Introduction
Pulmonary arterial hypertension (PAH) is a condition
of varying etiologies that results in an abnormally elevated mean
pulmonary arterial pressure (PAP). PAH is characterized by multiple
abnormalities, including endothelial cell proliferation,
dysfunction of the peripheral pulmonary arterioles, thrombotic
obliteration of the vascular lumen, abnormal vasomotor control and
chronic remodeling of the vascular wall. In untreated cases, PAH
can lead to severe right heart dysfunction and mortality (1). Although the availability of multiple
therapeutic agents has improved the long-term survival for patients
with PAH, the 5-year survival rate remains low (2).
Hydroxymethylglutaryl coenzyme A reductase
inhibitors, or statins, are widely used for the treatment of
hypercholesterolemia. Previous experiments and clinical trials have
indicated that statins also exert a beneficial impact on PAH beyond
their lipid-lowering effects, and may form the basis of potential
future therapeutic strategies (3–5). The
fungal-derived statin simvastatin can suppress the abnormal
proliferation of pulmonary vascular smooth muscle cells, and induce
the apoptosis of pathological vascular smooth muscle cells in a
pneumonectomized and monocrotaline injured PAH model (6). Furthermore, it was reported that
statins can exert protective effects against hypoxia-induced PAH
(7). However, the mechanisms
underlying the beneficial effects of statins have not yet been
elucidated. It is well recognized that the intermolecular
differences between statins can contribute to distinct additional
pharmacological actions in PAH (8,9).
Notably, a report by Satoh et al (10) indicated that the hydrophilic statin
pravastatin can contribute to the development of pulmonary
hypertension. The aim of the present study was to determine whether
the synthetic statin fluvastatin could suppress the progression of
experimental PAH. Caveolin-1 (cav-1) expression is depleted in the
plexiform lesions and muscularized pulmonary arterioles that occur
in patients with PAH (11), and
may represent a potential upstream regulatory pathway of PAH
development (12). Thus, potential
associations between cav-1 expression and fluvastatin were also
investigated in the PAH animal model.
Materials and methods
Animal and experimental design
A total of 136 male Wistar rats (Experimental Animal
Center of Hubei Province, Wuhan, China) (250–300 g, 12 weeks old)
were used in the present study. All experiment procedures were
approved by the Animal Use Committee of Wuhan University (Wuhan,
China) in accordance with the institutional guidelines that comply
with national and international regulations.
Rats were randomly assigned to groups and received
either a single subcutaneous injection of monocrotaline (MCT; 60
mg/kg; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany; MCT group,
n=66), or a single subcutaneous injection of MCT (60 mg/kg)
followed by oral gavage of fluvastatin (MedChem Express, Monmouth
Junction, NJ, USA) with 10 mg/kg once daily until day 42 (M + F
group, n=42). MCT was prepared as previously described (12). Rats in the MCT group received an
equivalent volume of saline following the MCT injection. An
additional group of rats were given the equivalent volume of saline
as a control measure (saline group, n=28). Rats were housed in a
controlled environment under a 12-h light/dark cycle, with free
access to standard rat chow and water at a controlled temperature
of 20–22°C for 2 weeks prior to and throughout the experimental
period. Morphometric changes, PAP, percent wall thickness (PWT),
right ventricular hypertrophy index (RVHI) and cav-1 protein
expression levels were measured at 0, 2, 4 and 6 week time
intervals.
Hemodynamic measurement
At 0, 2, 4 and 6 week time intervals, rats were
anesthetized with an intraperitoneal injection of sodium
pentobarbital (60 mg/kg). Once stable anesthesia was obtained, a
skin incision was made on the neck to expose the trachea for
cannulation to a rodent ventilator. Rats were ventilated with room
air at a respiratory frequency of 70 breaths/min. Right ventricular
systolic pressure (RVSP) was investigated using the tip of an
intravenous trocar, which was pierced into the right ventricle. The
opposite end of the trocar was connected to a pressure sensor to
determine the RVSP (BL-420F; Chengdu Techman Software Co., Ltd.,
Chengdu, China). The left femoral artery was isolated and
cannulated to a polyethylene catheter to record the systemic
arterial pressure (SAP). Blood was sampled for hematocrit (HCT)
measurements prior to the aforementioned recording.
Morphometric analysis
Following hemodynamic measurements, rats were
euthanized by exsanguination, and the heart and lungs were removed
en bloc. The heart was dissected and any excess blood was
dried. The right ventricle (RV) free wall was separated from the
left ventricle and septum (LV+S) to determine the wet weight of
each section separately. RVHI was assessed by dividing the ratio of
the RV by the sum of the septum plus LV weight (g). The right lower
lobe of the lung was subsequently isolated, placed in liquid
nitrogen and stored at −80°C until use in western blot analysis.
The presence of pulmonary edema was evaluated by measuring the
ratio of dry/wet lung weight as described previously (13).
Histological examination of the
lung
A single lobe was excised from the lungs and fixed
by perfusion with 4% (weight/volume) paraformaldehyde through a
tracheal catheter at a transpulmonary pressure of 20 cm
H2O for 30 min. Following paraffin embedding, lungs were
sectioned into 5 µm thick sections and subjected to staining using
0.2% hematoxylin for 5 min at room temperature and 0.5% eosin for 3
min at room temperature. Finally, 3 fields of view were analyzed
and photographs were taken using a light microscope (Olympus
Corporation, Tokyo, Japan; magnification, ×20). Pulmonary arterial
area (diameter, 100–150 µm) was measured. To assess the degree of
pulmonary vascular remodeling, the PWT formula was applied
(14): PWT (%)=[(external
area-internal area)/external area] ×100, in which the external and
internal areas of the pulmonary arterioles are the areas bound by
the external elastic lamina and lumen, respectively.
