Introduction
Due to the outstanding performance with respect to
immune response and anti-inflammatory effect, glucocorticoids (GCs)
are routinely prescribed to treat autoimmune and noninfectious
inflammatory diseases. However, the long-term and excessive
administration of GCs can lead to many serious complications, such
as osteoporosis. GC-induced osteoporosis (GIOP), which is
considered to be the most common form of secondary osteoporosis
(1,2), increases the risk of fracture in
patients treated with GCs. Clinical studies have indicated that GCs
dysregulated bone metabolism and led to a rapid reduction in
trabecular bone mass within the first year of treatment (3,4).
However, this reduction in trabecular bone mass cannot entirely
explain the observed increase in fracture risk because the bone
mineral density (BMD) value of patients treated with GCs is higher
than those of women with postmenopausal osteoporosis (4). Therefore, it is necessary to more
comprehensively elucidate the pathology of GC-induced bone loss. A
better understanding of this process will help clinicians
effectively prevent and treat GIOP.
Osteoblasts, which are derived from mesenchymal
progenitor cells, are mainly responsible for bone formation and
development (5). Prior studies
have suggested that the administration of GCs induced the apoptosis
of osteoblasts and osteocytes, which was regarded as the main cause
of GIOP (6). Nevertheless, it is
well known that GCs are indispensable for normal bone metabolism
and development under physiological condition (7). A recent study demonstrated that
different dose of GCs led to different osteocyte fates (8). In addition, Shi et al
(9) concluded that GCs had
dose-related effect on osteoclast formation and function. Based on
these studies, GC dose is a crucial factor affecting the function
and viability of cells. Unfortunately, few studies have addressed
the relationship between osteoblast viability and GC dose,
particularly for low-GC dose.
Autophagy is a highly conserved intracellular
cleaning process in eukaryotic cells that is responsible for
removing damaged organelles and counteracting harmful stimuli
(10,11). Generally, physiological autophagy
exists in all cells to maintain normal cellular metabolism and
viability (12). Once cells
encounter stress stimuli, such as hypoxia (13), oxidative stress (14), or ER stress (15), autophagy is induced to prevent
cells from undergoing apoptosis by eliminating stress inducers.
Autophagy reportedly plays a protective role in maintaining
osteoblast viability by allowing cells to survive various stresses
(16,17).
Microtubule-associated protein light chain 3 (LC3)
and beclin 1 are important biological signal to identify autophagy
(18). In the process of
autophagy, cytoplasmic proteins, organelles and so on, are engulfed
by autophagosomes. LC3II is simultaneously formed by conjugating
LC3I with phosphatidylethanolamine, which is fixed on the membrane
of autophagosome. After the formation of autolysosome consisting of
lysosome and autophagosome, the cytoplasm in autophagosome is
degraded. Meanwhile, LC3II is also degraded in the autolysosome.
Therefore, the activity of autophagy could be characterized by
detecting activity of LC3II (18,19).
Beclin 1 is also an essential protein for the formation of
autophagosome. It can guide other autophagy-related proteins to
move into vacuoles, and thus regulates the formation of
autophagosome (20).
It is well-known that GCs can interrupt
mitochondrial function and thereby increase reactive oxygen species
(ROS) in many cells (21,22). Generally, ROS is produced by
mitochondria during cellular metabolism. However, ROS level is
significantly increased when cells are exposed to noxious stimuli,
such as cytotoxic drugs. At normal level, ROS plays a crucial role
in the energy cycling of cells; however, excessive ROS induces cell
apoptosis, even death, which are closely associated with many
diseases (23,24). Certain studies have indicated that
reduced bone formation and decreased BMD were induced by increased
oxidative stress in aged humans and mice (25,26).
