Introduction
Neuropathic pain refers to lesions of the
somatosensory system or pain caused by the disease (1). According to the survey, the incidence
of neuropathic pain is different, the British about 1% (2), about 1.5% in the United States
(3), Canada 17.9% (4), in China, the incidence rate is about
7%. More than 2/3 of patients can not effectively relieve pain,
which not only seriously affects the quality of life of patients
(5), but also increased the burden
on society (6). At present, we
mainly use analgesic drugs to treat neuropathic pain.
Unfortunately, pharmacological treatment of neuropathic pain is
more symptomatic, because the mechanisms responsible for the pain
remain unclear. Therefore, a better understanding of the mechanism
of neuropathic pain is important for the treatment of the disease.
Peripheral nerve injury-induced neuropathic pain have been related
to increased excitability occurring in peripheral and central
nervous systems as the product of enhanced synaptic excitation,
decreased synaptic inhibition, and increased neuronal
responsiveness. Pain of first with the participation and regulation
is the peripheral nervous system, including the DRG neurons. Pain
is initiated with the detection of noxious stimuli at terminals of
DRG neurons innervated in peripheral nervous system.
PAR2 is highly expressed in the peripheral and
central nervous system and belongs to the G protein coupled
receptors (GPCR) (7). Recent
research shows that PAR2 can promote neurogenic inflammation and
pain by stimulating sensory neurons (8), the activation of PAR2 increased the
release of IP3, further increased the intracellular
concentration of calcium ion (9),
the latter leads to calcium activated chloride channels (CaCCs)
opening. In the event of pain or inflammation, CaCCs can also
increase the excitability of DRG neurons, and then aggravate the
occurrence of neuropathic pain mediated by tissue injury (10,11).
In 2008, researchers found TMEM16A is the molecular
basis for CaCCs (12). It is a
kind of heat sensitive protein, which has been found to be involved
in many kinds of physiological functions, hyperalgesia and pain can
also be significantly reduced in TMEM16A knockout mice (13). In addition, the activation of PAR2
increased the release of IP3, further increased the
intracellular concentration of calcium ion (9). With the increase of intracellular
calcium in concentration, the activation threshold of TMEM16A was
decreased and the activating current of TMEM16A was enhanced
(14). These results suggest that
TMEM16A may participate in neuropathic pain.
We have shown that TMEM16A were widely expressed in
DRG neurons (15), however, we do
not know whether PAR2 were expressed in DRG neurons, whether the
PAR2 and TMEM16A are co-expression in DRG neurons, whether they
play the role in the molecular mechanism of neuropathic pain is
still unknown. Therefore, in this experiment, we suppose increasing
expression of PAR2 increased the release of IP3, further
increased the intracellular concentration of calcium ion, the
activation threshold of TMEM16A was decreased and the expression of
TMEM16A was increased. We observe the expression of PAR2 and
TMEM16A in DRG neurons and its changes with time in neuropathic
pain, then we detected the concentration of IP3 to
explore the relationship of PAR2 and TMEM16A in the development of
neuropathic pain.
Materials and methods
Animals
A total of 114 male Spraque-Dawley (SD) rats,
weighing 200–220 g, 8–12 weeks, provided by the Experimental Animal
Center of Xinjiang Medical University, Urumqi, China (certificate
no. SCXK 2003–0001), lived under standard laboratory conditions for
12 h light and 12 h darkness. Room temperature (20–22°C), humidity
(50–60%) and provide clean food and water. The present study was
conducted in accordance with approval from the Institutional Ethics
Review Board (IERB) at the First Affiliated Hospital of the Shihezi
University School of Medicine (IERB no. SHZ2010LL01). All
experiments were carried out according to the Ethical Guidelines
for Investigations of Experimental of International Association for
the Study of Pain. All experimental procedures minimized the
suffering and number of rats used, within the premises of achieving
the experimental purpose (16).
Experimental grouping and animal
modeling
Male SD rats were randomly divided into 3 groups,
sham group, CCI-D7 group and CCI-D14 group. In short, SD rats were
anesthetized with 10% chloral hydrate (0.3 g/kg) by intraperitoneal
injection and fixed on the operating table in prone position. For
skin preparation and disinfection of rats, open the left hind leg
skin with a knife, strip muscles and tissues with forceps, the
sciatic nerve of the left hind paw was exposed using blunt
dissection through the biceps femoris muscle at the midthigh, and
then mid-sciatic nerve was loosely tied using 4 ligatures (chromic
catgut, 4–0; Ethicon, Inc., Cincinnati, OH, USA) proximal to the
sciatic trifurcation at 1 mm spacing, which can induce mild
twitching leg strength. The rats in the sham group only exposed the
sciatic nerve without ligation. The wound after operation was
sutured using surgical thread (Mersilk, 5.0; Ethicon, Inc.) and
operation sterilization was performed using iodophor solution and
triple antibiotic ointment (Tricin; Merck KGaA, Darmstadt,
Germany). After surgery, the rats were housed in separate cages and
monitor recovery period. There were significant differences in
thermal withdrawal latency (TWL) between the contralateral side of
CCI group and sham group, and TWL of ipsilateral side in CCI group
were declined by at least 30% in contrast with sham group,
indicating CCI model was successfully established. Thermal
latencies of ipsilateral and contralateral paw withdrawal were
assessed before surgery (one day, baseline) and on days 1, 3, 5, 7,
10 and 14 after surgery in sham group and CCI group.
Hot-plate testing
CCI induced hyperalgesia was assessed by the hind
paw thermal withdrawal test using a hot-plate instrument (Hot
Plate; Ugo Basile Biological Research Apparatus, Ugo Basile,
Italy). A total of 24 rats were used to have the Hot-plate testing
in the result, 12 in the Sham group and 12 in the CCI group. Each
rat was placed in the plastic box on a hot plate at 58°C and
allowed to habituated for 30 min before testing. The duration from
onset of application of thermal stimulus to either hind-paw
lick/jump/withdraw was defined as withdrawal latency (TWL). In
order to avoid the damage of tissue, cut-off time for paw
withdrawal latency was maintained at 30 sec in all cases. The TWL
of each rat at each time point was recorded in sec and was repeated
three times with 4 min interval. The TWL was obtained by
calculating the mean of three withdrawal latencies. All the
subjects were tested in the morning from 8:00am to 10:00am.
Compared with the preoperative, TWL decreased more than 30% as a
model of success criteria (17).
