Introduction
Pulmonary hypertension (PH) is a chronic progressive
disease characterized by increased pulmonary vascular resistance
and pulmonary vasculature remodeling. Although PH has a high
incidence of mortality (1), the
exact etiology and pathogenesis remain to be elucidated. There is
increasing evidence that implicates abnormal endothelial cells
(ECs) apoptosis and ECs dysfunction to be involved in the
initiation and development of PH (2,3). ECs
isolated from the idiopathic pulmonary arterial hypertension
patients had disordered growth features (4). However, enhanced EC growth and
survival, associated with reduced EC apoptosis may prevent the
development of PH induced by monocrotaline (5,6).
Additionally, dysfunction of ECs may be a consistent marker of PH
in rodents and humans (4). In
hypoxic PH, the dysfunction of ECs was believed to lead to the
reduction in the endothelium-derived nitric oxide (NO) production
(7).
NO, working as a potent vasodilator has become an
important therapeutic target for hypoxic PH. Endogenous NO is
produced by nitric oxide synthase (NOS), which has been efficiently
phosphorylated by serine/threonine-specific protein kinase (Akt)
activation (8,9). However, the expression of endothelial
NOS (eNOS) is reduced (10),
whilst the production of endogenous NOS inhibitors, such as
asymmetrical dimethylarginine (ADMA) and symmetrical
dimethylarginine (SDMA) is enhanced in PH (11). These findings indicated that
protecting EC from abnormal apoptosis and normalization of the
dysregulated NO-mediated signal in ECs may be potential therapeutic
strategies in patients with hypoxic PH.
Asiaticoside (AS) is a saponin monomer extracted
from a medicinal plant termed Centella asiatica. It has been
previously documented to have multiple biological effects, such as
antioxidant (12),
anti-inflammatory (13),
anti-hepatofibrotic (14) and
acting as a neuroprotector against transient cerebral ischemia and
reperfusion (15). However,
further investigation that highlights its protective effects on ECs
is required. Our previous study revealed rudimentary understanding,
that AS may prevent the development of PH by attenuating pulmonary
cardiovascular remodeling in hypoxia-induced PAH rats, which may be
mediated by blocking the hypoxia-induced over activity of
transforming growth factor (TGF)-β1/SMAD family member (SMAD) 2/3
signaling (16). However, whether
AS may attenuate established hypoxic PH and its effects on the
vitality and function of ECs remains to be determined.
Therefore, the present study compared the effect of
AS on hypoxic PH rats with different treatment strategies.
Subsequently, the effect of AS on EC function was examined by
evaluation of NO production and AKT/eNOS activation in vivo
and in vitro and investigated its effects on EC survival and
apoptosis in vitro. The present study indicated that AS may
prevent the development of hypoxic PH and attenuate established
hypoxic PH, which may be primarily due to the enhanced NO-mediated
signal and the reduced apoptosis of EC under hypoxia.
Materials and methods
Animal experimental protocols
Animal experiments were approved by the
Institutional Animal Ethics Committee for Experimentation on
Animals of Wenzhou Medical University (Wenzhou, China) and followed
the National Institutes of Health Guide for the Care and Use of
Laboratory Animals. A total of 40 Adult male Sprague-Dawley rats
with a body weight of 180–200 g (SLAC Laboratory Animal Co., Ltd.,
Shanghai, China) were used in experiments and they were kept at
25°C, 21% O2, a 12 h/12 h light/dark cycle and free
access to food and water. The rats were then separated randomly
into four groups, and each group contained 10 rats: i) Control rats
raised in normoxia for 4 weeks (Nox); ii) control rats raised in
hypoxia for 4 weeks (Hx); iii) rats raised in hypoxia and received
AS (diluted with normal saline; Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany) from the same day for 4 weeks (HP); and iv)
rats raised in hypoxia for 2 weeks, then received the 4-week AS
treatment, i.e., continued another 2 weeks' treatment after
finishing the 4-week hypoxia exposure (HT). Hypoxia-induced PH was
developed by keeping rats in a sealed, but ventilated hypoxic
chamber (9% O2, YPC-160D; Changsha Huaxiao Electronic
Technology Co., Ltd., Changsha China) as previously described
(17). The treatment groups, HP
and HT group received AS (50 mg/kg), while Nox and Hx groups
received vehicle (normal saline, 1.5–2 ml) administration as
control. AS and vehicle were administered daily through
intragastric administration.
Examination of mean pulmonary artery
pressure and right ventricular hypertrophy
After treatment, rats were weighed and received
sodium pentobarbital (Sigma-Aldrich; Merck Millipore)
anesthetization by intraperitoneal injection (35 mg/kg). Invasive
hemodynamic measurements, including mean pulmonary arterial
pressure (mPAP) and mean carotid arterial pressure (mCAP) were
examined as described previously (18). Following the measurement, the right
ventricular (RV) wall was separated from the left ventricular (LV)
wall, and the interventricular septum (S) and weighed. An index of
right ventricular hypertrophy (RVH) was calculated by right
ventricle to left ventricle plus septum ratio.
Morphometric analysis of pulmonary
artery
Hematoxylin and eosin (H&E) staining
(hematoxylin for 5 min and eosin for 30 sec at 25°C) was performed
and the images (×400 magnification) of the lung tissue (5-µm-thick
sections) and pulmonary arterioles were captured with a microscope
digital camera (BX51 microscope; Olympus Corporation, Tokyo,
Japan). The percent of medial wall thickness was determined by
Image-Pro Plus 6.0 as previously described (19). A small part of flesh lung tissue
was maintained in glutaraldehyde. The 3-µm-thick sections of each
sample were fixed first with 25% glutaraldehyde 2 h at 4°C, then
incubated with 1% osmic acid 1 h at 25°C, stained with uranyl
acetate for 1 h and lead citrate for 1 h at 25°C, embedded with
ethoxyline resin 24 h at 60°C, and then observed under transmission
electron microscope detection (H-600 transmission electron
microscope; Hitachi, Ltd., Tokyo, Japan).
Measurement of endothelin (ET)-1,
prostacyclin (PGI2), cyclic guanosinc monophosphate (cGMP) and NO
in vivo
The 1 ml blood samples were taken from each rat's
heart after hemodynamic measurements to obtain serum at a volume of
150 ml, which was centrifuged at 200 × g, 8 min at 25°C. The levels
of ET-1 in the serum of rats were determined by enzyme-linked
immunosorbent assay (ELISA) kit (cat. no. ABP52878; Abcam,
Cambridge, UK). PGI2 concentration was also tested by ELISA kit
(cat. no. MBS266717; MyBioSource, Inc., San Diego, CA, USA). cGMP
levels in lung tissue was quantified by the cGMP Direct Immunoassay
kit (cat. no. 581022-96; Abcam). The level of NO measured by
quantitating total nitrate/nitrite using nitric oxide colorimetric
assay kit (cat. no. K205-100; BioVision, Inc., Milpitas, CA, USA).