Cav-1 western blot analysis
Pulmonary expression of cav-1 was detected by
western blot analysis. Lung samples from each group were
homogenized in homogenization buffer (50 mM hydroxyethyl
piperazine-ethanesulfonic acid, pH 7.55; 10% glycerol; 0.1% Triton
X-100 and 1 mM dithiothreitol). Lysates were subsequently
centrifuged at 7,000 × g for 10 min in order to remove the
insoluble debris. Protein concentration in the supernatant was
determined using a Bradford assay. Equal quantities of protein (20
µg) were separated by 12% SDS-PAGE and transferred onto
nitrocellulose membranes. The membranes were incubated at room
temperature for 30 min in 5% bovine serum albumin (Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China) and
subsequently washed with TBST (0.15 M NaCl; 0.05% Tween-20; 20 mM
Tris-HCl, pH 8.0). They were then incubated with a rabbit
polyclonal anti-cav-1 primary antibody (cat. no. ab2910; 1:1,000;
Abcam, Cambridge, UK) and mouse monoclonal β-actin (cat. no.
BM0627; 1:2,000; Wuhan Boster Biological Technology Ltd., Wuhan,
China), at room temperature for 1 h. Following this, membranes were
incubated with horseradish peroxidase-conjugated secondary
antibodies (cat. nos. BA1054 and BA1058; 1:2,000; Wuhan Boster
Biological Technology Ltd.) for 1 h at room temperature and the
antibody complexes were detected using enhanced chemiluminescence
reagents (GE Healthcare, Chicago, IL, USA). An image analysis
device (FAS-100; Toyobo Life Science, Osaka, Japan) was used to
detect cav-1 immunoreactivity. The relative levels of proteins were
then quantified using Image J 1.46 software (National Institutes of
Health, Bethesda, MD, USA). The blots were stripped (cat. no.
PS107; Epizyme, Inc., Cambridge, MA, USA) re-blotted for β-actin as
a control and to ensure that equal amounts of proteins were
loaded.
Reverse
transcription-semi-quantitative polymerase chain reaction
(sqPCR)
Total RNA was extracted from lung tissue using the
TRIzol reagent (Invitrogen; Thermo Fisher Scientific Inc., Waltham,
MA, USA) according to the manufacturer's instructions and
quantified using an ultraviolet spectrophotometer. RNA samples were
reverse transcribed into cDNA using a reverse transcriptase kit
(Takara Biotechnology Co., Ltd., Dalian, China). The cav-1 primer
sequences were as follows: Forward, 5′-CAGTTGAGCGCCCACGCCAG-3′ and
reverse, 5′-GCGGCCTTCACCATCTTCTT-3′. The β-actin primer sequences
were as follows: Forward, 5′-ATCCTGTTTCCGACCTTCAACA-3′ and reverse,
5′-CATCTCTTCCACGAAGAGCA-3′ The reaction began with denaturation for
1 min at 94°C, followed by 30 cycles of replication (20 sec at 98°C
and 10 min at 68°C) and a final extension at 72°C for 10 min using
a GeneAmp PCR System 2400 (PerkinElmer, Inc., Waltham, MA, USA).
The PCR products of cav-1 and β-actin mRNA were subsequently
electrophoresed through a 2% agarose gel and stained with ethidium
bromide (5 mg/l). The photograph of the ethidium bromide staining
gel was taken using Gel-Doc 2000 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The expression of cav-1 mRNA was represented by
the relative yield compared with β-actin mRNA. Semi-quantitative
analyses were quantified by using Image J software (National
Institutes of Health).
Survival analysis
A total of 36 rats from the M + F (n=15), MCT (n=15)
and saline (n=6) groups were followed for 6 weeks to assess
survival rates. The day of MCT injection was defined as day 0.
Statistical analysis
Data are presented as mean ± standard error of mean.
Student's t-test was used for comparison between 2 groups. For
multiple group comparisons, analysis of variance was performed
followed by the Fisher's least significant difference post hoc
test. Survival analysis was performed using the Kaplan-Meier method
with a log-rank test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effects of fluvastatin on the
development of MCT-induced PAH
Rats had a significantly elevated RVSP (19.3±2.5
mmHg), RVHI (0.29±0.01) and PWT (48.7±2.31) following 2 weeks of
MCT treatment in the MCT group. However, M + F rats did not have a
significant increase in the aforementioned parameters when compared
with those of saline treated rats following 2 weeks of treatment
(RVSP, 16.3±2.5 mmHg; RVHI, 0.27±0.02 and PWT, 33.7±3.2%). PAH and
right ventricular hypertrophy progressed further in the MCT group
in the following 4 weeks. As presented in Fig. 1, MCT rats exhibited a further
increase in RVSP (MCT, 30.0±5.0 mmHg; saline, 14.6±1.1; P<0.01),
the RV/(LV+S) ratio (MCT, 0.34±0.03; saline, 0.27±0.01; P<0.01)
and PWT (MCT, 68.0±7.0%; saline, 34.3±3.2%; P<0.001) in week 6.
M + F rats had a significant elevation in RVSP and RVHI at week 6
when compared with the saline group. No differences in PWT were
observed between M + F and the saline rats throughout the 6 week
observation period (M + F, 37.7±3.2; saline, 32.3±2.3; P>0.05).