DRG harvesting
Rats of all groups were anesthetized with 10%
chloral hydrate (0.3 g/kg) by intraperitoneal injection. The skin
overlying the lumbar region was cut away. A laminectomy of the
lumbar region was then carried out. After exposing sciatic nerve,
attached ipsilateral lumbar DRGs (L4–6) were carefully
dissected and rapidly transferred into liquid nitrogen and kept at
−80°C. Ipsilateral L4-6 DRGs were collected from animals on days 7
and 14 following CCI, when neuropathic pain was developed. The
L4–6 DRGs of sham group were harvested after two days of
surgery.
Immunofluorescence
Immunofluorescent staining of PAR2 and TMEM16A in
DRGs were performed according to Miao et al (18) with some modification 36 rats were
randomly divided into 3 groups. Rats from sham group (n=12) and
days 7 (n=12) and 14 (n=12) post-CCI were anaesthetized with 10%
chloral hydrate (0.3 g/kg), and then perfused through the aorta
with 0.9% normal saline, followed by fresh 4% paraformaldehyde in
PBS for 10 min for tissue fixation. The L4–6 DRGs were
removed rapidly and placed in 4% paraformaldehyde in PBS for 24 h.
Then the L4–6 DRGs were sectioned (thickness, 5 µm) by a
freezing microtome (18). Sections
were washed and incubated with blocking buffer (4% BSA in PBS
buffer with Tween-20) for 30 min. It was incubated with
anti-TMEM16A (1:200; S-20, no. sc-69343; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) and anti-PAR2 (1:200; ab180953; Abcam,
Cambridge, MA, USA) in a wet box overnight at 4°C. The sections
were then washed 4 times with 0.01M PBS, 5 min each time. Then it
was incubated with the solution containing donkey anti-rabbit
IgG-FITC (1:200; Santa Cruz Biotechnology, Inc.) and donkey
anti-goat IgG-TRITC (1:200; Santa Cruz Biotechnology, Inc.) at 37°C
for 2 h. Slides were then examined by confocal microscopy (LSM710;
Carl Zeiss AG, Oberkochen, Germany). Immunofluorescence
quantification for PAR2 and TMEM16A expression in the dorsal root
ganglion were performed by measuring the mean absorbance following
laser confocal microscopy with the use of analysis software (ZEN
2009 Light Edition; Carl Zeiss AG).
Western blot analysis
We harvested the L4-6 DRGs from sham group (n=12)
and days 7 (n=12) and 14 (n=12) post-CCI. The homogenates were
incubated at 4°C for 30 min and centrifuged at 12,000 g for 40 min
at 4°C. The supernatant was collected, and the protein
concentration in the supernatant was estimated by the BCA protein
assay. Each samples with equal amounts of protein (20 µg/lane) were
separated by 10% SDS-PAGE electrophoresis. The resolved proteins
were then transferred to a PVDF membrane (EMD Millipore, Billerica,
MA, USA). The membranes were blocked with 5% non-fat milk in TBST
buffer (pH 8.0, 10 mmol/l Tris-HCl, 150 mmol/l NaCl and 0.2% Tween
20) for 1 h at room temperature, and then probed with various
primary antibodies [anti-PAR2 antibody (1:250, ab180953; Abcam);
anti-TMEM16A antibody (1:1,000, ab53212; Abcam); anti-β-actin
antibody (1:5,000; Merck KGaA)] overnight at 4°C. After incubation
of primary antibodies, the blots were washed three times with TBST,
5 min each time and incubated with secondary antibody [1:10,000;
horseradish peroxidase-conjugated goat anti-rabbit or goat
anti-mouse secondary antibodies (Alpha Diagnostic, San Antonio,
Texas, USA)] for 2 h at room temperature. The blots were finally
washed six times with TBST, 5 min each time and visualized on the
X-ray film using the ECL chemiluminescence reagent (GE Healthcare,
Chicago, IL, USA). The optical density of each target protein band
was assessed with Quantity one software (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) and normalized using corresponding β-actin
bands in the same sample (18).
Detection of IP3 in dorsal
root ganglion by ELISA
We harvested the L4-6 DRGs from sham group (n=6) and
days 7 (n=6) and 14 (n=6) post-CCI. The homogenates were
centrifuged at 5,000 g for 5 min. According to the IP3
test kit (CEC037Ge 96T; Cloud-Clone Corp., Wuhan, Hubei, China)
instructions, we leave the supernatant to test the concentration of
IP3 (pg/ml) in the dorsal root ganglion of each group
(19).
Statistical Analysis
Data were analyzed with SPSS v19.0 software (SPSS,
Inc., Chicago, IL, USAs) and presented as the mean ± standard error
of the mean. A homogeneity test for variance was performed followed
by one-way analysis of variance with with post hoc
Student-Newman-Keuls tests. Two-group comparison was conducted
using the least significant difference t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Decreased TWL in Hot-plate
testing
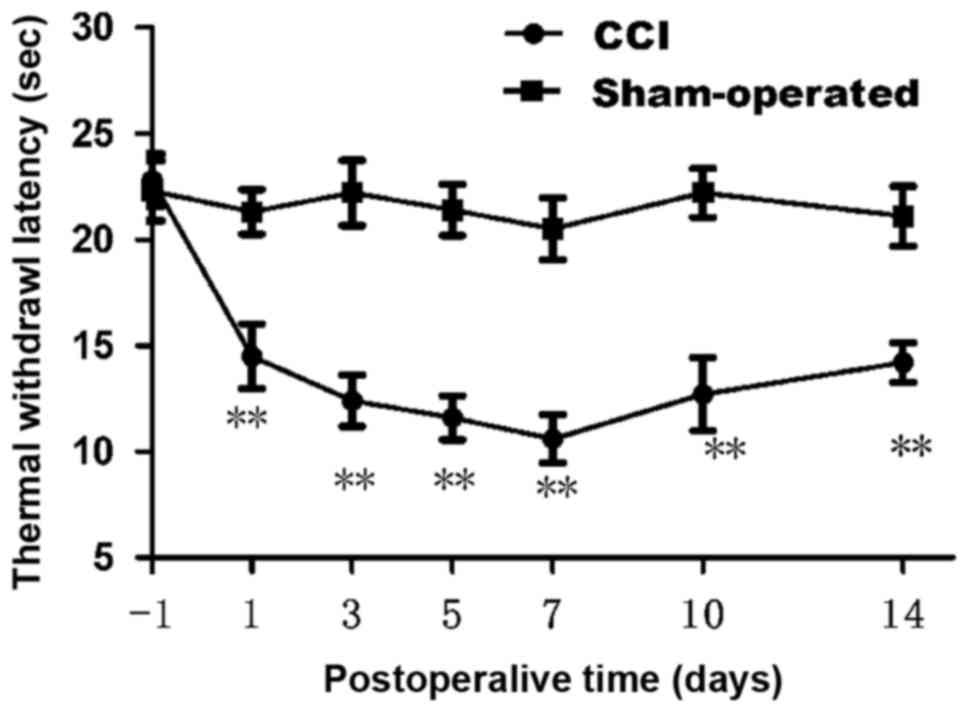
We first confirmed whether chronic constriction
injury of the sciatic nerve cause hyperalgesia or allodynia in rats
by withdrawal responses to thermal stimulation. The thermal
withdrawal latency from radiant heat was measured at time points of
1, 3, 5, 7, 10 and 14 day after chronic constriction injury. As
shown in Fig. 1, starting from
fist day after surgery, apparent dramatically decrease (n=12;
P<0.01) in the thermal withdrawal latency of ipsilateral paws
was observed in CCI model, reflecting a state of persistent
hyperalgesia. Comparing with the sham group (21.30±1.05 at 1 day;
21.10±1.41 at 14 day), the TWL of CCI group was decreased
significantly from 1 day (14.50±1.53) to 14 days (14.20±0.92).