All the procedures were completed following to the manufacturer's
instructions. Data were quantified using a standard curve of known
concentrations. Each sample was evaluated in triplicate.
Cell treatment and NO production of
HPAECs
According to the results of cell viability, HPAECs
were divided into the following four groups: i) Nox, cells were
cultured under normoxia (21% O2, 5% CO2); ii)
H0, cells were cultured under hypoxia (5% O2, 5%
CO2); iii) H50, cells were cultured with AS (50 µg/ml)
under hypoxia; iv) HL, cells were cultured with AS (50 µg/ml) and
LY294002 (20 µmol/l) under hypoxia, 3 wells were used per group.
All cells were cultured at 37°C for 24 h. The production of NO in
HPAECs was quantified by the same colorimetric assay used for the
detection of NO levels in rat serum as aforementioned.
Apoptosis detection
After treatment, the cells were detected by the
terminal deoxynucleotidyl-transferase-mediated dUTP nick end
labeling (TUNEL) assay kit (Roche Applied Science, Penzberg,
Germany). A total of four groups were fixed of air-dried cell
samples with a freshly prepared fixation solution for 1 h at 25°C,
then incubated with blocking solution and permeabilization solution
sequentially for 10 min and 2 min at 25°C respectively after
rinsing slides with PBS in between. The TUNEL reaction mixture was
prepared and 50 µl was added on sample and incubate for 60 min at
37°C. Samples at last analyzed in a drop of PBS under a
fluorescence microscope (CX21FS1; Olympus) with the wavelength of
550 nm. TUNEL-positive cells were calculated as they represent
apoptosis cells. For further confirmation, the activity of
caspase-3 was quantified using a Caspase-3/CPP32 Colorimetric Assay
kit (BioVision, Inc.). Caspase-3 is a key in apoptosis, as its
activity reflects the intensity of apoptosis. Cytosolic extracts
were incubated in 96-well plates at 37°C for 1–2 h with DEVD-pNA
substrate (200 µM final concentration). Absorbance was detected in
a microtiter plate reader at 400 nm. Results were calculated by the
equation obtained from a standard curve.
Western blot analysis
The protein expression of Akt, phosphorylated
(p)-Akt at Ser473, eNOS, and p-eNOS at
Ser1177 in lung tissue of rats was detected by western
blot analysis. Frozen lung tissue was prepared and homogenized in
lysis buffer (Beyotime Institute of Biotechnology, Haimen, China),
sonicated twice and then centrifuged for 20 min at 10,000 × g. The
expression of these proteins in HPAECs was also quantified 24 h
after the aforementioned treatments. Cell proteins were isolated
from HPAECs by centrifuging at 4°C for 5 min at 12,000 × g and
using lysis buffer (Beyotime Institute of Biotechnology). To
determine the protein concentration of the lysate, the Bradford
method was used with bovine serum albumin (ScienCell Research
Laboratories, Inc., San Diego, CA, USA) as the standard. Proteins
from each sample with equal amounts (25 µg) were resolved by
SDS-PAGE (12% separation gel) and then transferred onto
polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA,
USA). Western blots were blocked by incubating with PBS containing
5% skimmed milk for 1 h at room temperature. The membrane was then
incubated with the primary antibodies overnight at 4°C. The primary
antibodies used were as follows: Anti-eNOS (cat. no. 5880, 1:1,000;
Cell Signaling Technology, Inc., Danvers, MA, USA), anti-p-eNOS
(cat. no. 9571, 1:1,000; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA), anti-Akt (cat. no. 2920, 1:1,000), anti-p-Akt (cat. no.
4051, 1:1,000) (both from Cell Signaling Technology, Inc.), and
anti-GAPDH (cat. no. G8795, 1:1,000; Sigma-Aldrich; Merck
Millipore). Subsequently, the blots were incubated with peroxidase
conjugated goat anti-rabbit secondary antibody (cat. no. sc-2004,
1:2,000; Santa Cruz Biotechnology, Inc.) for 1 h. Subsequently,
peroxidase labeling was visualized via enhanced chemiluminescence
reagent provided by Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). By scanning the X-ray film (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), densitometry results were analyzed using
Image-Pro Plus 6.0 (Media Cybernetics, Inc., Rockville, MD, USA)
software (National Institutes of Health, Bethesda, MD, USA). All
experiments were repeated at least three times.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Between-group mean comparisons were performed using one-way
analysis of variance followed by a Student-Newman-Keuls test using
R version 3.3.2. P<0.05 were considered to indicate a
statistically significant difference.