Significantly lower values of RVSP (M + F, 22.3±3.5 mmHg; MCT,
30.0±5.0 mmHg; P<0.01) and PWT (M + F, 40.3±3.5% MCT, 68.0±7.0%;
P<0.05) were observed in the M + F group compared with the MCT
group at 6 weeks (Fig. 1).
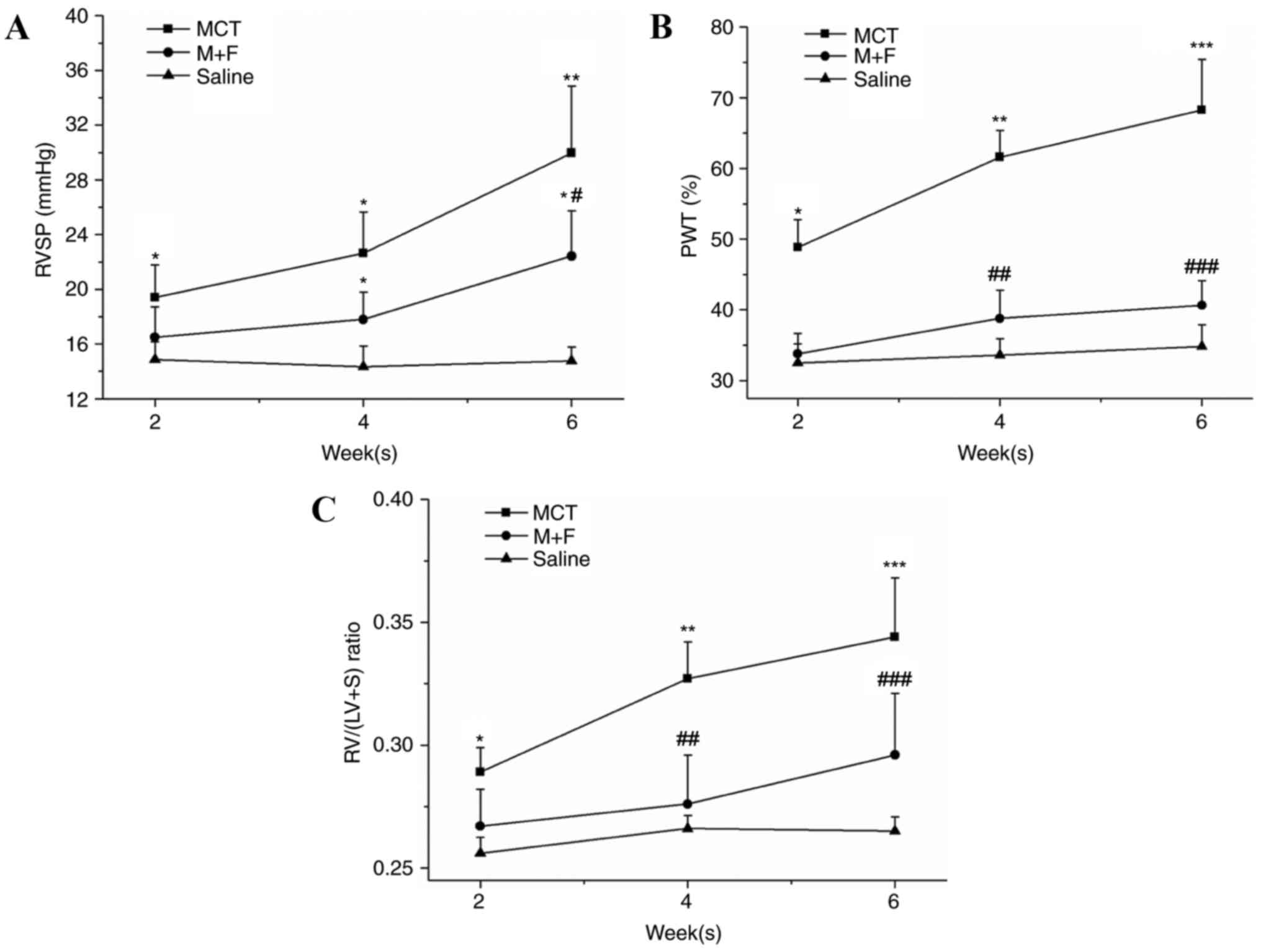 | Figure 1.(A) RVSP, (B) PWT and (C) RV/(LV+S)
weight ratio were examined every 2 weeks in the saline, M + F and
MCT groups, respectively. Data are presented as the mean ± standard
deviation of five measurements at various times. *P<0.05,
**P<0.01 and ***P<0.001, vs. saline group;
#P<0.05, ##P<0.01 and
###P<0.001 vs. MCT group. RVSP, right ventricular
systolic pressure; PWT, percent wall thickness; RV/(LV+S), right
ventricle (left ventricle + septum); MCT, monocrotaline; M + F, MCT
+ fluvastatin. |
Effects of fluvastatin on PAH
associated variables
PAH associated variables were measured to observe
the progression of MCT-induced PAH at 6 weeks (Table I). MCT rats developed a