Moreover, a recent study demonstrated that the GC-induced
upregulation of ROS could lead to osteoporosis by inducing
osteoblast apoptosis (2). However,
studies have claimed that ROS, as a source of oxidative stress,
also contributed to the initiation of autophagy to prevent
osteoblasts from undergoing apoptosis (17). We hypothesize that the biphasic
effect of ROS on osteoblast viability may be attributable to the
difference in ROS level, which is associated with GC dose. In other
words, GC dose determine osteoblast fate by modulating
intracellular ROS levels. However, this hypothesis must be verified
by additional research.
The primary aim of this study is to investigate the
effect of GC dose on the viability of osteoblasts, assess whether
autophagy is involved in this process, and examine the relationship
between ROS and autophagy.
Materials and methods
Reagents and antibodies
Anti-LC3 and anti-beclin 1 antibodies were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA). Rapamycin
(Rap, autophagy agonists), 3-methyladenine (3-MA, autophagy
inhibitor) were purchased from Selleck Chemicals (Houston, TX,
USA). Dexamethasone (DEX) was purchased from Sigma-Aldrich; Merck
KGaA (St. Louis, MO, USA). The ROS scavenger catalase was purchased
from Beyotime Institute of Biotechnology (Nantong, China).
Cell culture and treatment
The human fetal osteoblastic cell line hFOB 1.19
cells were kindly provided by Dr. M. Subramaniam. The cells were
cultured in a 1:1 mixture of DMEM/F-12 supplemented with 10% fetal
bovine serum and 0.3 g/l G418 under 5% CO2 humidified
atmosphere at 33.5°C. After the cells reached a steady-state of
exponential growth in the media, they were exposed to variable
concentrations of DEX for specific time. Rap (3 µM), 3-MA (5 mM),
and catalase (500 U/ml) were added to cultures, respectively where
indicated.
Cell viability assay
The hFOB 1.19 cells were seeded into 96-well plates
(4×103 cells/well). When 60% confluence was achieved,
cells were treated with indicated treatments. Cell viability was
determined by 3-[4,5-dimethylthiazol-2-y]-2,5-diphenyltetrazolium
bromide (MTT; Keygen, Nanjing, China) assay following the
manufacturer's instructions.
Cell apoptosis analysis
After treated with indicated treatment for 24 h,
cells were collected and stained with the Annexin V-FITC Apoptosis
Detection kit (Keygen) according to manufacturer's instructions,
and the apoptosis was analyzed by the flow cytometer equipped with
Modfit LT 3.0 (BD Biosciences, Franklin Lakes, NJ, USA). Different
subpopulation was distinguished using the following criteria: Q3,
viable cells (TITC−/PI−); Q4, early apoptotic
cells (FITC+/PI−); Q2, late apoptotic cells
(FITC+/PI+); Q1, necrotic cells
(FITC−/PI+). The apoptotic rate was
determined as the percentage of Q2 + Q4.
Intracellular ROS measurement
The ROS level of osteoblasts was monitored using a
ROS detection kit (Beyotime Institute of Biotechnology). Cells in
6-well plates were incubated with variable concentration
(10−4, 10−6, 10−8 M) of DEX for 6
h. Cells were incubated with 10 mM of DCFH-DA at 37°C for 20 min,
and then washed with PBS. Intracellular ROS level was measured by
an inverted fluorescence microscope (IX71; Olympus, Tokyo, Japan).
All images were captured under the same conditions, and the
fluorescence was quantified by ImageJ software (National Institutes
of Health, Bethesda, MD, USA). Average fluorescence intensity was
calculated based on the data from the six fields of each treatment.
Three independent experiments were conducted.
Protein extraction and western blot
analysis
The treated hFOB 1.19 cells were collected and
lysed. The extract was centrifuged (20,000 × g, at 4°C for 20 min),
and the supernatant was collected to harvest total protein. The
protein concentration in the extract was determined using the BCA
protein assay (Wuhan Boster Biological Technology, Ltd., Wuhan,
China). An equal amount of protein was resolved using SDS-PAGE and
electroblotted onto a PVDF membrane (Millipore, Billerica, MA,
USA). The membrane was then incubated overnight at 4°C with primary
antibody against LC3 (1:1,000), beclin 1 (1:1,000) and GAPDH
(1:2,000), respectively. Next, the membrane was incubated with goat
anti-rabbit secondary antibody conjugated to horseradish peroxidase
(1:5,000; ZSGB-BIO, Beijing, China) for 1 h at room temperature.