Results
AS inhibits the development of hypoxic
PH, cardiovascular remodeling and endothelial cell injury in
hypoxic PH
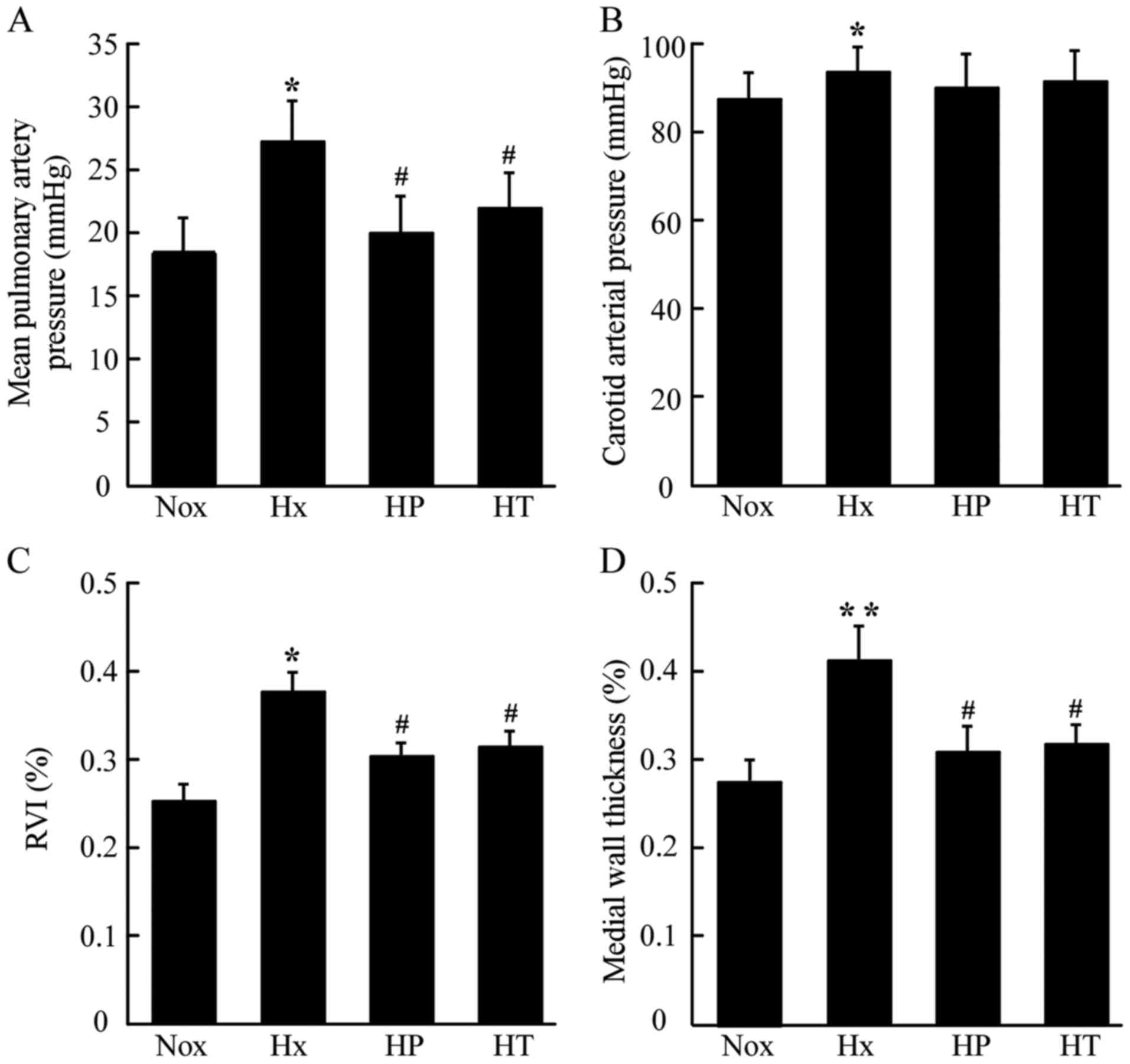
As presented in Fig.
1A, mPAP in Hx group increased compared with that in the Nox
group. AS administered at 50 mg/kg daily for 4 weeks in prevention
and treatment groups (HP group and HT group, respectively)
inhibited the elevation of mPAP induced by hypoxia. There was no
significant difference between the HP and HT groups. AS seemed to
have no impact on the systemic blood pressure, as carotid arterial
pressure (CAP), although there was an increased level of CAP in Hx
group compared with the Nox group. (Fig. 1B). After exposure to hypoxia, rats
exhibited more severe right ventricular hypertrophy
(RVI=RV/LV+Sep), which was relieved in both the HP and HT groups
(Fig. 1C). Analyses of medial wall
thickness of pulmonary arterioles revealed that the Hx group had
much higher medial wall thickness than the Nox group, whereas the
HT groups showed reduced higher medial wall thickness than Hx
(Fig. 1D).
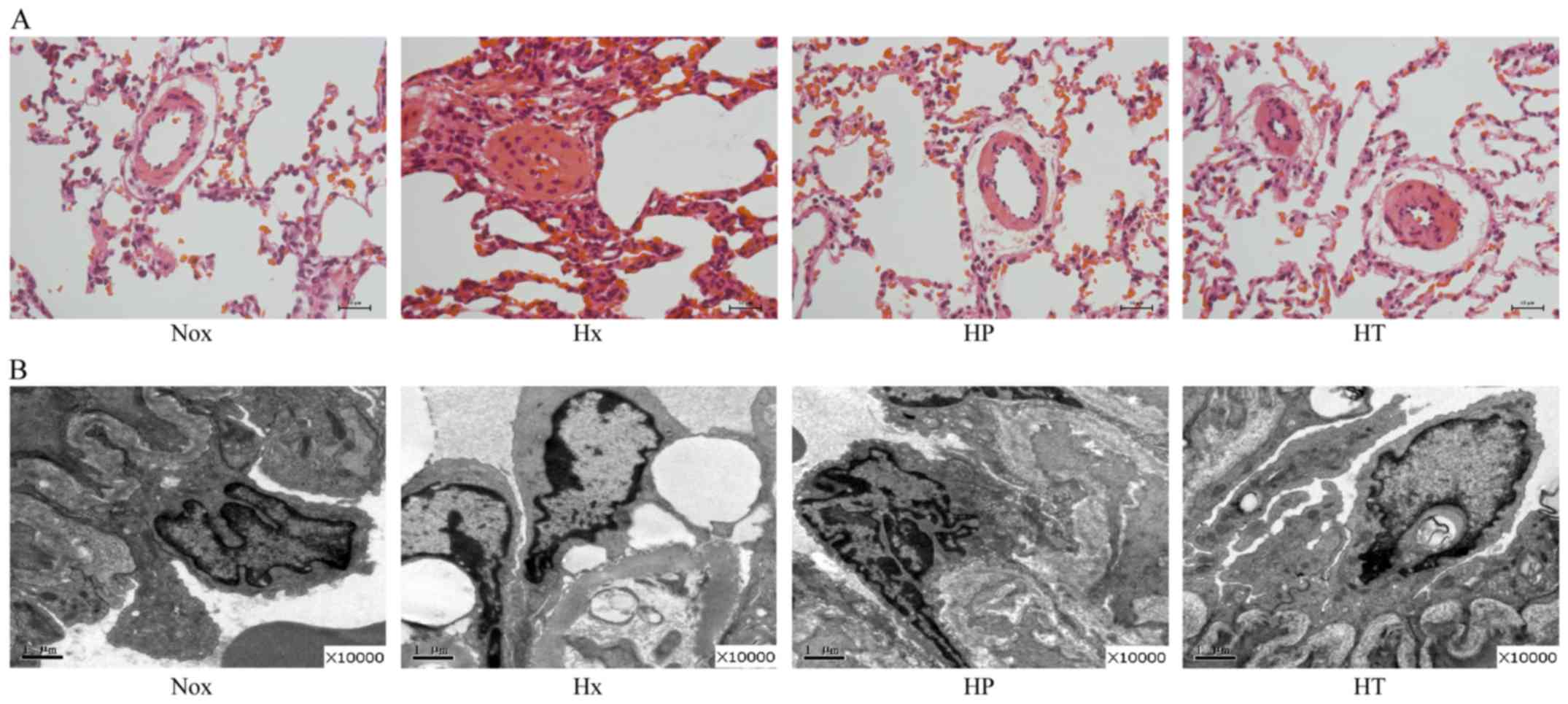
Compared with the Nox group, the lumina of pulmonary
arterioles in Hx group was reduced, with inflammatory cells were
located along vascular walls (Fig.