significantly reduced HCT percentage and an increased lung weight
in comparison with the saline-treated rats (wet and dry). However,
heart rate, SAP and dry/wet lung weight ratio were not
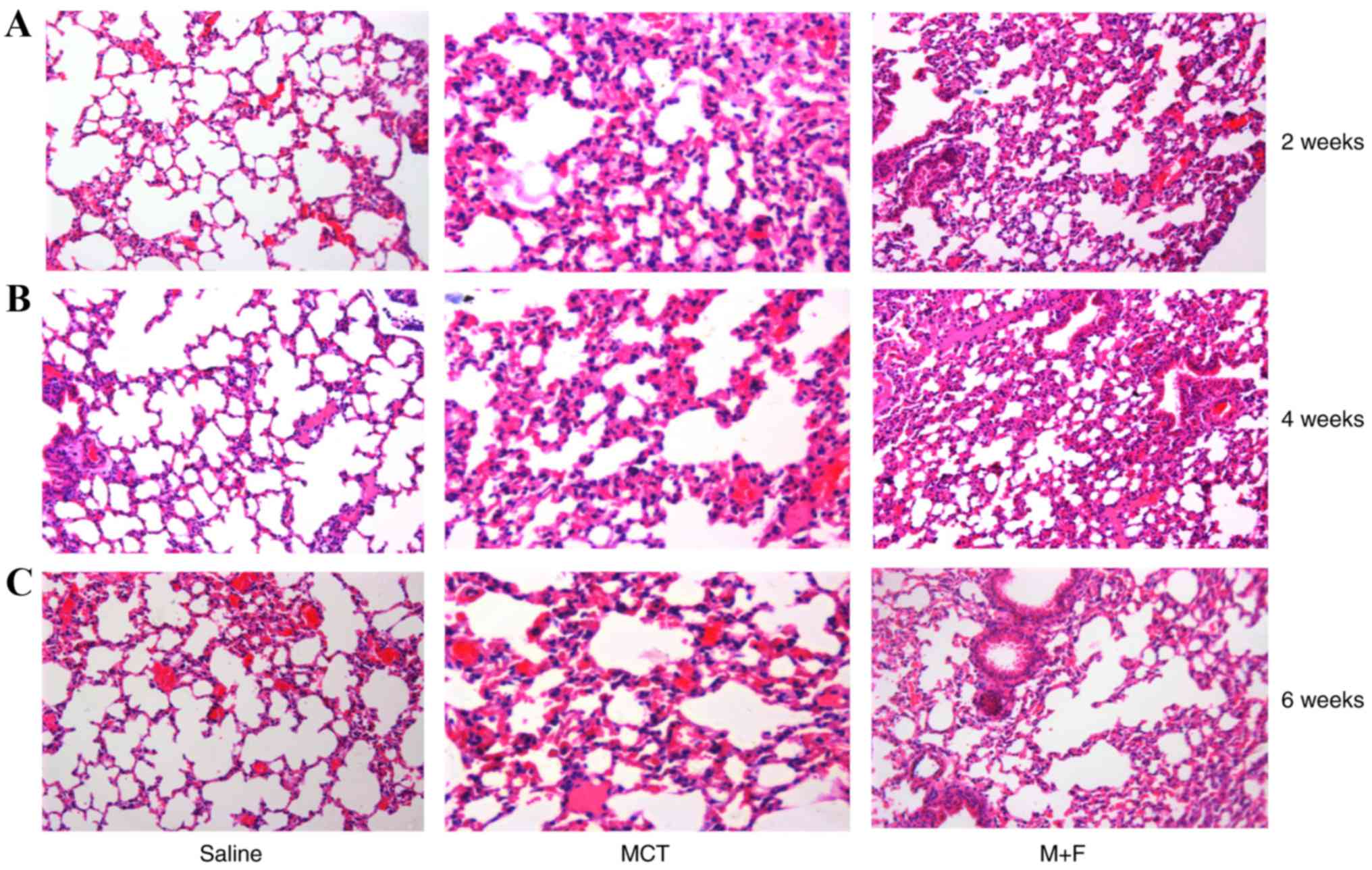
significantly different between the groups. In comparison with
saline treated rats, histological examination indicated marked
pulmonary interstitial hyperplasia and inflammatory cell
infiltration in the MCT group (Fig.
2), with a notably thickened alveolar duct and alveolar wall at
6 weeks following MCT injection (Fig.
2C). M + F rats also developed a moderate thickening of the
alveolar septum (Fig. 2).
 | Table I.Effects of fluvastatin on pulmonary
arterial hypertension associated variables in rats treated with
saline, M + F and MCT. |
Table I.
Effects of fluvastatin on pulmonary
arterial hypertension associated variables in rats treated with
saline, M + F and MCT.
