophila eyes absent c.966+5G>A
mutation. |
Table I.
Phenotypic features identified in
patients with the Drosophila eyes absent c.966+5G>A
mutation.
| Author, year | Patient | Hearing loss | Ear anomalies | Branchial
anomalies | Renal
anomalies | Other features | (Refs.) |
|---|
| Kause et
al | I.2 | Unilateral inner
ear (unspecified) | Middle ear | Unilateral fistula
(ear) | – | – | Present study |
| Kause et
al | II.2 | – | – | Bilateral fistula
(ear), unilateral fistula (neck) | – | – | Present study |
| Kause et
al | II.3 | – | – | Bilateral fistula
(ear), bilateral fistula (neck) | – | Esophageal atresia
(Vogt 3b) | Present study |
| Stockley et
al, 2009 | 8 | Mild | – | Not specified
fistula and cyst, bilateral preauricular pit | URA | – | (14)a |
| Stockley et
al, 2009 | 9 | Yes,
unspecified | – | n/a | URA | – | (14)a |
| Stockley et
al, 2009 | 10 | Mild-to-moderate
(mixed) | Cup shaped ears,
posteriorly rotated | Not specified
fistula and cyst, unilateral preauricular pit | URA, VUR | – | (14)a |
| Krug et al,
2011 | 1,291 | – | – | Yes
(unspecified) | – | Bilateral
cataract | (15)a |
| Song et al,
2013 | 7 | Bilateral (mixed,
unspecified) | Cochlear hypoplasia
(bi), dilated vestibule (bi), enlarged vestibular aqueduct (bi);
middle ear: Ossicular anomaly (bi) and deviated facial nerve (bi);
enlarged endolymphatic sac (l) and duct (bi) | – | n/a | – | (16)b |
| Bekheirnia et
al, 2017 | Family 3 | – | – | – | VUR, multicystic
dysplastic kidney | – | (17)a |
WES and segregation of identified
variants
In the context of the index patient reported here,
Eisner et al (13) were
recently able to show that several of the EA/TEF-associated SHH
pathway genes GLI1, GLI2, and GLI3 interact with the BO
syndrome-associated EYA1-SIX1 pathway genes. Hence, we
performed whole-exome sequencing (WES) in the index patient (i) to
identify disease causing variants in EYA1-SIX1 pathway genes
(ii) and to identify variants in EA/TEF-associated SHH pathway
genes (13). Mutation analysis was
performed on our patient with WES and the applied filtering
identified more than 50 variants (data not shown). From these, one
obvious genetic variant explains most of the congenital anomalies
seen in the family. An EYA1 mutation (c.966+5G>A,
according to ENSEMBL transcript ENST00000340726, with the A of the
start methionine as no. 1) was present in the donor splice site of
exon 10 in the index patient. Sanger sequencing confirmed the
mutation in the patient as well as two other affected family
members (sister: II.2, and the mother, I.2; Fig. 1 and Table I). This mutation is known to cause
exon skipping with a premature termination codon in the resultant
mRNA (14).
Our second analysis of the index patient's WES
dataset focused on candidate variants with an allele frequency of
<0.01 in SHH signaling pathway genes with special emphasis on
GLI1, GLI2, GLI3, SUFU, NRP1, NRP2, and
SMO. In this context, we detected additional heterozygous
variants in GLI1 (p.Thr176Met), GLI3 (splice variant
c.1028+3A>G, according to ENSEMBL transcript ENST00000395025,
with the A of the start methionine as no.1), NRP1
(p.Asp601Asn) and SMO (p.Arg168His) (Table II). Apart from the four variants
in GLI1, GLI3, NRP1 and SMO, WES did
not detect any further variations that might be attributable to
EA/TEF in our patient. Sanger sequencing confirmed all four
variants in the index patient (Fig.
1). Two of the four variants in GLI1 and SMO were
transmitted from the EYA1 carrying mother and the other two
variants in GLI3 and NRP1 were transmitted from the
healthy father (Fig. 1). Since the
mother did not present with EA/TEF we excluded the two variants in
GLI1 and SMO as EA/TEF disease causing. As the
variant in NRP1 is located in a region of low conservation
(Table II), it was also excluded.