2A). These abnormalities were partly ameliorated in the HP and
HT groups, with the HP group that started treatment at beginning of
hypoxia exposure having reduced pulmonary arteriole wall thickening
and inflammatory cell infiltration. At ultrastructure level
(Fig. 2B), the pulmonary arteries
of the Hx group compared with Nox group showed swelling of
endothelial, edema of mitochondria, increased vacuoles, destructive
cell junction and exfoliation of basement membrane. Additionally,
clustered collagen fibers deposited in adventitia pulmonary
arterial walls were observed. Conversely, reduced morphological
abnormality of endothelial cells was identified in the HP and HT
groups.
AS modulates dysregulation of ET-1,
PGI2, cGMP and NO in hypoxic PH
The circulating concentration of ET-1 was markedly
elevated in the Hx group compared with the Nox group, whereas it
was lower in the HP and HT groups compared with the in Hx group
(Fig. 3A). The serum level of NO
which was represented by nitric products was reduced in the Hx
group compared with the Nox group. The level of NO was restored in
HP group, whereas no significant difference was identified in the
HT group (Fig. 3B). The serum
levels of PGI2 showed no significant differences among these groups
(Fig. 3C). The cGMP concentrations
in lung tissue demonstrated similar pattern as the NO levels in the
serum. The HP group had higher cGMP concentration compared with the