| Group | Saline | M + F | MCT |
|---|
| N number of rats | 5 | 6 | 6 |
| HR (beats/min) | 271±5 | 269±9 | 275±4 |
| SAP (mmHg) | 82.3±5.8 | 84.2±6.7 | 85.3±9.2 |
| HCT (%) | 48±3 | 45±3 | 40±2a |
| DLW (g) | 11.7±1.8 | 13.5±2.3c | 17.2±2.6a |
| WLW (g) | 55.6±3.6 | 64.4±4.7a,c | 84.7±7.8b |
| DLW/WLW (%) | 21.3±2.3 | 20.9±3.5 | 21.2±3.1 |
Effect of fluvastatin treatment on
cav-1 mRNA and protein expression
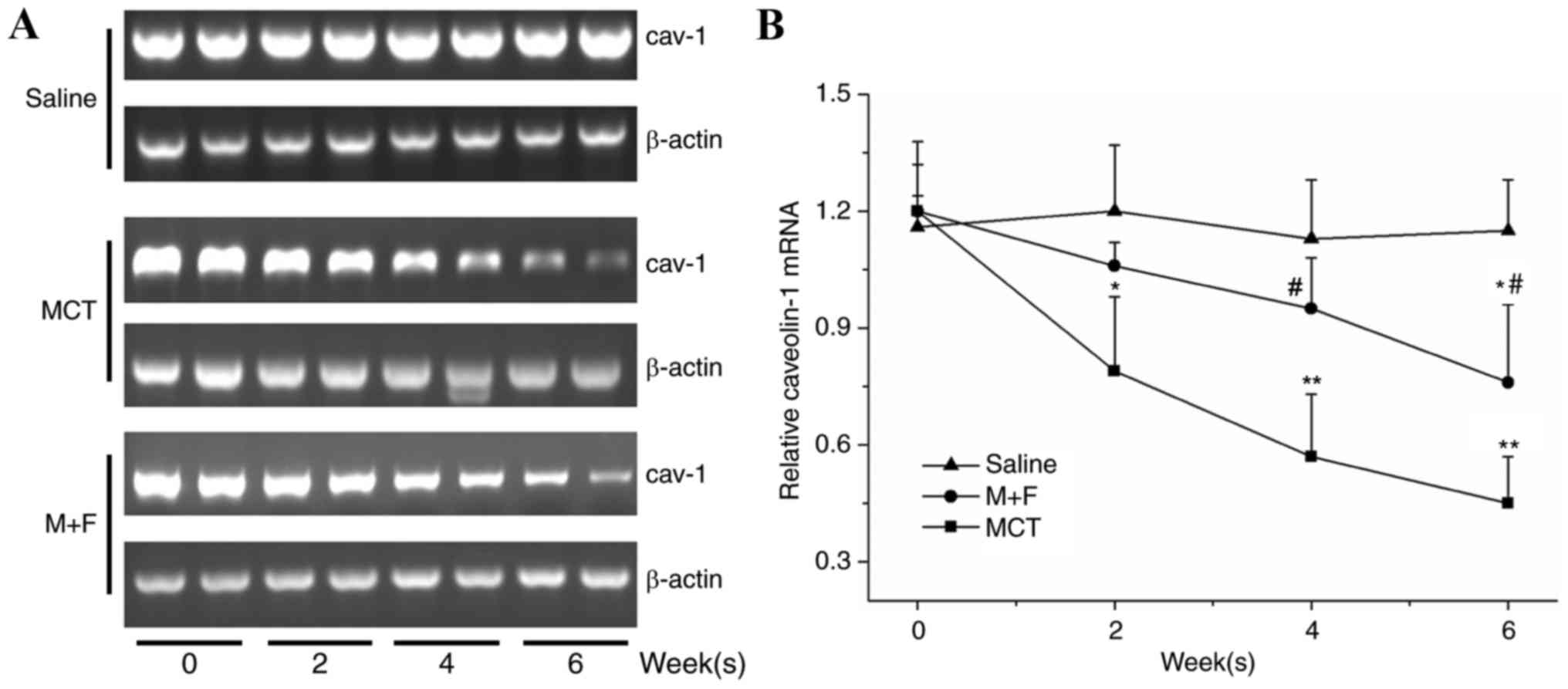
The relative expression of cav-1 mRNA in saline
treated rats at day 0 was 1.17±0.15 and no significant changes
developed throughout the experimental period. MCT-injected rats
exhibited significantly reduced cav-1 mRNA expression at the 2, 4
and 6 week time intervals compared with the saline group. In the M
+ F group, a significant downregulation of cav-1 mRNA expression
was observed at 4 and 6 week time intervals when compared with the
saline group (P<0.05; Fig. 3).
Furthermore, the M + F group exhibited a significant increase in
cav-1 expression at 4 and 6 week time intervals compared with
MCT-injected rats (Fig. 3).
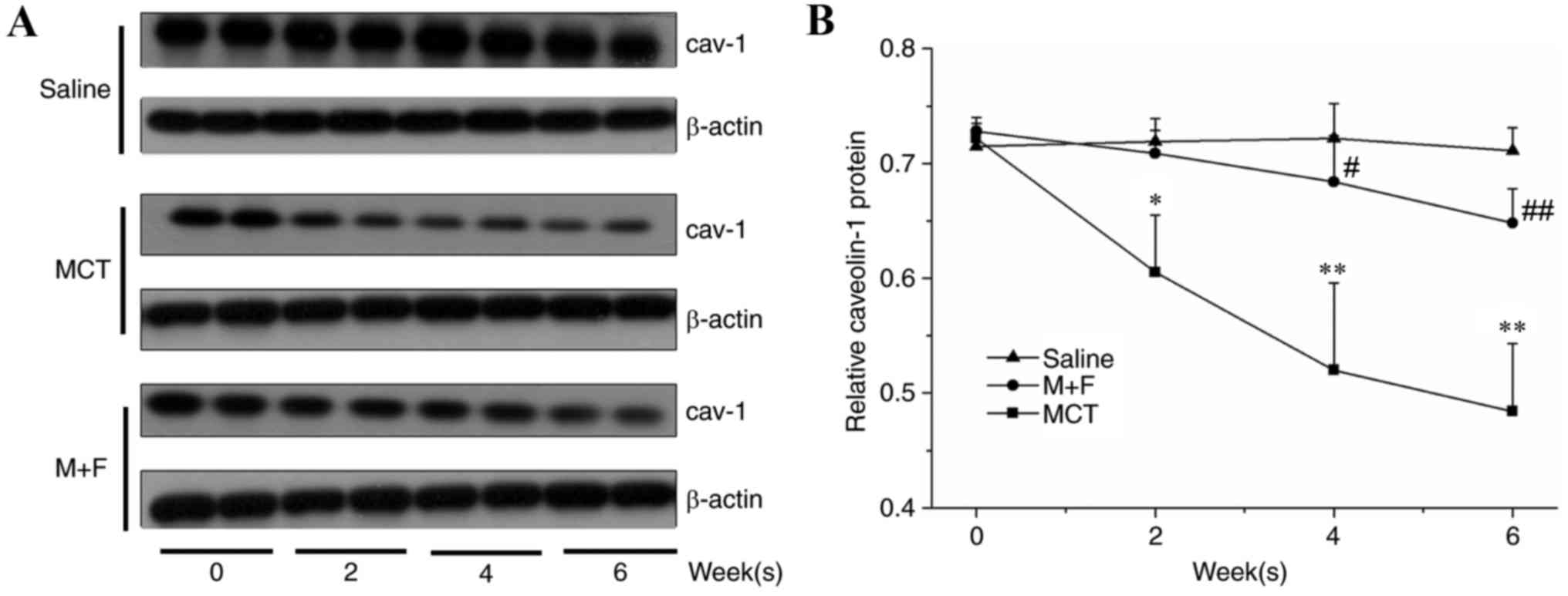
Cav-1 protein expression in the lung tissue was
significantly reduced in the MCT group at the 2, 4 and 6 week time
intervals (Fig. 4). By contrast,
the M + F group did not exhibit a significant reduction in cav-1
protein expression within 6 weeks compared with the saline group;
however, the M + F group exhibited significantly enhanced cav-1
protein expression at 4 and 6 week time intervals compared with
MCT-injected rats (Fig. 4).