The protein signal was enhanced using a chemiluminescene system
(DNR MF-ChemiBIS 3.2). The band density was quantified using the
ImageJ image processing program.
Immunofluorescence staining
For immunofluorescence detection, 8×103
hFOB 1.19 cells were seeded in a 24-well plate and treated with
indicated drug for 6 h. The hFOB 1.19 cells were then incubated
with anti-LC3 antibody (1:200) at 4°C overnight. The secondary
antibody, Alexa Fluor® 488 Goat Anti-Rabbit IgG
(ZSGB-BIO), was applied at room temperature for 1 h, followed by
staining the nuclei with DAPI (Beyotime Institute of
Biotechnology). Cells were photographed using the inverted
fluorescence microscope. LC3-positive cells, defined as cells with
visible LC3 foci, were quantified by manually in three
randomly-selected fields; nuclei were enumerated by counting
DAPI-stained nuclei in the same field. The number of LC3-positive
cells in each microscopic field was divided by the number of nuclei
in the same field, and was regarded as the rate of LC3-positive
cells.
Transmission electron microscopy
Following the incubation with indicated drug for 6
h, the cells were collected and fixed in 5% (v/v) glutaraldehyde.
Subsequently, the cells were conventionally dehydrated, embedded,
sectioned, and stained. The formation of autophagosomes was
observed using a transmission electron microscopy (TEM; H-7650;
Hitachi, Ltd., Tokyo, Japan). The number of intracellular
autophagosomes in every 10 fields was counted.
Statistical analysis
All the experiments were conducted in triple.
Quantitative data were presented as mean ± SD. All statistical
analyses were conducted using one-way analysis of variance (ANOVA)
by SPSS 24.0 software (IBM, Armonk, NY, USA). Statistical
significance was determined using the t-test (P<0.05).
Results
Dose-dependent effect of DEX on the
viability of hFOB1.19 osteoblasts
To clarify the effect of GCs on osteoblast
viability, we used the hFOB1.19 cell line as a cellular model due
to these cells' isogeny to human osteoblasts and their strong
proliferation ability (27). We
treated these cells with different concentrations of DEX
(10−4-10−8 M) for different time (0, 24, 48,
and 72 h) and then assessed cell viability via MTT analysis.
Interestingly, our results showed that DEX had a biphasic effect on
the viability of hFOB1.19 cells. High-dose DEX (≥10−6 M)
continuously and significantly inhibited cell viability. In
contrast, the viability of hFOB1.19 cells treated with a low dose
of DEX (10−8, 10−9 M) was increased at 24 h,
and there was no significant difference (P>0.05) in viability
between the DEX (10−8, 10−9 M) groups and the
control group at 48 h (Fig.
1A).