Decrease in the thermal pain threshold indicates the success of the
CCI model. Low tolerance and paw flinching suggest neuropathic pain
in rats. Rats in CCI group presented a dramatic decrease in thermal
pain threshold on 7 day after operation. Data are expressed as mean
± SME (n=12; P<0.01; Fig.
1).
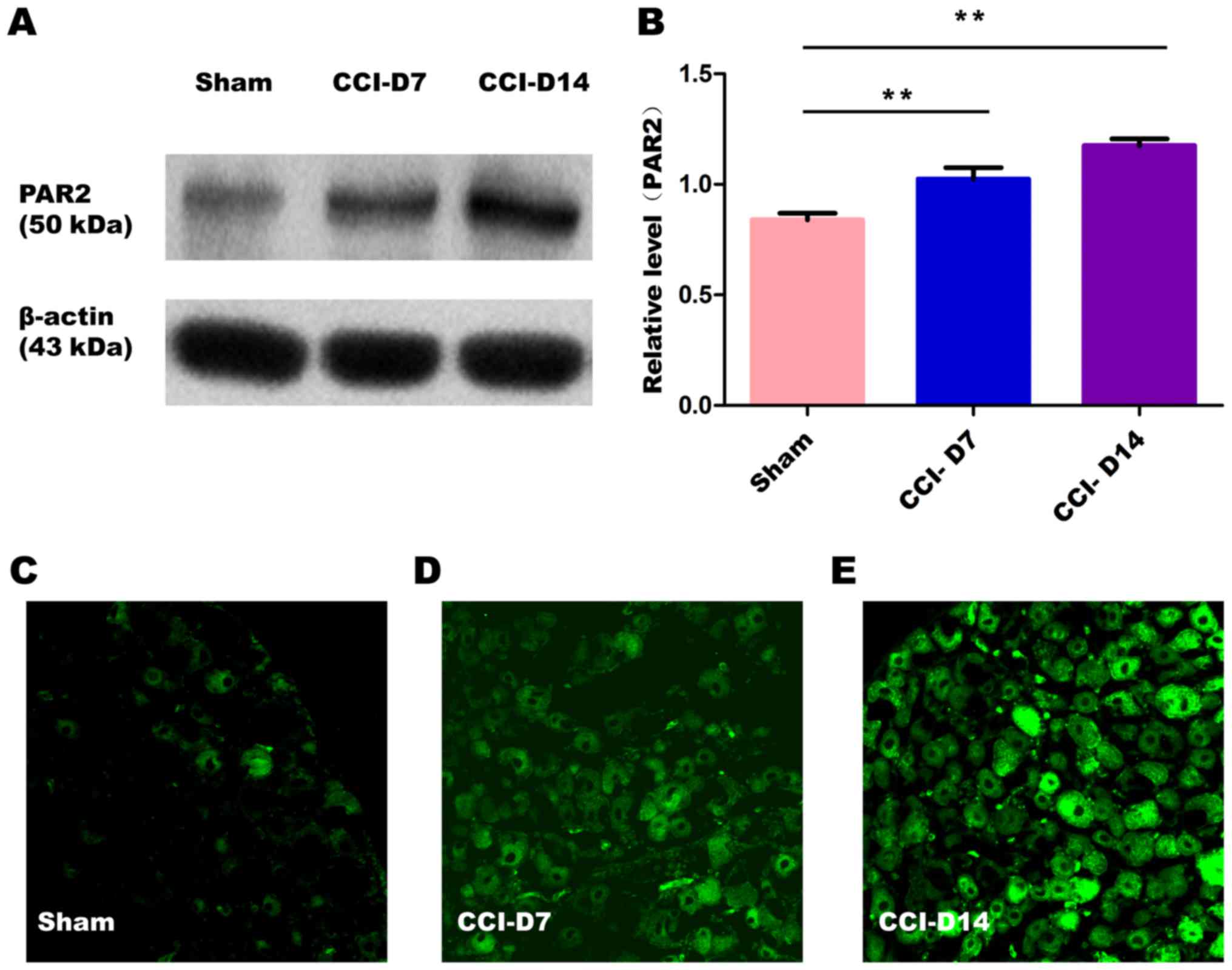
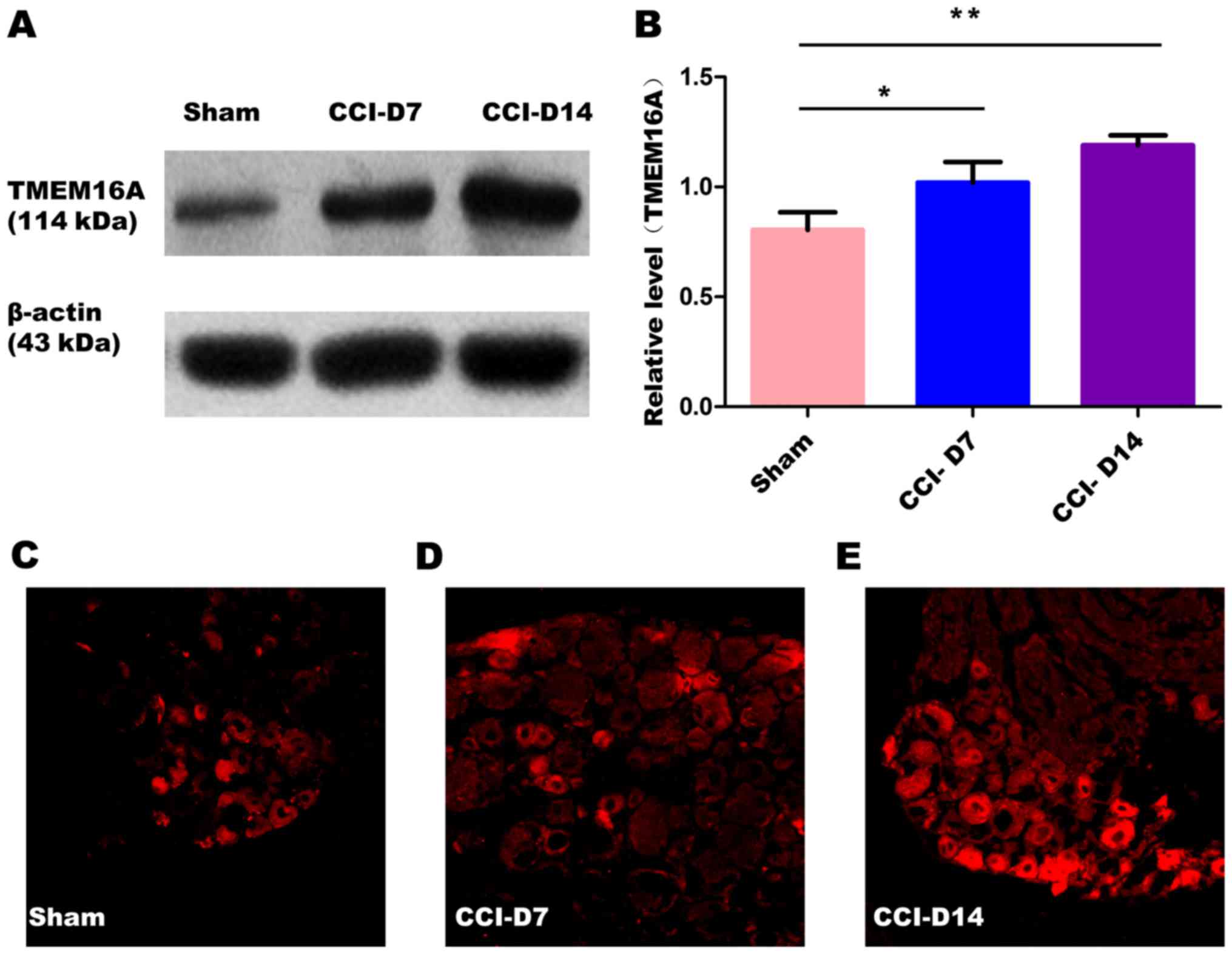
The changes in the expression of PAR2 and TMEM16A
after CCI. To confirm effects of neuropathic pain on the PAR2
and TMEM16A protein level, we performed immunoblotting on L4-6 DRGs
in sham group, CCI-D7 group and CCI-D14 group. We collected DRG
neurons between three groups and analyzed the proteins of PAR2 and
TMEM16A. Compared with the sham group, we found that the expression
of PAR2 (Fig. 2) and TMEM16A
(Fig. 3) protein in the CCI-D7