The remaining GLI3 splice site variation, c.1028+3A>G, is
an extremely rare variant (rs368499795), observed only once in the
ExAc database (n=121,314 alleles). According to Shapiro and
Senapathy (11), the A-to-G
substitution slightly reduces the consensus value (CV) for splice
site recognition from a CV of 0.887 for the wildtype sequence to a
CV of 0.854 for the mutant one. As Human Splicing Finder 3.0
(12) also predicts this variation
as most probably affecting splicing, these data imply that this
GLI3 variant interferes to a certain extent with correct
mRNA processing.
| Table II.Gene-prediction of the exonic
variants with an amino-acid substitution and their conservation
status. |
Table II.
Gene-prediction of the exonic
variants with an amino-acid substitution and their conservation
status.
| Gene | Variation | Substitution | Mm | Dr | Gg | Xt | Polyphen | SIFT | MutPred
(probability of deleterious mutation) | Variation
frequency |
|---|
| GLI1 | c.527C>T | T176M | T | T | T | T | Probably
damaging | Damaging | 0.290 | rs755035040
(5.912×10−5) |
| GLI3 | c.1028+3A>G | – | – | – | – | – | n/a | n/a | n/a | rs368499795
(8.24×10−6) |
| NRP1 | c.1801G>A | D601N | D | – | D | A | Possibly
damaging | Damaging | 0.326 | rs145594886
(4.97×10−5) |
| SMO | c.503G>A | R168H | R | K | K | K | Possibly
damaging | Damaging | 0.524 |
rs61746143 (0.009458) |
To elucidate a more common involvement of EYA1 in
the etiology of EA/TEF, we screened a further 18 patients with
EA/TEF and BO syndrome-associated anomalies, such as hearing loss
or malformation of the ears, for variants in EYA1. Sanger
sequencing revealed 15 intronic and exonic common SNPs (allele
frequencies all >0.08) and three further intronic variants with
no influence on a splice site or a branch point (data not
shown).
Discussion
The initial objective of the present study was to
identify a genetic etiology of BO syndrome and EA/TEF in the index
patient. Initially, WES demonstrated a heterozygous splice mutation
in EYA1. To date, this c.966+5G>A mutation has been
reported in nine other unrelated patients (Table I) (14–17),
where it caused a pleiotropic spectrum of features. Stockley et
al (14) reported the
c.966+5G>A mutation in three BO syndrome patients, who each
presented with the most severe renal phenotype in their cohort.
However, it was associated without branchial and renal anomalies in
a patient reported by Song et al (16), and only with branchial anomalies
and congenital cataract in another patient (15). Most recently, Bekheirnia et
al (17) detected the
c.966+5G>A mutation in a patient solely affected with a renal
phenotype, i.e., vesicoureteral reflux and multicystic dysplastic
kidney. In the patient's family, the EYA1 mutation caused
branchial anomalies in the index patient (II.3) and all other
affected subjects (I.2, II.2) as well as additional unilateral
hearing loss in the mother (I.2). The older brother (II.1) of the
index patient only presented with unilateral preauricular tag, a
common benign congenital malformation of the external ear (18) possibly attributable to BO syndrome.
Consequently, he was negative for the EYA1 mutation. In
conclusion, the detected EYA1 mutation should explain all of
the BO features observed in the index patient and the other family
members.
Our second analysis of the index patient's WES
dataset focused on candidate variants with an allele frequency of
<0.01 in SHH signaling pathway genes. Evaluation of prioritized
genes revealed the presence of an additional potential pathogenic
GLI3 splice variant (c.1028+3A>G) in the index case.
Heterozygous mutations in GLI3 are a most likely cause of
Greig cephalopolysyndactyly syndrome (GCPS; OMIM #175700) and
Pallister-Hall syndrome (PHS; OMIM #146510), both inherited as an
autosomal dominant trait (19,20).
Both disorders manifest polyaxial polydactyly with other
overlapping features. However, neither a literature review nor the
reviews of 174 GCPS/PHS patients, provided by Johnston et al
(19,20), revealed the presence of our
GLI3 splice variant or EA/TEF in these patients. Yet, Yang
et al (21) reported a
de novo missense GLI3 variant (p.M111T) in a patient
with EA with hemivertebrae, resembling the phenotypic spectrum in
murine models as reported by Motoyama et al (10).