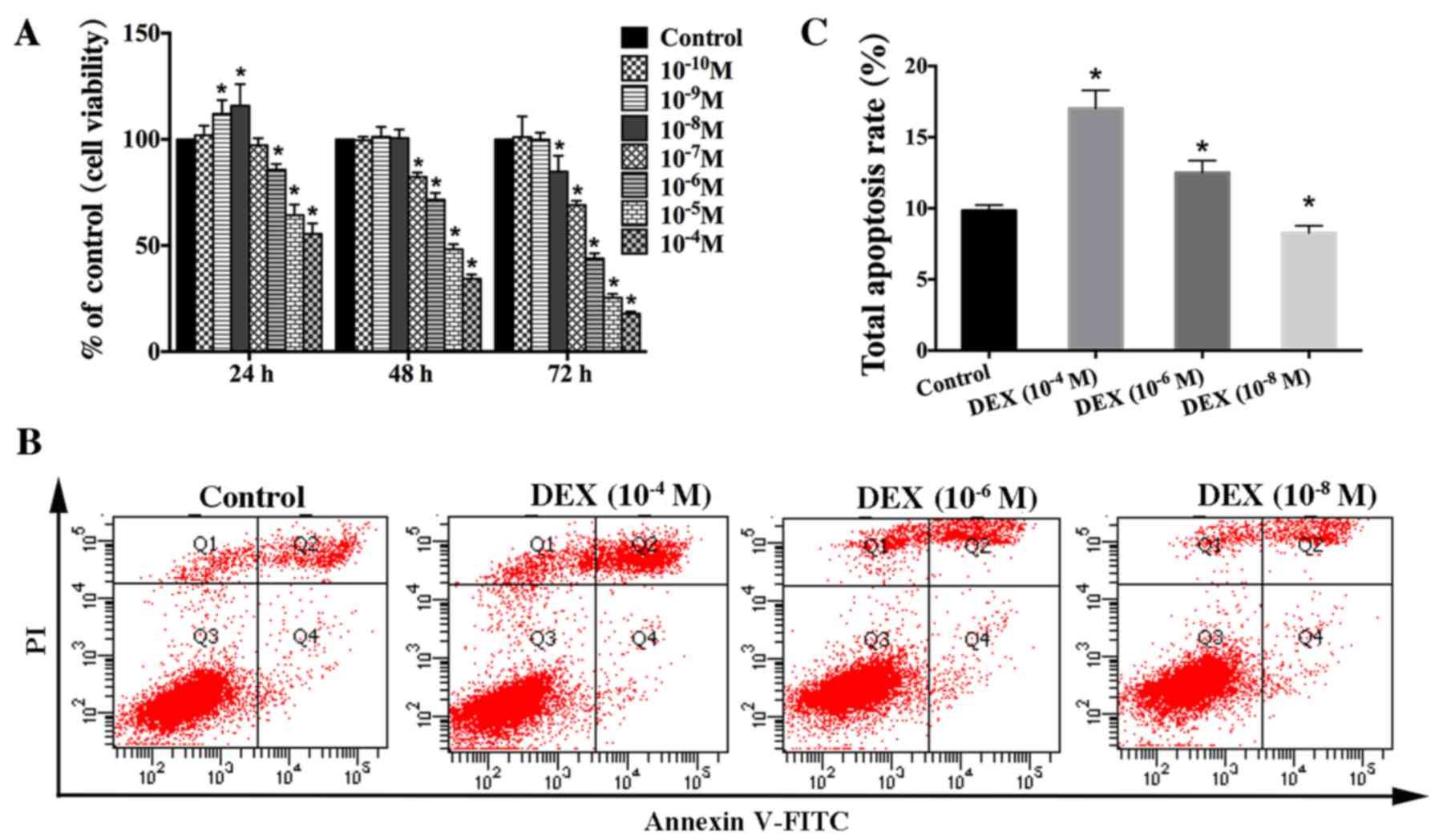 | Figure 1.Effect of DEX on the viability of
hFOB 1.19 cells. (A) Cells were treated with various concentration
of DEX (10−4-10−10 M) for different times (0,
24, 48 and 72 h). Cell viability was estimated using an MTT assay.
(B) Apoptosis among cells treated with various concentration of DEX
(10−4, 10−6 or 10−8 M) for 24 h
was estimated using Annexin V-FITC/PI staining. (C) Quantification
analysis of apoptotic cells. Viable cells, early apoptotic cells,
late apoptotic cells and necrotic cells appear in the bottom left
quadrant (Q3), bottom right quadrant (Q4), top right quadrant (Q2)
and top left quadrant (Q1), respectively. The apoptotic rate was
determined as the percentage of Q2 + Q4. Values are presented as
the mean ± standard deviation from three independent experiments.