group and CCI-D14 group increased significantly (n=12; P<0.05).
At the level of protein expression, the relative level of band
intensity of PAR2 protein in sham, CCI-D7 and CCI-D14 groups were
0.84, 1.02 and 1.18 respectively (Fig.
2B), the relative level of band intensity of TMEM16A protein in
Sham, CCI-D7 and CCI-D14 groups were 0.80, 1.02, and 1.19
respectively (Fig. 3B). In
addition, the fluorescence intensity of PAR2 and TMEM16A were
increased with the progress of neuropathic pain (Figs. 2C-E; Fig. 3C-E). Furthermore, the changes in
protein expression of PAR2 and TMEM16A are consistent with the
fluorescence intensity of PAR2 and TMEM16A (Figs. 2, 3). Indeed, these results indicate that
CCI induced neuropathic hyperalgesia can up-regulate protein
expression of PAR2 and TMEM16A.
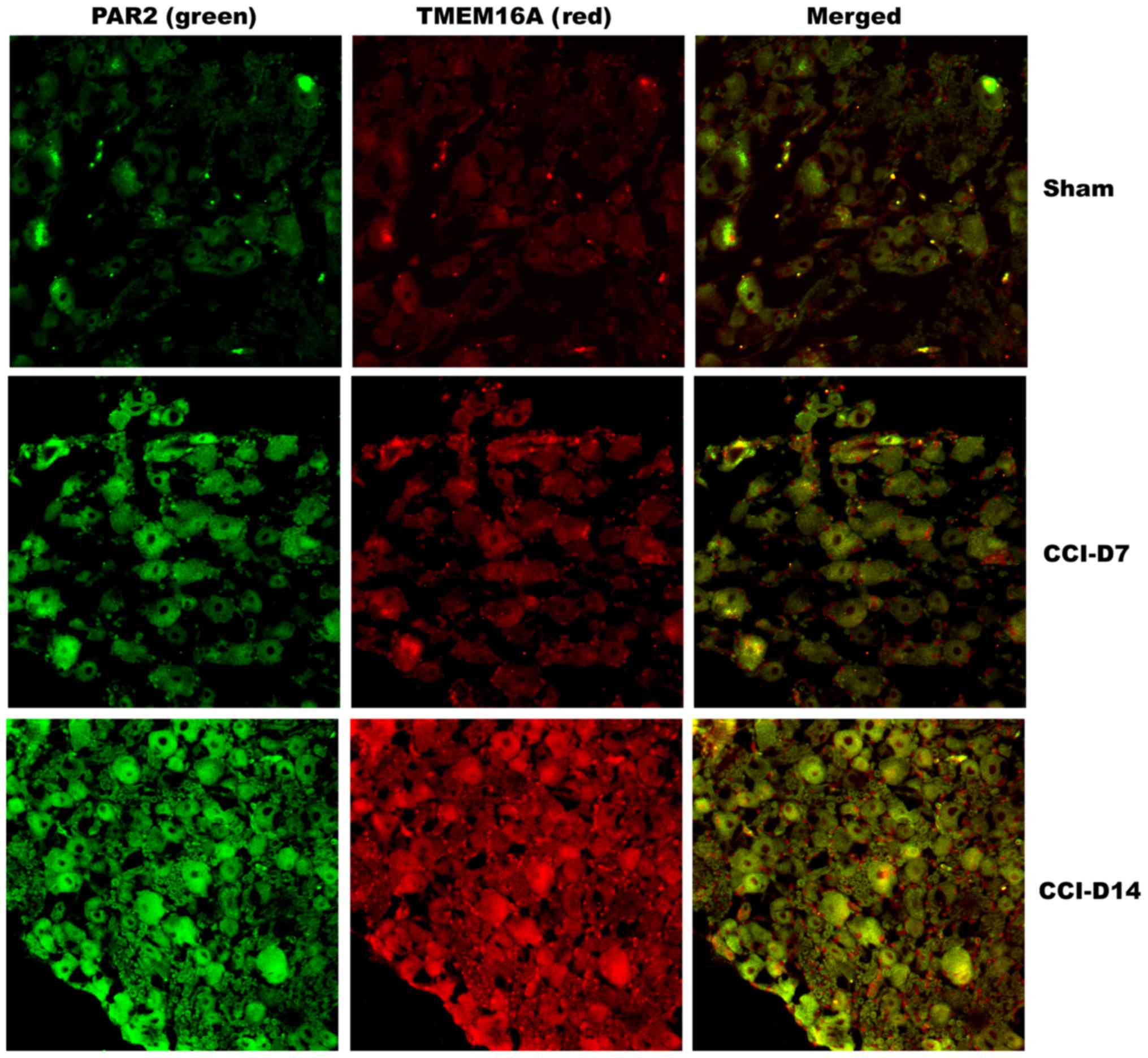
Co-expression of PAR2 and TMEM16A
proteins on DRG neurons in neuropathic pain rats
To clarify the localization of PAR2 and TMEM16A on
DRG neurons, we performed immunofluorescence double staining
experiments to identify the expression of the two proteins in DRG
neurons. As shown in Fig. 4,
immunofluorescence double staining experiments showed that PAR2 and
TMEM16A were co-expressed in each group of DRG. As shown in
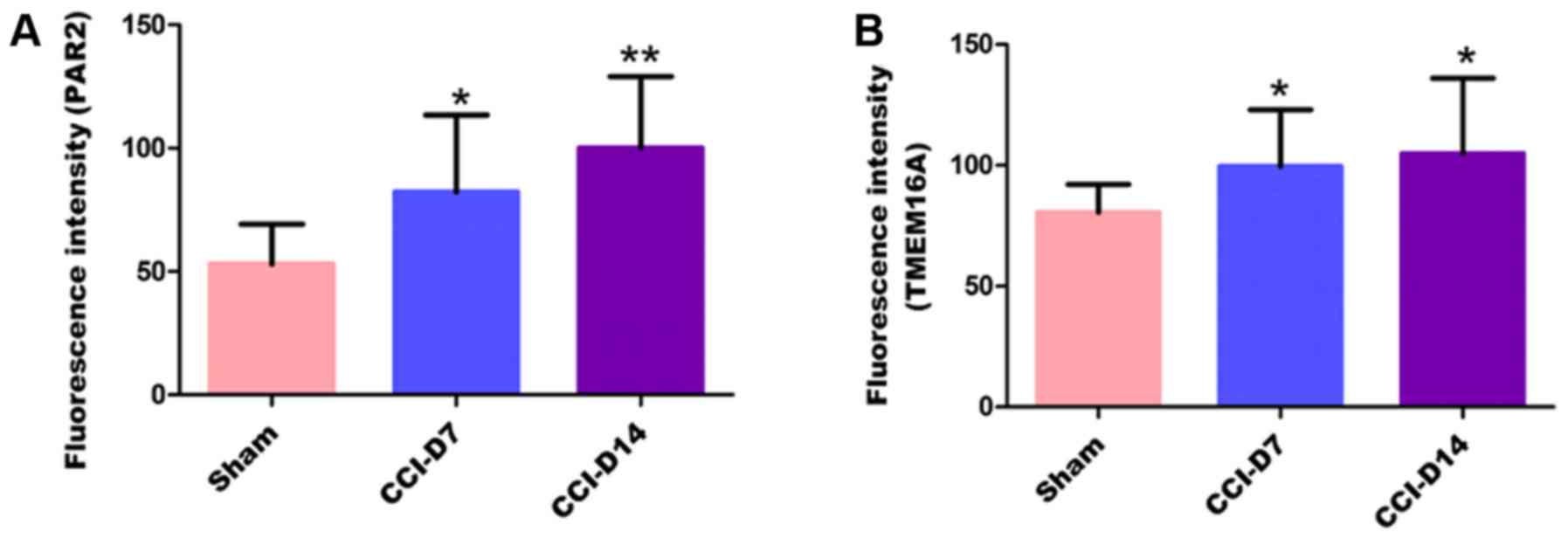
Fig. 5, compared with the sham
group, the fluorescence intensity of PAR2 in CCI-D7 group was
increased (n=12; P<0.05) and in CCI-D14 group was increased
significantly (n=12; P<0.01) (Fig.
5A); the fluorescence intensity of TMEM16A in CCI-D7 group and
CCI-D14 group was increased too (n=12; P<0.05) (Fig. 5B).