Human Splicing Finder 3.0 (12), predicted the consequence of the
c.1028+3A>G variant as most probably affecting splicing.
However, according to Shapiro and Senapathy (11), the A-to-G substitution only
slightly reduces the CV for splice site recognition, suggesting
formation of a relevant amount of normally spliced mRNA, thereby
avoiding GLI3 functional haploinsufficiency. This would explain the
absence of typical phenotypic features caused by autosomal dominant
GLI3 mutations, as observed in patients with Pallister-Hall
syndrome, Greig cephalopolysyndactyly syndrome or different forms
of polydactyly (22). However, a
small decrease in the formation of correct GLI3 transcripts
may interfere with the fine-tuning of the Eya1-Six1-SHH
pathway. In mutant mice lungs, Lu et al (23) have shown that Six1 and
Eya1 act together to regulate SHH/Gli3 signaling
activity. Lewandowski and coworkers reported that more than 40 GLI
target genes in the mammalian limb bud are predominantly regulated
by GLI3, but show a different spatiotemporal requirement for
SHH signaling (24). Moreover, it
has been reported that in murine peri-cloacal mesenchyme,
Six1 and Eya1 functionally interact with the SHH
pathway and that both these transcripts are down regulated in SHH
mutants (25). Based on these
observations, and since segregation analysis revealed the
inheritance of the GLI3 splice variant from the unaffected
father, one may speculate about a digenic inheritance model
involving EYA1 and GLI3, where the effect of the
GLI3 variant emerges only in the background of the
EYA1 defect.
However, the recent work of Eisner et al
(13), who described Eya1
and Six1 as key components of the Shh transcriptional
network with Eya1 and Six1 as co-regulators of
Gli transcriptional activators during normal organ
development, and several other findings are suggestive of a direct
involvement of EYA1/Eya1 in esophageal development in
vertebrates. In mice, Eya1 has been shown to play a critical
role in epithelial, mesenchymal and vascular morphogenesis of the
embryonic lung as an upstream coordinator of SHH fibroblast growth
factor 10 (Fgf10) signaling (26).
It has been shown that the foregut epithelium gives rise to the
esophagus, trachea, lungs, thyroid, stomach, liver, pancreas, and
hepatobiliary system and there is experimental evidence that they
are derived from a common progenitor cell population in the ventral
foregut (27). Hence, in case of
EYA1 haploinsufficiency, impairment of this SHH-FGF10
cascade might also interfere with correct esophageal development.
In zebra fish, requirement of Shh and Fgf10 for esophageal
morphogenesis has been reported (28) and similarly, disruption of the
Fgf10 gene during the critical period of separation of the
trachea and esophagus caused tracheo-esophageal malformations in a
mouse model (29). Moreover, it
has been shown in mice that the Shh-Fgf10 cascade controls the
patterning of the tracheal cartilage rings (30), and that defective Shh and Fgf
signaling plays a role in the pathogenesis of EA/TEF (31). Here, the coexistence of the
EYA1 mutation and the additional variant in trans in
GLI3 of our patient is suggestive of a possible digenic mode
of inheritance and might explain the co-occurrence of BO syndrome
and EA/TEF in our patient. Screening of 18 EA/TEF patients with BO
syndrome-associated phenotypic features did not reveal any
additional EYA1 mutation. While investigations of larger
EA/TEF cohorts with BO syndrome-associated phenotypic features are
warranted, our present approach to elucidate the coincidence of BO
syndrome and EA/TEF in the index patient did not imply trio-based
WES analysis. Hence, we cannot exclude any other possibly disease
causing de novo mutations as the cause of EA/TEF in our
patient.
Acknowledgements
We thank the family for their invaluable help. Pia
Uerdingen is acknowledged for excellent assistance. F.K. was
supported by the BONFOR program of the University of Bonn (grant
no. O-149.0096). H.R. was supported by a grant from the Else
Kröner-Fresenius-Stiftung (EKFS; grant no. 2014_A14) and by two
grants from the German Research Foundation (Deutsche
Forschungsgemeinschaft, DFG; grant nos. RE 1723/1-1 and RE
1723/2-1). M.L was supported by a grant from the German Research
Foundation (DFG; grant no. LU 731/3-1).