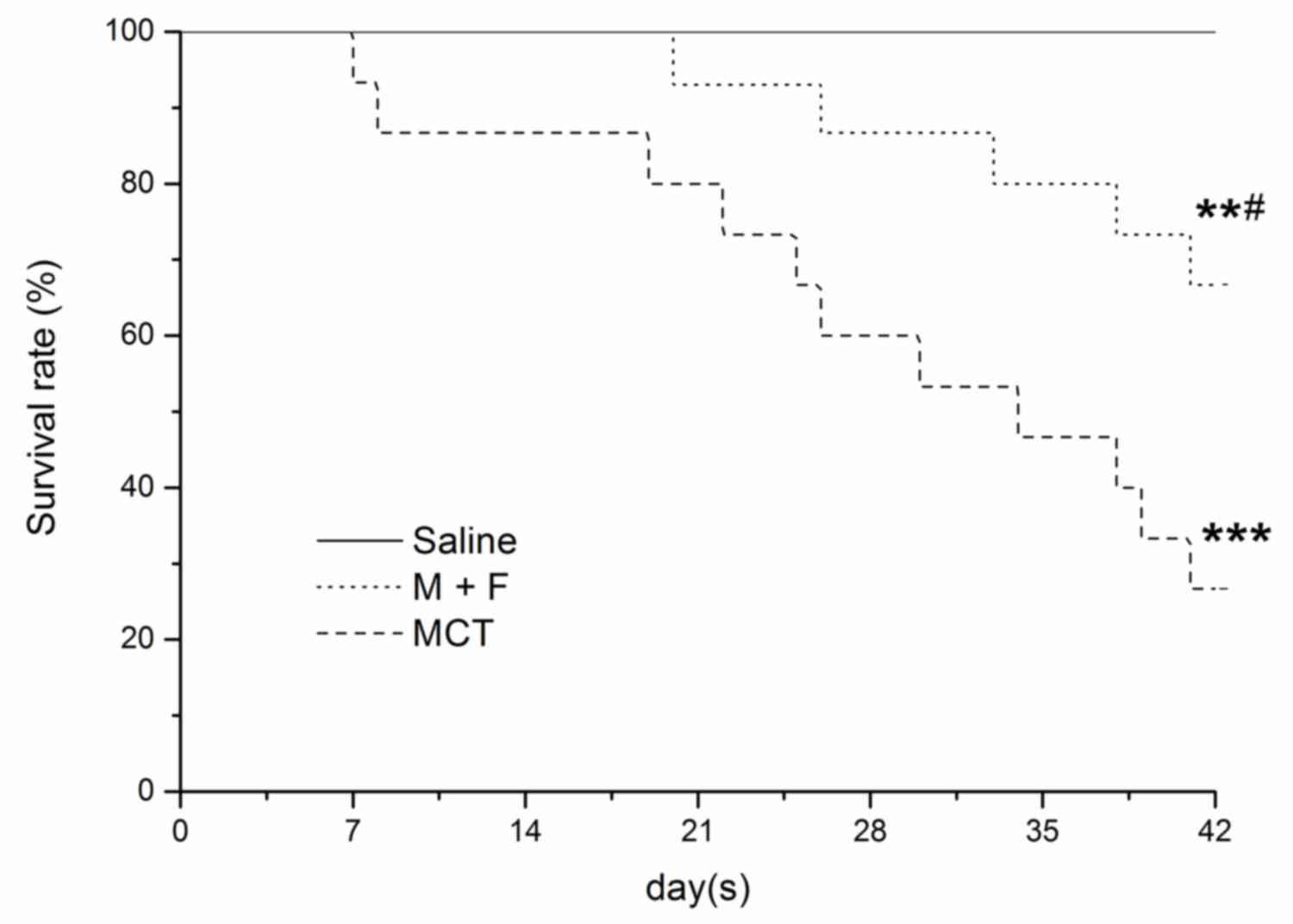
Survival analysis
Survival was measured from the date of MCT injection
until mortality or 6 weeks following the MCT injection. During the
6 week period, there were 15 mortalities in the 36 rats studied,
with 11 in the MCT group and 4 in the M + F group. The survival
rate at 6 weeks was significantly higher in the M + F group
compared with the MCT group (M + F, 66.7%; MCT, 26.7%; P<0.05;
Fig. 5). There were no mortalities
in the saline treated rats. All 15 mortalities were caused by
progressive right heart failure with ascites, and pericardial and
pleural effusions.