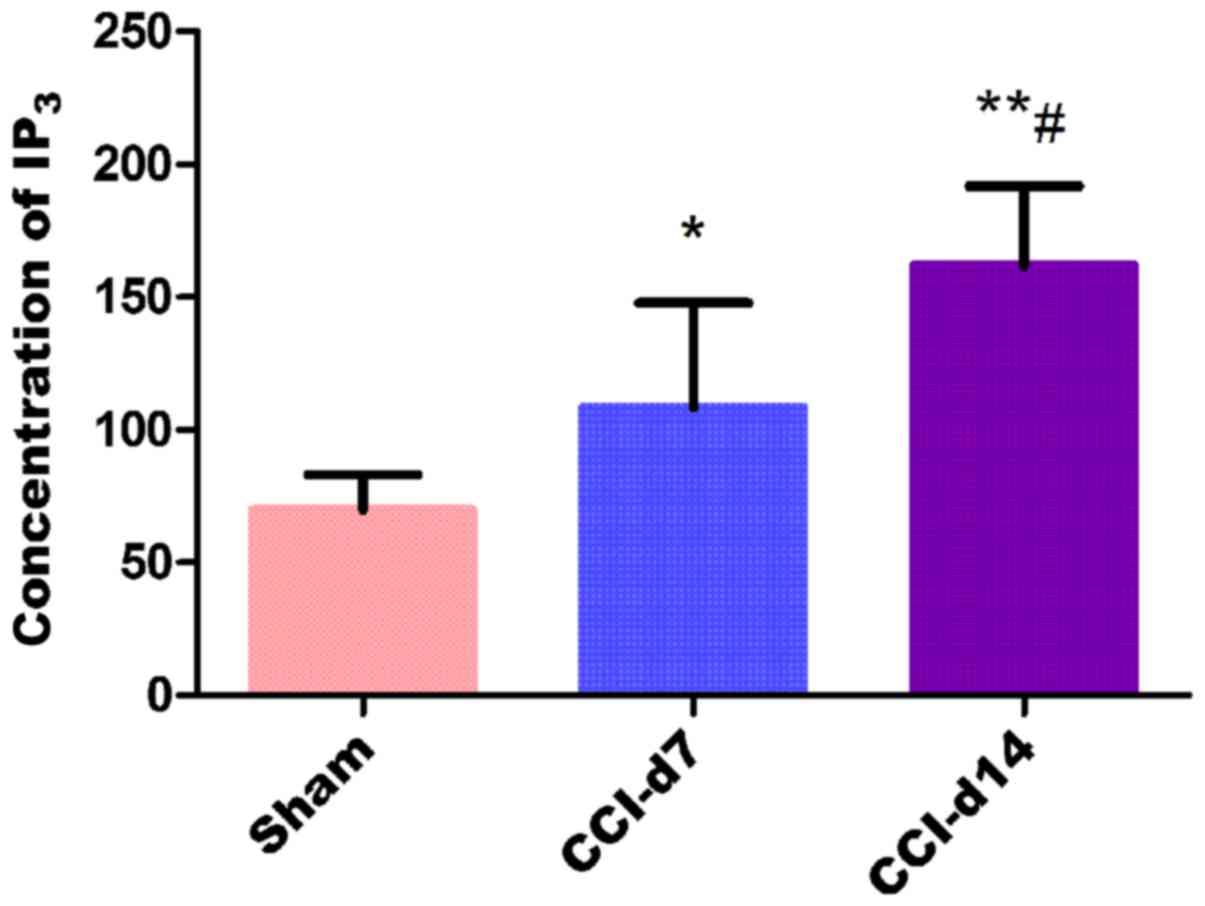
Detection of IP3
concentration in dorsal root ganglion by ELISA
The experimental results by enzyme linked
immunosorbent assay shows that compared with the sham group, the
concentration of IP3 in CCI-D7 group (n=6; P<0.05)
and CCI-D14 group (n=6; P<0.01) were increased; compared with
CCI-D7 group, the concentration of IP3 in CCI-D14 group
was increased significantly (n=6; P<0.01) (Fig. 6).
Discussion
Management of neuropathic pain is a major medical
issue. In the last decades, great progress have been made in
understanding the molecular mechanisms involved in pain generation
and perception. Key cellular components for neuropathic pain
generation are nociceptors. Nociceptive neural impulses originate
in primary afferent neurons in dorsal root ganglia, which then
activate neurons in the spinal cord and in specific nuclei in the
brain to induce the perception of pain. There are many mechanisms
for neuropathic pain. The present study showed that some factors
expressed in damaged tissues such as histamine, prostaglandins,
neuropeptides, cytokines, growth factors, 5-HT, ATP and BK, these
substances could increase excitability and result in pain
sensitization when the nerve fibers were stimulated (20). Increasing evidence suggests that
differential expression of PAR2 and TMEM16A protein results in the
development of chronic neuropathic pain induced hyperalgesia. In
the present study, we characterized the effects of PAR2 and TMEM16A
on the excitability of DRG neurons during CCI induced hyperalgesia.
The effects of neuropathic pain on PAR2 and TMEM16A protein
expression were evaluated with immunocytochemistry and western
blot. The aim of our study was to investigate the expression of
PAR2 and TMEM16A in the dorsal root ganglion neurons of rats with
neuropathic pain model (CCI), and to explore its possible role in
neuropathic pain.
CCI, as a novel model of peripheral neuropathic
pain, which analogous to the human beings and the symptoms in this
rat model. According to Bennett and Xie (21) method, we successfully established
animal model of CCI, the main performance is: The left hind limb
paw slight adduction deformity and limp, licking the left foot and
left hanging leg protective posture. The thermal withdrawal
latency, CCI group began to decrease after 1 day, reaching a
minimum after 7 days (Fig. 1). The
above described is basically the same as Bennett and Xie (21) method.
Previous studies have confirmed that PAR2 were
detected in the rat hippocampus, amygdala, thalamus, cortex,
hypothalamus, striatum and dorsal root ganglia (22). In our experiment, we found the
expression of PAR2 on DRG neurons in the CCI group was gradually
increased (Fig. 2A and B),
suggesting that PAR2 may be involved in the mechanism of
inflammatory or neuropathic pain. Activation of PAR2 may be due to
the activation of protein kinase A and protein kinase C signaling
pathway (23). Activation of PAR2
may increase the neurons excitability which dependented on cGMP
activity and cAMP activity and the application of PAR2 inhibitors
could inhibit activation of c-AMP and protein kinase A signaling
pathway, finally inhibited the hyperalgesia and inflammation
(9). During Inflammation, BDNF was
also released through the activation of PAR2 signal pathway in
microglia (24). At the same time,
studies have shown that activation of PAR2 could activate TRPV1,
then stimulated the release of neuropeptides SP and CGRP, which can
cause the occurrence of hyperalgesia (25–27).
In addition, in the PAR2 deficient mice, due to the disappearence
of serine protease and depletion of mast cells in mice, pain
significantly reduced, so the activation of PAR2 is considered to
be the new mechanism of abnormal pain associated with cancer
(28), inhibiting the activation
of PAR2 may become a new target for the development and treatment
of bone cancer pain (29). All of
the above studies indicated that PAR2 may be closely related to
pain.