References
|
1
|
Fraser FC, Sproude JR and Halal F:
Frequency of the branchio-oto-renal (BOR) syndrome in children with
profound hearing loss. Am J Med Genet. 7:341–349. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abdelhak S, Kalatzis V, Heilig R, Compain
S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M, et
al: A human homologue of the Drosophila eyes absent gene underlies
Branchio-Oto-Renal (BOR) syndrome and identifies a novel gene
family. Nat Genet. 15:157–164. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vincent C, Kalatzis V, Abdelhak S, Chaib
H, Compain S, Helias J, Vaneecloo FM and Petit C: BOR and BO
syndromes are allelic defects of EYA1. Eur J Hum Genet. 5:242–246.
1997.PubMed/NCBI
|
|
4
|
Ruf RG, Xu PX, Silvius D, Otto EA,
Beekmann F, Muerb UT, Kumar S, Neuhaus TJ, Kemper MJ, Raymond RM
Jr, et al: SIX1 mutations cause branchio-oto-renal syndrome by
disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci USA.
101:pp. 8090–8095. 2004; View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hoskins BE, Kramer CH, Silvius D, Zou D,
Raymond RM, Orten DJ, Kimberling WJ, Smith RJ, Weil D, Petit C, et
al: Transcription factor SIX5 is mutated in patients with
branchio-oto-renal syndrome. Am J Hum Genet. 80:800–804. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kumar S, Deffenbacher K, Marres HA,
Cremers CW and Kimberling WJ: Genomewide search and genetic
localization of a second gene associated with autosomal dominant
branchio-oto-renal syndrome: Clinical and genetic implications. Am
J Hum Genet. 66:1715–1720. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vogt EC: Congenital esophageal atresia. Am
J Roentgenol. 22:463–465. 1929.
|
|
8
|
Depaepe A, Dolk H and Lechat MF: The
epidemiology of tracheo-esophageal fistula and oesophageal atresia
in Europe. EUROCAT Working Group. Arch Dis Child. 68:743–748. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
De Jong EM, Douben H, Eussen BH, Felix JF,
Wessels MW, Poddighe PJ, Nikkels PG, de Krijer RR, Tibboel D and de
Klein A: 5q11.2 deletion in a patient with tracheal agenesis. Eur J
Hum Genet. 18:1265–1268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Motoyama J, Liu J, Mo R, Ding Q, Post M
and Hui CC: Essential function of Gli2 and Gli3 in the formation of
lung, trachea and oesophagus. Nat Genet. 20:54–57. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shapiro MB and Senapathy P: RNA splice
junctions of different classes of eukaryotes: Sequence statistics
and functional implications in gene expression. Nucleic Acids Res.
15:7155–7174. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Desmet FO, Hamroun D, Lalande M,
Collod-Béroud G, Claustres M and Béroud C: Human splicing finder:
An online bioinformatics tool to predict splicing signals. Nucleic
Acids Res. 37:e672009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eisner A, Pazyra-Murphy MF, Durresi E,
Zhou P, Zhao X, Chadwick EC, Xu PX, Hillman RT, Scott MP, Greenberg
ME and Segal RA: The eya1 phosphatase promotes shh signaling during
hindbrain development and oncogenesis. Dev Cell. 33:22–35. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stockley TL, Mendoza-Londono R, Propst EJ,
Sodhi S, Dupuis L and Papsin BC: A recurrent EYA1 mutation causing
alternative RNA splicing in branchio-oto-renal syndrome:
Implications for molecular diagnostics and disease mechanism. Am J
Med Genet A. 149A:1–327. 2009. View Article : Google Scholar
|
|
15
|
Krug P, Moriniére V, Marlin S, Koubi V,
Gabriel HD, Colin E, Bonneau D, Salomon R, Antignac C and Heidet L:
Mutation screening of the EYA1, SIX1 and SIX5 genes in a large
cohort of patients harboring branchio-oto-renal syndrome calls into
question the pathogenic role of SIX5 mutations. Hum Mutat.