*P<0.05 vs. control group. FITC, fluorescein isothiocyanate; PI,
propidium iodide; DEX, dexamethasone. |
To verify that DEX impacts osteoblast viability in a
dose-dependent manner, apoptosis of hFOB1.19 cells was estimated
via Annexin V-FITC/PI staining after treatment with various
concentration of DEX (10−4, 10−6, or
10−8 M) for 24 h. The results indicated that high dose
of DEX (≥10−6 M) significantly increased cell apoptosis,
which was not significantly elevated in the low-dose
(10−8 M) group compared with that seen in the control
group (Fig. 1B and C). Based on
these results, we suggest that DEX impacts osteoblast viability in
a dose-dependent manner in vitro. High-dose DEX promotes
osteoblast apoptosis, whereas the low-dose DEX increases osteoblast
viability.
Autophagy is involved in the effect of
DEX on osteoblast viability
To investigate whether increased viability of
hFOB1.19 cells induced by a low dose of DEX (10−8 M) is
associated with autophagy, cells were pretreated with 3-MA, an
autophagy inhibitor, to inhibit cell autophagy. The results showed
that 3-MA significantly inhibited the viability-promoting effect of
DEX (10−8 M) for hFOB1.19 cells (Fig. 2A). Meanwhile, the results of total
apoptotic cells showed that the decrease in apoptotic cells induced
by low-dose DEX (10−8 M) was inhibited by 3-MA (Fig. 2B and C). Therefore, we hypothesize
that the dose-dependent effect of DEX on osteoblast viability is
closely related to autophagy. To verify this hypothesis, we treated
osteoblasts with the specific autophagy agonist Rap. It was
observed that the decrease in the viability of hFOB1.19 cells
induced by a higher dose of DEX (10−6 M) was ameliorated
(Fig. 2F). In addition, the
increased level of apoptotic cells induced by a high dose of DEX