Hx group, which was significantly reduced compared with Nox group
(Fig. 3D). However, no significant
difference was identified between the Hx and HT group.
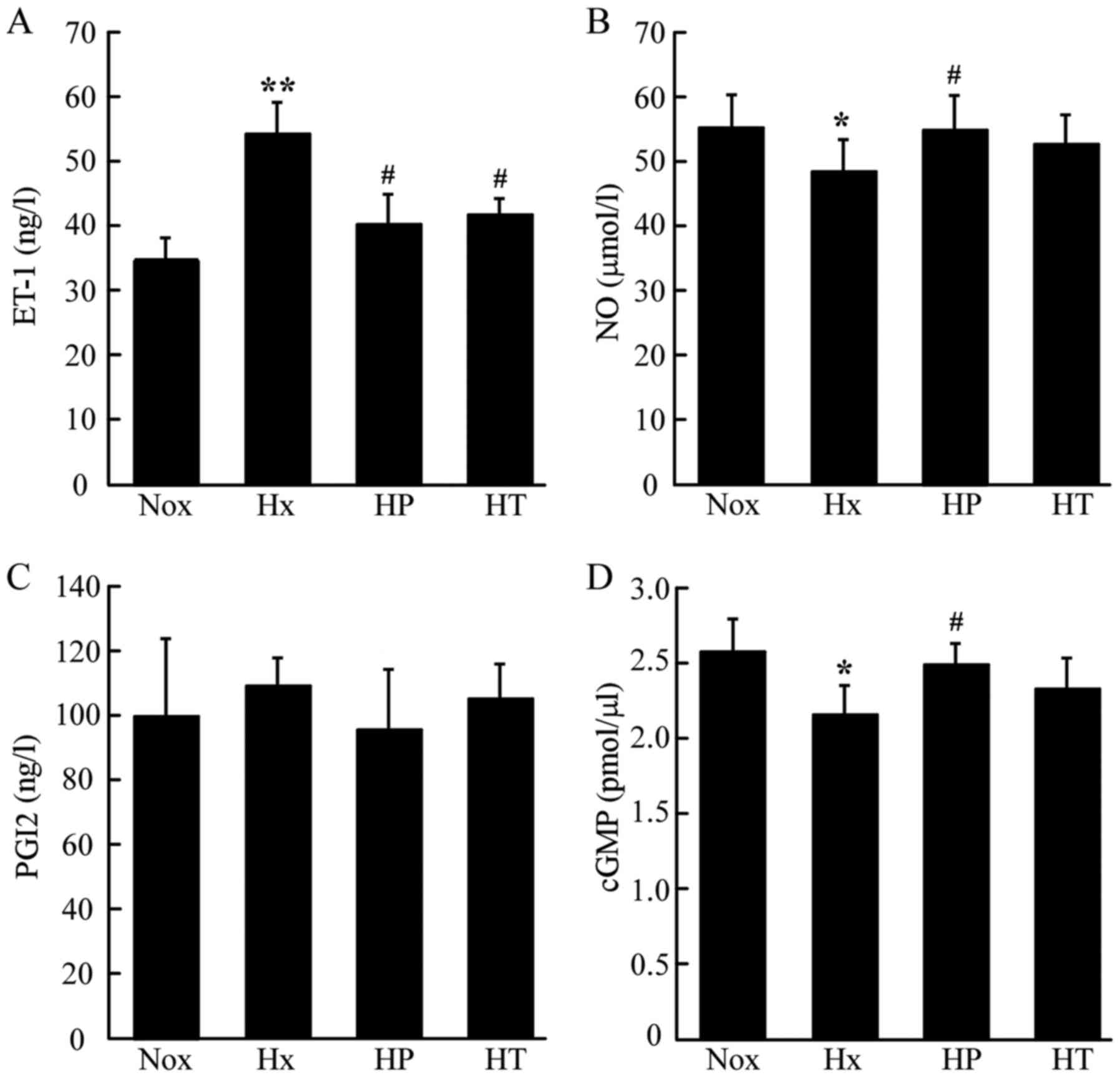 | Figure 3.Effect of AS on concentrations of
vascular activators in rats tested by ELISA kits. (A)
Concentrations of (A) ET-1 (B) NO (C) PGI2 in serum and (D) cGMP in
lung tissue. *P<0.05, **P<0.01 vs. Nox group;
#P<0.05 vs. Hx group, n=10. AS, asiaticoside; ET-1,
endothelin-1, NO, nitric oxide; PGI2, prostacyclin; cGMP, cyclic
guanosinc monophosphate; Nox, control rats raised in normoxia for 4
weeks; Hx, control rats raised in hypoxia for 4 weeks; HP, rats
raised in hypoxia and received AS from the same day for 4 weeks;
HT, rats raised in hypoxia for 2 weeks, then received the 4-week AS
treatment. |
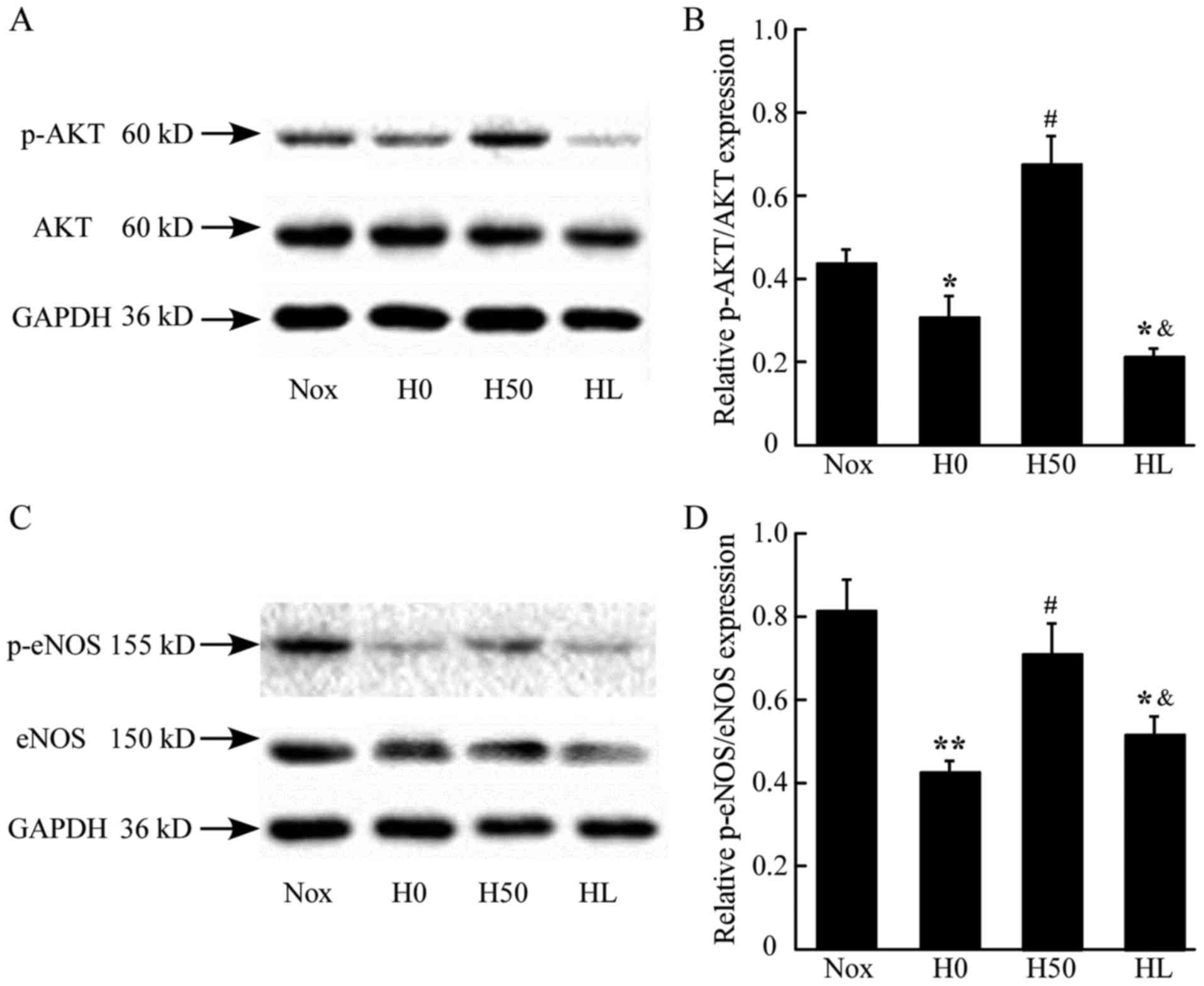
AS upregulates activation of Akt and
eNOS in lung tissue
To examine the effect of AS on the activation of Akt
and eNOS in vivo, the present study assessed the expression
of Akt and eNOS, and their phosphorylated products, p-Akt at
Ser473 and p-eNOS at Ser1177 in lung tissues.
The relative ratio of p-Akt/AKT and p-eNOS/eNOS represent the
activation of the key proteins in NO-mediated signaling. The
phosphorylation of AKT was elevated in the Hx, HP and HT groups.
The HP and HT groups had higher levels of activation of AKT
compared with the Nox group (Fig. 4A
and B). The phosphorylation of eNOS decreased significantly in