Our present study also found that TMEM16A was
expressed widely in DRG neurons, and positive cells were evenly
distributed in the cytoplasm and cell membrane, the nucleus no
mark. In the experimental process, the expression of TMEM16A in the
CCI group of DRG neurons was gradually increased compared with the
sham group (Fig. 3A and B),
suggesting that the expression of TMEM16A is closely related to the
progression of neuropathic pain. TMEM16A is not only the molecular
basis of calcium activated chloride channels, but also a presence
in the nociceptor heat sensitive protein, which mediates many
physiological functions. Moreover, hyperalgesia and allodynia were
significantly reduced in TMEM16A knockout mice (30). Knockdown of TMEM16A from DRG
neurons also attenuated thermal hyperalgesia in both neuropathic
pain and inflammatory models (31). In addition, a recent research also
suggested that activated pain response could be suppressed by the
TMEM16A inhibitor A01 (32). These
results suggest that TMEM16A may play a role in neuropathic
pain.
Studies have shown that TMEM16A can be activated by
an increase in the intracellular Ca2+ concentration
through ion channels (33). Recent
research have found that TMEM16A can be regulated by mechanical
stimuli (34), Protons (35), cholesterol (36,37),
calmodulin (38–40) and IP3 (41,42).
In our experiment, we found that the concentration of
IP3 in dorsal root ganglia was increased following the
experimental process (Fig. 6),
protein expression of PAR2 and TMEM16A were increased in the same
trend, therefore, we assume that increasing expression of two
protains may be related to the release of IP3. That is
to say, the increasing expression of PAR2 protein caused the
increased release of IP3, and the increasing release of
IP3 induced the activation of TMEM16A. Recent studies
have shown that activation of PAR2 increased calcium ion
concentration by release IP3 (9), accompanied by an increase in
intracellular calcium concentration, TMEM16A activation threshold
reduced and activated current enhanced (14).
In conclusion, these data suggest that PAR2 and
TMEM16A were co-expression on DRGs of the primary afferent neurons,
which were sensitive to CCI-induced neuropathic pain. CCI-induced
the up-regulation expression of PAR2 and TMEM16A may maintain
hyperalgesia or allodynia. In addition, this experiment confirmed
that expression of PAR2 and TMEM16A were gradually increased with
the progression of neuropathic pain duration, and the concentration
trend of IP3 were consistent with the changes in protein expression
of PAR2 and TMEM16A. But the relationship between PAR2 and TMEM16A
in the development of neuropathic pain, and the specific mechanism
needs further verification in future experiments.
Acknowledgements
The authors would like to express their thanks to
all those who helped in the writing of this paper. And thanks to
the project supported by the National Natural Science Fund (no.
81560175).
References
|
1
|
Jensen TS, Baron R, Haanpää M, Kalso E,
Loeser JD, Rice AS and Treede RD: A new definition of neuropathic
pain. Pain. 152:2204–2205. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bowsher D: Neurogenic pain syndromes and
their management. Br Med Bull. 47:644–666. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carter GT and Galer BS: Advances in the
management of neuropathic pain. Phys Med Rehabil Clin N Am.
12:447–459. 2001.PubMed/NCBI
|
|
4
|
Toth C, Lander J and Wiebe S: The
prevalence and impact of chronic pain with neuropathic pain
symptoms in the general population. Pain Med. 10:918–929. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mcdermott AM, Toelle TR, Rowbotham DJ,
Schaefer CP and Dukes EM: The burden of neuropathic pain: Results
from a cross-sectional survey. Eur J Pain. 10:127–135. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
O'Connor AB: Neuropathic pain:
Quality-of-life impact, costs and cost effectiveness of therapy.
Pharmacoeconomics. 27:95–112. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park GH, Jeon SJ, Ryu JR, Choi MS, Han SH,
Yang SI, Ryu JH, Cheong JH, Shin CY and Ko KH: Essential role of
mitogen-activated protein kinase pathways in protease activated
receptor 2-mediated nitric-oxide production from rat primary
astrocytes. Nitric Oxide. 21:110–119. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bao Y, Hou W and Hua B: Protease-activated
receptor 2 signalling pathways: A role in pain processing. Expert
Opin Ther Targets. 18:15–27. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang ZJ, Li HC, Cowan AA, Liu S, Zhang YK
and Song XJ: Chronic compression or acute dissociation of dorsal
root ganglion induces cAMP-dependent neuronal hyperexcitability
through activation of PAR2. Pain. 153:1426–1437. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee B, Cho H, Jung J, Yang YD, Yang DJ and
Oh U: Anoctamin 1 contributes to inflammatory and nerve-injury
induced hypersensitivity. Mol Pain. 10:52014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
García G, Martínezrojas VA, Rochagonzález
HI, Granadossoto V and Murbartián J: Evidence for the participation
of Ca(2+)-activated chloride channels in formalin-induced acute and
chronic nociception. Brain Res. 1579:35–44. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang YD, Cho H, Koo JY, Tak MH, Cho Y,
Shim WS, Park SP, Lee J, Lee B, Kim BM, et al: TMEM16A confers
receptor-activated calcium-dependent chloride conductance. Nature.
455:1210–1215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cho H, Yang YD, Lee J, Lee B, Kim T, Jang
Y, Back SK, Na HS, Harfe BD, Wang F, et al: The calcium-activated
chloride channel anoctamin 1 acts as a heat sensor in nociceptive
neurons. Nat Neurosci. 15:1015–1021. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsuba S, Niwa S, Muraki K, Kanatsuka S,
Nakazono Y, Hatano N, Fujii M, Zhan P, Suzuki T and Ohya S:
Downregulation of Ca2+-activated Cl-channel TMEM16A by the
inhibition of histone deacetylase in TMEM16A-expressing cancer
cells. J Pharmacol Exp Ther. 351:510–518. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao L, Li LI, MA KT, Wang Y, Li J, Shi
WY, Zhu HE, Zhang ZS and Si JQ: NSAIDs modulate GABA-activated
currents via Ca2+-activated Cl-channels in rat dorsal root ganglion
neurons. Exp Ther Med. 11:1755–1761. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li L, Zhao L, Wang Y, Ma KT, Shi WY, Wang
YZ and Si JQ: PKCε mediates substance P inhibition of GABAA
receptors-mediated current in rat dorsal root ganglion. J Huazhong
Univ Sci Technolog Med Sci. 35:1–9. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han YY, Huang Z, Wang YP, et al: The study
of the expression of potassium sodium chloride transporter on DRG
neurons in CCI model rat. Chinese Pain Med. 22:583–90. 2016.(In
Chinese).