Discussion
Evidence from a variety of experimental and clinical
studies has demonstrated that statins can lower serum cholesterol
and also exert additional beneficial effects (15,16).
Simvastatin has been reported to attenuate PAH through the
regulation of the bone morphogenetic protein receptor type-2
(BMPR-2) signaling pathway in pulmonary artery endothelial cells
(17,18). Simvastatin may also exert
protective effects on endothelial nitric oxide synthase activity
(7). Previous studies have also
indicated that statins exert beneficial effects in PAH (19,20)
forming the basis for a potential future therapeutic strategy
against PAH in humans. Previous in vitro and in vivo
studies concerning statins and PAH have been largely focused on
potential therapeutics against established lesions in PAH (10,21,22).
The present study demonstrated that fluvastatin can reduce PAH
development and ameliorate MCT-induced inhibition of cav-1
expression in rats.
MCT-induced PAH is an artificial model of pulmonary
hypertension that is acknowledged to satisfactorily reproduce the
processes occurring secondary to the dysfunction of the pulmonary
arteries (23). In the present
study, a daily gavage of 10 mg/kg fluvastatin was administered to M
+ F rats immediately following a single subcutaneous MCT injection,
at which time PAH was not yet established. PAH evaluation was
performed every 2 weeks during the experimental period to reduce
the number of experimental animals required. Although it could not
be concluded that M + F rats were completely protected from the
development of PAH, development was delayed ≥2 weeks, as indicated
by the results of hemodynamic and morphometric analysis. Pulmonary
vascular remodeling is a hallmark pathological feature of PAH
(24); PWT can be measured to
evaluate the extent of this remodeling. In the present study, no
significant changes in PWT were observed in M + F rats at 6 weeks,
indicating that early fluvastatin administration may exert
effective protection against pulmonary vascular remodeling; this
may contribute to the reduced PAH development observed in M + F
rats.
Cav-1 is a major component of the coat proteins that
surround caveolae, which are flask-shaped plasma membrane
invaginations that have important functions in vesicular transport
and signal transduction (25).
Knockout mice develop severe lung abnormalities characterized by
hypercellularity, interstitial fibrosis and alveolar septa
thickening, indicating that cav-1 may be involved in the
development of normal lung vasculature (26). In the present study, pulmonary
cav-1 protein expression was reduced in MCT rats, which is in
accordance with other reported data (11,27).
By contrast, significant differences in cav-1 protein expression in
the lungs of M + F rats were not observed throughout the
experimental period. The difference in cav-1 expression between the
MCT and M + F groups was largely attributed to the 6 week
fluvastatin intervention. However, a significant downregulation of
cav-1 mRNA expression in the M + F group was observed at 6 weeks
when compared with the saline group, indicating that fluvastatin
confers only partial protection to cav-1 expression. A complete
inhibition of cav-1 reduction in the M + F group within 6 weeks did
not result in an expected complete inhibition of PAH development.
RVSP and RVHI were moderately elevated, suggesting cav-1 was not
the determining factor in this animal model. Survival analysis also
revealed a significantly increased survival benefit in fluvastatin
treated rats. Taken together, these results suggest that
fluvastatin is effective in slowing PAH progression in an
MCT-induced model, at least in part through the protection of cav-1
expression in rats.
Patients with congenital heart disease, BMPR-2
mutations or sickle cell anemia are particularly vulnerable to PAH
development (28,29). Thus, the findings of the present
study may be of clinical interest in respect to the development of
therapeutic strategies aimed at the prevention of PAH in at-risk
individuals. Furthermore, future research should explore the
potential beneficial effects of other statins in models of PAH, as
only fluvastatin was investigated in the present study.
There are several limitations to the findings of the
present study. Interactions between fluvastatin and MCT were not
investigated, which may potentially influence the successful
establishment of the PAH animal model. Additionally, cav-1
expression cannot be concluded to be the main contributor to PAH
development, as the expression of cav-2, cav-3 and other
potentially involved proteins were not evaluated. Furthermore, as
the established animal model may not accurately reproduce the
pathological modifications present in various forms of PAH in
humans, the findings of this pre-clinical study require
confirmation in clinical trials.
Acknowledgements
The present study was supported by The Science and
Development Research Institute of Wuhan University (grant no.
303274009).
References
|
1
|
Kanemoto N: Natural history of primary
pulmonary hypertension elucidated by pulmonary hemodynamics. J
Cardiol. 18:757–763. 1988.(In Japanese). PubMed/NCBI
|
|
2
|
Larsen CM, McCully RB, Murphy JG, Kushwaha
SS, Frantz RP and Kane GC: Usefulness of high-density lipoprotein
cholesterol to predict survival in pulmonary arterial hypertension.
Am J Cardiol. 118:292–297. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen D, Zhou D, Qian J, Chen F, Guan L,
Dong L and Ge J: Atorvastatin prevents dehydromonocrotaline-induced
pulmonary hypertension in beagles. Exp Lung Res. 38:333–343. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maron DJ, Fazio S and Linton MF: Current
perspectives on statins. Circulation. 101:207–213. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li XL, Guan RJ and Li JJ: Attenuation of
monocrotaline-induced pulmonary arterial hypertension in rats by
rosuvastatin. J Cardiovasc Pharmacol. 60:219–226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nishimura T, Vaszar LT, Faul JL, Zhao G,
Berry GJ, Shi L, Qiu D, Benson G, Pearl RG and Kao PN: Simvastatin
rescues rats from fatal pulmonary hypertension by inducing
apoptosis of neointimal smooth muscle cells. Circulation.