the Hx group compared with the Nox group. The HP and HT groups were
increased in the phosphorylation of eNOS compared with Hx group,
whereas the HP group had a higher phosphorylation level compared
with the HT group (Fig. 4C and D),
which indicated that early treatment of AS contributes to higher
phosphorylation levels in hypoxia.
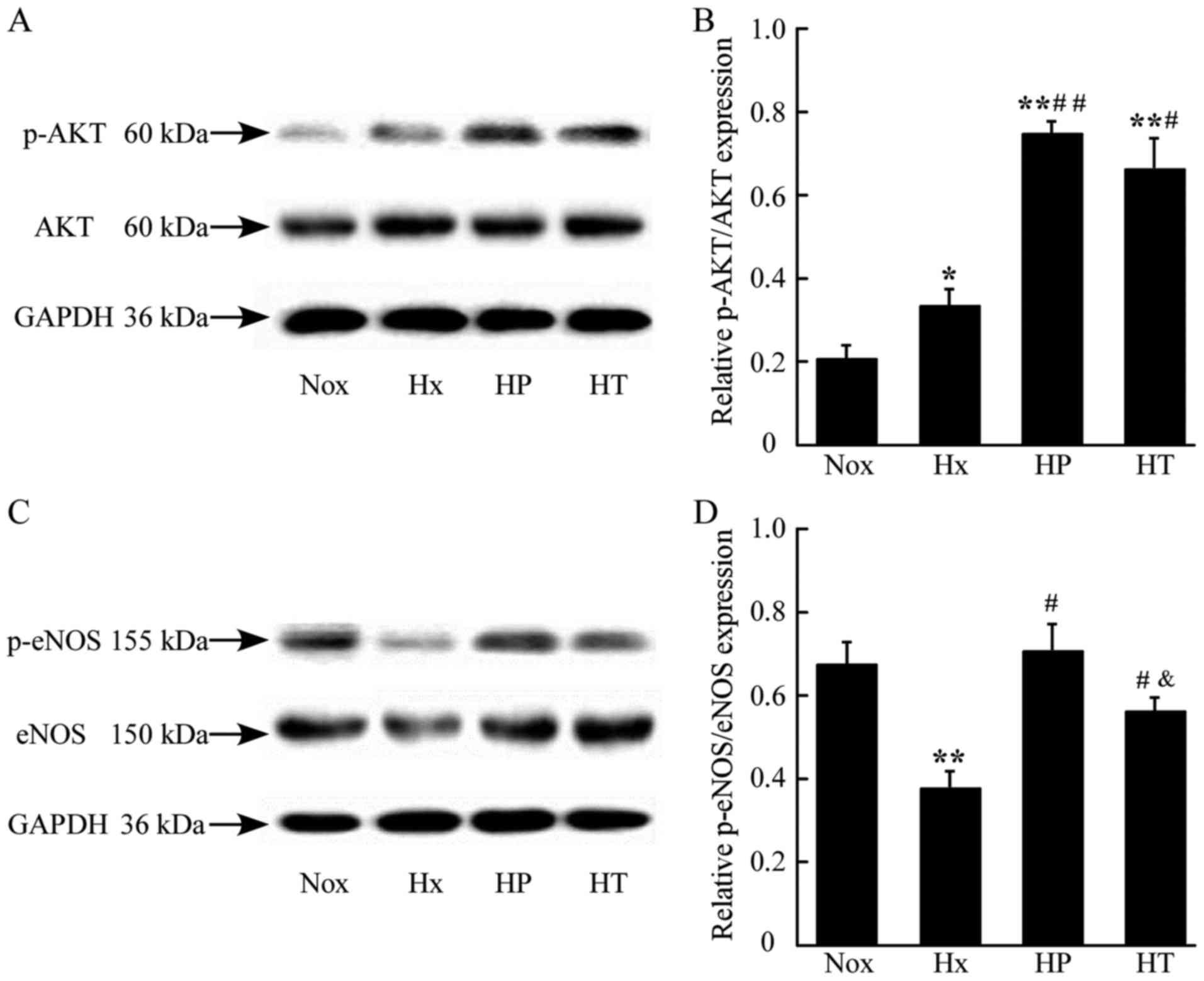 | Figure 4.Effect of AS on the activation of eNOS
and Akt in lungs of rats assessed by western blotting. (A)
Demonstrative immunoblot of Akt and p-Akt in rat lung tissue. (B)
Densitometric analysis of expression of p-Akt/Akt ratio. (C)
Demonstrative immunoblot of eNOS and p-eNOS in rat lung tissue. (D)
Densitometric analysis of expression of p-eNOS/eNOS ratio.
*P<0.05, **P<0.01 vs. Nox group; #P<0.05,
##P<0.01 vs. Hx group; &P<0.05 vs.
HP group, n=3. AS, asiaticoside; Akt, serine/threonine-specific
protein kinase; p-Akt, phosphorylated-Akt; eNOS, endothelial nitric
oxide synthase; Nox, control rats raised in normoxia for 4 weeks;
Hx, control rats raised in hypoxia for 4 weeks; HP, rats raised in
hypoxia and received AS from the same day for 4 weeks; HT, rats
raised in hypoxia for 2 weeks, then received the 4-week AS
treatment. |
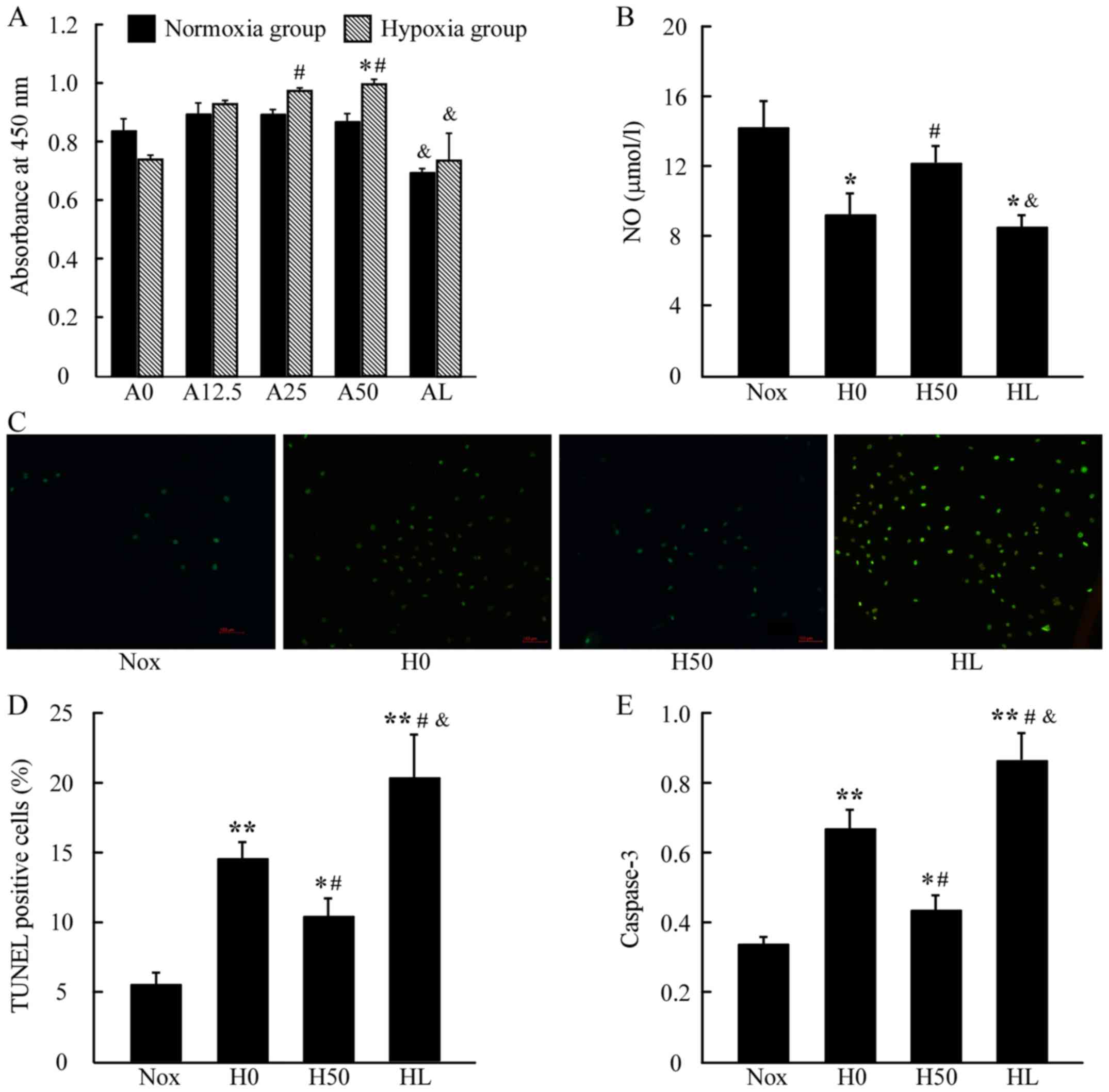
AS prevents endothelial cells from
hypoxia-induced inhibition of cell viability and NO production
To evaluate whether AS protects HPAECs from
hypoxia-induced damage in vitro, cell viability was
quantified using CCK-8 tests (Fig.