32:183–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song MH, Kwon TJ, Kim HR, Jeon JH, Baek
JI, Lee WS, Kim UK and Choi JY: Mutational analysis of EYA1, SIX1
and SIX5 genes and strategies for management of hearing loss in
patients with BOR/BO syndrome. PLoS One. 8:e672362013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bekheirnia MR, Bekheirnia N, Bainbridge
MN, Gu S, Coban Akdemir ZH, Gambin T, Janzen NK, Jhangiani SN,
Muzny DM, Michael M, et al: Whole-exome sequencing in the molecular
diagnosis of individuals with congenital anomalies of the kidney
and urinary tract and identification of a new causative gene. Genet
Med. 19:412–420. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Firat Y, Şireci Ş, Yakinci C, Akarçay M,
Karakaş HM, Firat AK, Kizilay A and Selimoğlu E: Isolated
preauricular pits and tags: Is it necessary to investigate renal
abnormalities and hearing impairment? Eur Arch Otorhinolaryngol.
265:1057–1060. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Johnston JJ, Olivos-Glander I, Killoran C,
Elson E, Turner JT, Peters KF, Abbott MH, Aughton DJ, Aylsworth AS,
Bamshad MJ, et al: Molecular and clinical analyses of greig
cephalopolysyndactyly and pallister-hall syndromes: Robust
phenotype prediction from the type and position of GLI3 mutations.
Am J Hum Genet. 76:609–622. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Johnston JJ, Sapp JC, Turner JT, Amor D,
Aftimos S, Aleck KA, Bocian M, Bodurtha JN, Cox GF, Curry CJ, et
al: Molecular analysis expands the spectrum of phenotypes
associated with GLI3 mutations. Hum Mutat. 31:1142–1154. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang L, Shen C, Mei M, Zhan G, Zhao Y,
Wang H, Huang G, Qiu Z, Lu W and Zhou W: De novo GLI3 mutation in
esophageal atresia: Reproducing the phenotypic spectrum of Gli3
defects in murine models. Biochim Biophys Acta. 1842:1755–1761.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Al-Qattan MM, Shamseldin HE, Salih MA and
Alkuraya FS: GLI3-related polydactyly: A review. Clin Genet. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu K, Reddy R, Berika M, Warburton D and
El-Hashash AH: Abrogation of Eya1/Six1 disrupts the saccular phase
of lung morphogenesis and causes remodeling. Dev Biol. 382:110–123.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lewandowski JP, Du F, Zhang S, Powell MB,
Falkenstein KN, Ji H and Vokes SA: Spatiotemporal regulation of GLI
target genes in the mammalian limb bud. Dev Biol. 406:92–103. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang C, Gargollo P, Guo C, Tang T, Mingrin
G, Sun Y and Li X: Six1 and Eya1 are critical regulators of
peri-cloacal mesenchyme progenitors during genitourinary tract
development. Dev Biol. 360:186–194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
El-Hashash AH, Al Alam D, Turcatel G,
Bellusci S and Warburton D: Eyes absent 1 (Eya1) is a critical
coordinator of epithelial, mesenchymal and vascular morphogenesis
in the mammalian lung. Dev Biol. 350:112–126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zaret KS: Genetic programming of liver and
pancreas progenitors: Lessons for stem-cell differentiation. Nat
Rev Genet. 9:329–340. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Korzh S, Winata CL, Zheng W, Yang S, Yin
A, Ingham P, Korzh V and Gong Z: The interaction of epithelial Ihha
and mesenchymal Fgf10 in zebrafish esophageal and swimbladder
development. Dev Biol. 359:262–276. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hajduk P, Murphy P and Puri P: Fgf10 gene
expression is delayed in the embryonic lung mesenchyme in the
adriamycin mouse model. Pediatr Surg Int. 26:23–27. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sala FG, Del Moral PM, Tiozzo C, Al Alam
D, Warburton D, Grikscheit T, Veltmaat JM and Bellusci S: FGF10
controls the patterning of the tracheal cartilage rings via Shh.
Development. 138:273–282. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Spilde T, Bhatia A, Ostlie D, Marosky J,
Holcomb G III, Snyder C and Gittes G: A role for sonic hedgehog
signaling in the pathogenesis of human tracheoesophageal fistula. J
Pediatr Surg. 38:465–468. 2003. View Article : Google Scholar : PubMed/NCBI
|