108:1640–1645. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Murata T, Kinoshita K, Hori M, Kuwahara M,
Tsubone H, Karaki H and Ozaki H: Statin protects endothelial nitric
oxide synthase activity in hypoxia-induced pulmonary hypertension.
Arterioscler Thromb Vasc Biol. 25:2335–2342. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mason RP, Walter MF, Day CA and Jacob RF:
Intermolecular differences of 3-hydroxy-3-methylglutaryl coenzyme a
reductase inhibitors contribute to distinct pharmacologic and
pleiotropic actions. Am J Cardiol. 96:11F–23F. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rakotoniaina Z, Guerard P, Lirussi F,
Goirand F, Rochette L, Dumas M and Bardou M: The protective effect
of HMG-CoA reductase inhibitors against monocrotaline-induced
pulmonary hypertension in the rat might not be a class effect:
Comparison of pravastatin and atorvastatin. Naunyn Schmiedebergs
Arch Pharmacol. 374:195–206. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Satoh K, Fukumoto Y, Nakano M, Sugimura K,
Nawata J, Demachi J, Karibe A, Kagaya Y, Ishii N, Sugamura K and
Shimokawa H: Statin ameliorates hypoxia-induced pulmonary
hypertension associated with down-regulated stromal cell-derived
factor-1. Cardiovasc Res. 81:226–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Achcar RO, Demura Y, Rai PR,
Taraseviciene-Stewart L, Kasper M, Voelkel NF and Cool CD: Loss of
caveolin and heme oxygenase expression in severe pulmonary
hypertension. Chest. 129:696–705. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chan SY and Loscalzo J: Pathogenic
mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol.
44:14–30. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang L, Zhao B, Chen Y, Ma L, Chen EZ and
Mao EQ: Inflammation and edema in the lung and kidney of
hemorrhagic shock rats are alleviated by biliary tract external
drainage via the heme oxygenase-1 pathway. Inflammation.
38:2242–2251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zuckerbraun BS, Chin BY, Wegiel B, Billiar
TR, Czsimadia E, Rao J, Shimoda L, Ifedigbo E, Kanno S and
Otterbein LE: Carbon monoxide reverses established pulmonary
hypertension. J Exp Med. 203:2109–2119. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Adam O and Laufs U: Antioxidative effects
of statins. Arch Toxicol. 82:885–892. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Steinberg D: The statins in preventive
cardiology. N Engl J Med. 359:1426–1427. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nishimura T, Faul JL, Berry GJ, Vaszar LT,
Qiu D, Pearl RG and Kao PN: Simvastatin attenuates smooth muscle
neointimal proliferation and pulmonary hypertension in rats. Am J
Respir Crit Care Med. 166:1403–1408. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu H, Sung A, Zhao G, Shi L, Qiu D,
Nishimura T and Kao PN: Simvastatin enhances bone morphogenetic
protein receptor type II expression. Biochem Biophys Res Commun.
339:59–64. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kao PN: Simvastatin treatment of pulmonary
hypertension: An observational case series. Chest. 127:1446–1452.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pei Y, Ma P, Wang X, Zhang W, Zhang X,
Zheng P, Yan L, Xu Q and Dai G: Rosuvastatin attenuates
monocrotaline-induced pulmonary hypertension via regulation of
Akt/eNOS signaling and asymmetric dimethylarginine metabolism. Eur
J Pharmacol. 666:165–172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yao J, Xiong M, Tang B, Chen G, Liang M,
Ma X, Wang Z and Wu Z: Simvastatin attenuates pulmonary vascular
remodelling by down-regulating matrix metalloproteinase-1 and −9
expression in a carotid artery-jugular vein shunt pulmonary
hypertension model in rats. Eur J Cardiothorac Surg. 42:e121–e127.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carlin CM, Celnik DF, Pak O, Wadsworth R,
Peacock AJ and Welsh DJ: Low-dose fluvastatin reverses the hypoxic
pulmonary adventitial fibroblast phenotype in experimental
pulmonary hypertension. Am J Respir Cell Mol Biol. 47:140–148.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Campian ME, Hardziyenka M, Michel MC and
Tan HL: How valid are animal models to evaluate treatments for
pulmonary hypertension? Naunyn Schmiedebergs Arch Pharmacol.
373:391–400. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jeffery TK and Morrell NW: Molecular and
cellular basis of pulmonary vascular remodeling in pulmonary
hypertension. Prog Cardiovasc Dis. 45:173–202. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
van Deurs B, Roepstorff K, Hommelgaard AM
and Sandvig K: Caveolae: Anchored, multifunctional platforms in the
lipid ocean. Trends Cell Biol. 13:92–100. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Razani B, Engelman JA, Wang XB, Schubert
W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, et
al: Caveolin-1 null mice are viable but show evidence of
hyperproliferative and vascular abnormalities. J Biol Chem.
276:38121–38138. 2001.PubMed/NCBI
|
|
27
|
Mathew R, Huang J, Shah M, Patel K, Gewitz
M and Sehgal PB: Disruption of endothelial-cell
caveolin-1alpha/raft scaffolding during development of
monocrotaline-induced pulmonary hypertension. Circulation.
110:1499–1506. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bouzas B and Gatzoulis MA: Pulmonary
arterial hypertension in adults with congenital heart disease. Rev
Esp Cardiol. 58:465–469. 2005.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Machado RF: Sickle cell anemia-associated
pulmonary arterial hypertension. J Bras Pneumol. 33:583–591.
2007.(In English, Portuguese). View Article : Google Scholar : PubMed/NCBI
|