|
|
18
|
Miao XR, Gao XF, Wu JX, Lu ZJ, Huang ZX,
Li XQ, He C and Yu WF: Bilateral downregulation of Nav1.8 in dorsal
root ganglia of rats with bone cancer pain induced by inoculation
with Walker 256 breast tumor cells. BMC Cancer. 10:2162010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang W, Si-yuan L, Ke-tao M, Jun-qiang S,
Lei Z, Zhong-Shuang Z, He Z and Li L: Changes in presynaptic
inhibition and the second message system of neuropathic pain model
in rats. China J Mod Med. 22:9–14. 2012.(In Chinese).
|
|
20
|
Mcmahon SB, Bennett DLH and Bevan S:
Inflammatory mediators and modulators of pain. Churchill
Livingstone (Elsevier Health Sciences). 1–72. 2006.
|
|
21
|
Bennett GJ and Xie YK: A peripheral
mononeuropathy in rat that produces disorders of pain sensation
like those seen in man. Pain. 33:87–107. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Striggow F, Riekburchardt M, Kiesel A,
Schmidt W, Henrich-Noack P, Breder J, Krug M, Reymann KG and Reiser
G: Four different types of protease-activated receptors are widely
expressed in the brain and are up-regulated in hippocampus by
severe ischemia. Eur J Neurosci. 14:595–608. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen Y, Yang C and Wang ZJ:
Proteinase-activated receptor 2 sensitizes transient receptor
potential vanilloid 1, transient receptor potential vanilloid 4 and
transient receptor potential ankyrin 1 in paclitaxel-induced
neuropathic pain. Neuroscience. 193:440–451. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fan Y, Chen J, Ye J, Yan H and Cai Y:
Proteinase-activated receptor 2 modulates corticotropin releasing
hormone-induced brain-derived neurotrophic factor release from
microglial cells. Cell Biol Int. 38:92–96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Spicarova D, Nerandzic V and Palecek J:
Update on the role of spinal cord TRPV1 receptors in pain
modulation. Physiol Res. 63 Suppl 1:S225–S236. 2014.PubMed/NCBI
|
|
26
|
Spicarova D, Adamek P, Kalynovska N,
Mrozkova P and Palecek J: TRPV1 receptor inhibition decreases
CCL2-induced hyperalgesia. Neuropharmacology. 81:75–84. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Uchytilova E, Spicarova D and Palecek J:
TRPV1 antagonist attenuates postoperative hypersensitivity by
central and peripheral mechanisms. Mol Pain. 10:672014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lam DK, Dang D, Zhang J, Dolan JC and
Schmidt BL: Novel animal models of acute and chronic cancer pain: a
pivotal role for PAR2. J Neurosci. 32:14178–14183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu S, Liu YP, Yue DM and Liu GJ:
Protease-activated receptor 2 in dorsal root ganglion contributes
to peripheral sensitization of bone cancer pain. Eur J Pain.
18:326–337. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stephan AB, Shum EY, Hirsh S, Cygnar KD,
Reisert J and Zhao H: ANO2 is the cilial calcium-activated chloride
channel that may mediate olfactory amplification. Proc Natl Acad
Sci USA. 106:pp. 11776–11781. 2009; View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu B, Linley JE, Du X, Zhang X, Ooi L,
Zhang H and Gamper N: The acute nociceptive signals induced by
bradykinin in rat sensory neurons are mediated by inhibition of
M-type K+ channels and activation of Ca2+-activated Cl-channels. J
Clin Invest. 120:1240–1252. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deba F and Bessac BF: Anoctamin-1 Cl(−)
channels in nociception: Activation by an N-aroylaminothiazole and
capsaicin and inhibition by T16A (inh)-A01. Mol Pain. 11:552015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ferrera L, Caputo A and Galietta LJ:
TMEM16A protein: A new identity for Ca(2+)-dependent Cl(−)
channels. Physiology (Bethesda). 25:357–363. 2010.PubMed/NCBI
|
|
34
|
Bulley S, Neeb ZP, Burris SK, Bannister
JP, Thomas-Gatewood CM, Jangsangthong W and Jaggar JH: TMEM16A
channels contribute to myogenic constriction in cerebral arteries.
Circ Res. 111:1027–1036. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chun H, Cho H, Choi J, Lee J, Kim SM, Kim
H and Oh U: Protons inhibit anoctamin 1 by competing with calcium.
Cell Calcium. 58:431–441. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sones WR, Davis AJ, Leblanc N and
Greenwood IA: Cholesterol depletion alters amplitude and
pharmacology of vascular calcium-activated chloride channels.
Cardiovasc Res. 87:476–484. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cipriani G, Serboiu CS, Gherghiceanu M,
Faussone-Pellegrini MS and Vannucchi MG: NK receptors, Substance
PAno1 expression and ultrastructural features of the muscle coat in
Cav-1(−/-) mouse ileum. J Cell Mol Med. 15:2411–2420. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tian Y, Kongsuphol P, Hug M, Ousingsawat
J, Witzgall R, Schreiber R and Kunzelmann K: Calmodulin-dependent
activation of the epithelial calcium-dependent chloride channel
TMEM16A. FASEB J. 25:1058–1068. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jung J, Nam JH, Park HW, Oh U, Yoon JH and
Lee MG: Dynamic modulation of ANO1/TMEM16A HCO3(−) permeability by
Ca2+/calmodulin. Proc Natl Acad Sci USA. 110:pp. 360–365. 2013;
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vocke K, Dauner K, Hahn A, Ulbrich A,
Broecker J, Keller S, Frings S and Möhrlen F: Calmodulin-dependent
activation and inactivation of anoctamin calcium-gated chloride
channels. J Gen Physiol. 142:381–404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tian Y, Schreiber R, Wanitchakool P,
Kongsuphol P, Sousa M, Uliyakina I, Palma M, Faria D,
Traynor-Kaplan AE, Fragata JI, et al: Control of TMEM16A by
INO-4995 and other inositolphosphates. Br J Pharmacol. 168:253–265.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pritchard HA, Leblanc N, Albert AP and
Greenwood IA: Inhibitory role of phosphatidylinositol
4,5-bisphosphate on TMEM16A-encoded calcium-activated chloride
channels in rat pulmonary artery. Br J Pharmacol. 171:4311–4321.
2014. View Article : Google Scholar : PubMed/NCBI
|