5A). A significant increase in absorbance was observed upon
treatment with AS under hypoxia exposure in a dose-dependent manner
between 12.5 and 50, and 25 and 50 µg/ml of AS (H25 and H50)
compared with control in hypoxia (H0), and their counterparts in
normoxia has no significant difference; however, AS (50 µg/ml)
together with LY294002 (20 µmol/l) significantly suppressed the
cell viability either under normoxia or hypoxia condition. As
presented in Fig. 5B, NO secretion
was reduced in the H0 group compared with the Nox group, whereas it
was significantly increased in the H50 group compared with the H0
group. Nevertheless, there was a reduced NO secretion in the HL
group compared with the Nox or H50 groups.
AS protects endothelial cells from
hypoxia-induced apoptosis
A TUNEL assay was used to determine whether AS
protects HPAECs by blocking hypoxia-induced apoptosis (Fig. 5C and D). As illustrated, the H0
group exhibited obvious signs of apoptosis compared with Nox,
whereas AS treatment (H50 group) prevented hypoxia-induced
apoptosis, restoring cell survival under hypoxic conditions.
However, the HL group presented a higher level of cell apoptosis
compared with the H0 group. Similar pattern was observed in the
activity of caspase-3 in these groups. Active caspase-3 was
noticeably increased in the H0 group compared to the Nox group, the
H50 group had reduced caspase-3 activity compared with the H0
group, whereas there was significantly increased level of caspase-3
activity in the HL group (Fig.
5E).
AS upregulated and phosphorylation of
AKT/eNOS in hypoxia-exposed HPAECs
Western blot analysis was performed to investigate
the effect of the AS treatment on the activation of Akt and eNOS in
HPAECs in vitro. The hypoxia stimulation of HPAECs induced
lower phosphorylation of AKT by detecting the p-AKT/AKT expression
ratio, which was significantly increased by AS treatment (50
µg/ml), whereas treatment combined with LY294002 (HL group)
markedly reduced the phosphorylation of AKT induced by the AS
treatment. Additionally, AS (50 µg/ml) upregulated the
phosphorylation of eNOS compared with the hypoxia or Nox group
(P<0.05), whereas LY294002 and AS combined (HL group)
downregulated the phosphorylation of eNOS (P<0.05). These
findings indicated that AS treatment may increase the
phosphorylation of AKT/eNOS in hypoxia conditions, whereas LY294002
may inhibit this effect (Fig. 6C and
D).
Discussion
Based on previous observations, the present study
determined that in addition to the preventive effect, AS performs a
beneficial role in established hypoxic PH as it was evident that AS
treatment may restore pulmonary artery pressure without causing
systemic hypotension, and attenuate RV hypertrophy, vascular
remodeling and ECs morphology changes induced by hypoxia in rats.
These findings indicate that AS is a potential option for the
prevention and treatment of hypoxic PH.
Endothelial damage is a key initial event in PH.
Hypoxia results in the reduction of NO production by interfering
the activation of eNOS (20). To
the best of our knowledge the present study was the first to
demonstrate clearly that AS is capable of maintaining the regular
morphology and vital functions of endothelial cells and correcting
the NO release in vivo. ET-1 is a potent vasoconstrictor,
whereas cGMP has vasodilatory and anti-platelet aggregation
properties (21). The
disequilibrium of those factors underpin various morphological and
hemodynamic changes in PH (22).
In addition to the ultrastructural restoration of ECs, misadjusted
levels of vascular mediators like ET-1, cGMP and NO were
ameliorated following AS treatment. It is of note, that the AS
treatment administered 2 weeks after the hypoxia exposure (HT
group) had reduced levels of cGMP and NO restoration in
vivo, which suggested that early treatment would bring about
improved outcomes. Increased NO production by AS was consistent
with the significant activated phosphorylation of eNOS at
Ser1177. Akt has been determined to activate the
phosphorylation of eNOS, inhibit apoptotic processes, thus
interfering with cellular survival pathways (23). The upregulation of Akt and eNOS
activation by AS treatment in the present study may contribute to
the normalization of NO-mediated signal and the preservation of
endothelial cell morphology and function, which inhibits the
progression of hypoxic PH in rats.
The present study confirmed the role of AS on the
PI3K/Akt signaling pathway by the experiments in HPAECs in
vitro using a specific PI3K inhibitor, LY294002. The PI3K/Akt
signaling pathway has a critical role in cell survival during
hypoxia, and phosphorylation of Akt protects cells against
hypoxia-induced apoptosis (24,25).
The present study determined that hypoxia decreased cell viability
and induced apoptosis in HPAECs, which is in accordance with a
previous study (26); however,
this was significantly reversed by AS (50 µg/ml) treatment, which
demonstrated the protective effects on HPAECs. However, the
cytoprotective effect of AS was reduced by PI3K inhibitors LY294002
which induced apoptosis and inhibited viability in ECs under
hypoxia. LY294002 may also reduce the effect of AS on the
activation of AKT and eNOS, which indicates that AS activated eNOS
through a PI3K/Akt-dependent mechanism in HPAECs, which eventually
resulted in the reduced generation of NO by ECs. To the best of our
knowledge for the first time, the present study demonstrated that
AS treatment upregulated the PI3K/Akt/eNOS pathway in vivo
and in vitro.
There are limitations of the present study. The rat
hypoxic PH model used cannot fully represent the molecular
complexity present in human patients. Additionally, AS may enhance
NO production via activation of the AKT/eNOS pathway, whereas the
precise molecular mechanisms underlying the process and their
cause-and-effect relationship remain to be elucidated. Additional
studies are required in order to resolve these questions.
In conclusion, the present findings suggest that
there are significant effects of AS on preventing and reversing
hypoxic PH. The present study revealed that the AS acted to promote
NO production in circulation of a hypoxic PH rat model, and in
HPAECs culture under hypoxia where it had an inhibitory effect on
hypoxia-induced apoptosis. The AS-mediated protective effect on ECs
was accompanied by phosphorylation of Akt and eNOS in vivo
and in vitro. In addition, this protective effect was
significantly inhibited by LY294002 treatment in vitro,
indicating that PI3K/Akt/eNOS signal pathways have key roles in
hypoxic PH and act as a possible target for AS treatment in hypoxic
PH. These findings imply that AS may be a potential therapeutic
option for hypoxic PH.
Acknowledgements
The present study was supported by the National
Science Foundation of China (grant nos. 81470250, 81473406 and
81700062), the Natural Science Foundation of Zhejiang Province
(grant no. LQ16H010003 and LY13H020005) and the Science and
Technology Project of Wenzhou (grant no. Y20140048).
References
|
1
|
Rubin LJ: Primary pulmonary hypertension.
N Engl J Med. 336:111–117. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Humbert M, Morrell NW, Archer SL, Stenmark
KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O,
Voelkel NF and Rabinovitch M: Cellular and molecular pathobiology
of pulmonary arterial hypertension. J Am Coll Cardiol. 43 12 Suppl
S:13S–24S. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shimoda LA, Sham JS and Sylvester JT:
Altered pulmonary vasoreactivity in the chronically hypoxic lung.
Physiol Res. 49:549–560. 2000.PubMed/NCBI
|
|
4
|
Xu W, Koeck T, Lara AR, Neumann D,
DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C,
et al: Alterations of cellular bioenergetics in pulmonary artery
endothelial cells. Proc Natl Acad Sci USA. 104:pp. 1342–1347. 2007;
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Campbell AI, Zhao Y, Sandhu R and Stewart
DJ: Cell-based gene transfer of vascular endothelial growth factor
attenuates monocrotaline-induced pulmonary hypertension.
Circulation. 104:2242–2248. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao YD, Campbell AI, Robb M, Ng D and
Stewart DJ: Protective role of angiopoietin-1 in experimental
pulmonary hypertension. Circ Res. 92:984–991. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Le Cras TD and McMurtry IF: Nitric oxide
production in the hypoxic lung. Am J Physiol Lung Cell Mol Physiol.
280:L575–L582. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fulton D, Gratton JP, McCabe TJ, Fontana
J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A and Sessa WC:
Regulation of endothelium-derived nitric oxide production by the
protein kinase Akt. Nature. 399:597–601. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ou J, Fontana JT, Ou Z, Jones DW, Ackerman
AW, Oldham KT, Yu J, Sessa WC and Pritchard KA Jr: Heat shock
protein 90 and tyrosine kinase regulate eNOS NO* generation but not
NO* bioactivity. Am J Physiol Heart Circ Physiol. 286:H561–H569.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Giaid A and Saleh D: Reduced expression of
endothelial nitric oxide synthase in the lungs of patients with
pulmonary hypertension. N Engl J Med. 333:214–221. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pullamsetti S, Kiss L, Ghofrani HA,
Voswinckel R, Haredza P, Klepetko W, Aigner C, Fink L, Muyal JP,
Weissmann N, et al: Increased levels and reduced catabolism of
asymmetric and symmetric dimethylarginines in pulmonary
hypertension. FASEB J. 19:1175–1177. 2005.PubMed/NCBI
|
|
12
|
Guo JS, Cheng CL and Koo MW: Inhibitory
effects of Centella asiatica water extract and asiaticoside on
inducible nitric oxide synthase during gastric ulcer healing in
rats. Planta Med. 70:1150–1154. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yun KJ, Kim JY, Kim JB, Lee KW, Jeong SY,
Park HJ, Jung HJ, Cho YW, Yun K and Lee KT: Inhibition of
LPS-induced NO and PGE2 production by asiatic acid via NF-kappa B
inactivation in RAW 264.7 macrophages: Possible involvement of the
IKK and MAPK pathways. Int Immunopharmacol. 8:431–441. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dong MS, Jung SH, Kim HJ, Kim JR, Zhao LX,
Lee ES, Lee EJ, Yi JB, Lee N, Cho YB, et al: Structure-related
cytotoxicity and anti-hepatofibric effect of asiatic acid
derivatives in rat hepatic stellate cell-line, HSC-T6. Arch Pharm
Res. 27:512–517. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen S, Yin ZJ, Jiang C, Ma ZQ, Fu Q, Qu R
and Ma SP: Asiaticoside attenuates memory impairment induced by
transient cerebral ischemia-reperfusion in mice through
anti-inflammatory mechanism. Pharmacol Biochem Behav. 122:7–15.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang XB, Wang W, Zhu XC, Ye WJ, Cai H, Wu
PL, Huang XY and Wang LX: The potential of asiaticoside for
TGF-β1/Smad signaling inhibition in prevention and progression of
hypoxia-induced pulmonary hypertension. Life Sci. 137:56–64. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Muramatsu M, Oka M, Morio Y, Soma S,
Takahashi H and Fukuchi Y: Chronic hypoxia augments endothelin-B
receptor-mediated vasodilation in isolated perfused rat lungs. Am J
Physiol. 276:L358–L364. 1999.PubMed/NCBI
|
|
18
|
Michelakis ED, McMurtry MS, Wu XC, Dyck
JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R and
Archer SL: Dichloroacetate, a metabolic modulator, prevents and
reverses chronic hypoxic pulmonary hypertension in rats: Role of
increased expression and activity of voltage-gated potassium
channels. Circulation. 105:244–250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zheng Y, Li M, Zhang Y, Shi X, Li L and
Jin M: The effects and mechanisms of mycophenolate mofetil on
pulmonary arterial hypertension in rats. Rheumatol Int. 30:341–348.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mukhopadhyay S, Xu F and Sehgal PB:
Aberrant cytoplasmic sequestration of eNOS in endothelial cells
after monocrotaline, hypoxia and senescence: Live-cell caveolar and
cytoplasmic NO imaging. Am J Physiol Heart Circ Physiol.
292:H1373–H1389. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wilkins MR: Pulmonary hypertension: The
science behind the disease spectrum. Eur Respir Rev. 21:19–26.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Coggins MP and Bloch KD: Nitric oxide in
the pulmonary vasculature. Arterioscler Thromb Vasc Biol.
27:1877–1885. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mangi AA, Noiseux N, Kong D, He H, Rezvani
M, Ingwall JS and Dzau VJ: Mesenchymal stem cells modified with Akt
prevent remodeling and restore performance of infarcted hearts. Nat
Med. 9:1195–1201. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Martorell L, Gentile M, Rius J, Rodríguez
C, Crespo J, Badimon L and Martínez-González J: The
hypoxia-inducible factor 1/NOR-1 axis regulates the survival
response of endothelial cells to hypoxia. Mol Cell Biol.
29:5828–5842. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pullamsetti SS, Savai R, Janssen W, Dahal
BK, Seeger W, Grimminger F, Ghofrani HA, Weissmann N and Schermuly
RT: Inflammation, immunological reaction and role of infection in
pulmonary hypertension. Clin Microbiol Infect. 17:7–14. 2011.
View Article : Google Scholar : PubMed/NCBI
|