(10−6 M) was alleviated by Rap (Fig. 2D and E). All of these findings
demonstrated that autophagy protected hFOB1.19 osteoblasts against
DEX by increasing viability and inhibiting apoptosis.
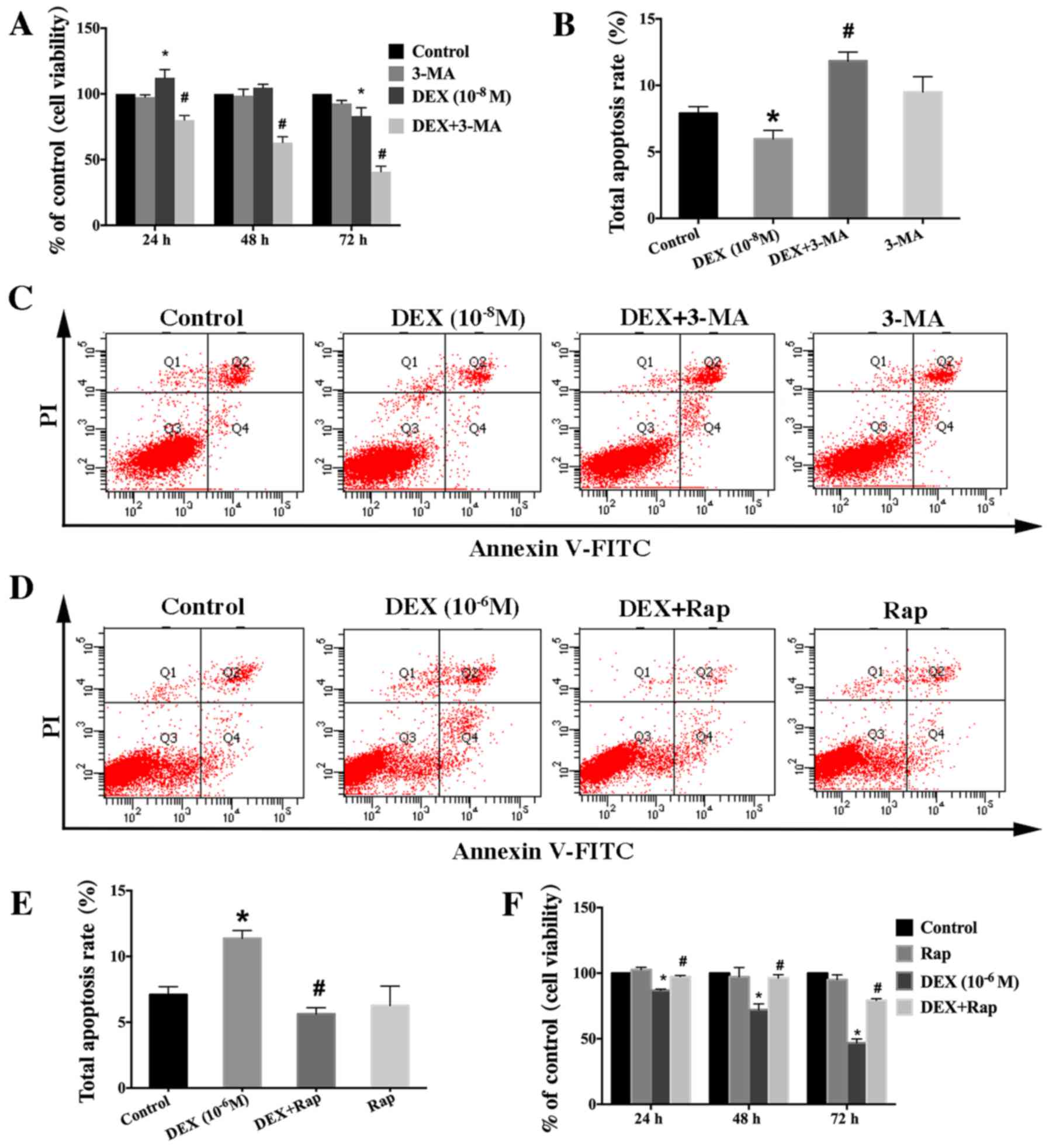 | Figure 2.Effect of autophagy modulators on the
viability of DEX-treated cells. (A) Cells were treated with DEX
(10−8 M) or DEX (10−8 M + 3-MA (5 mM) for 0,
24, 48 or 72 h, and cell viability was estimated using the MTT
assay. (B) Quantification analysis of apoptotic cells in 3-MA
studies. (C) Apoptosis among cells treated with DEX
(10−8 M) or DEX (10−8 M) + 3-MA (5 mM) for 24
h was estimated using Annexin V-FITC/PI staining. (D) Apoptosis
among cells treated with DEX (10−6 M) or DEX
(10−6 M) + Rap (3 µM) for 24 h was estimated using
Annexin V-FITC/PI staining. (E) Quantification analysis of
apoptotic cells in Rap studies. (F) Cells were treated with DEX
(10−6 M) or DEX (10−6 M) + Rap (3 µM) for 0,
24, 48 or 72 h, and cell viability was then estimated using the MTT
assay. Viable cells, early apoptotic cells, late apoptotic cells
and necrotic cells appear in the bottom left quadrant (Q3), bottom
right quadrant (Q4), top right quadrant (Q2) and top left quadrant
(Q1), respectively. The apoptotic rate was determined as the
percentage of Q2 + Q4. Values are presented as the mean ± standard
deviation from three independent experiments. *P<0.05 vs.
control group; #P<0.05 vs. DEX only group. FITC,
fluorescein isothiocyanate; PI, propidium iodide; DEX,
dexamethasone; Rap, rapamycin; 3-MA, 3-methyladenine. |
DEX induced autophagy of hFOB1.19
cells in a dose- and time-dependent manner
According to guideline for monitoring autophagy,
autophagy level can be accurately evaluated by detecting the
autophagic flux, which can be assessed based on protein level of
LC3II and beclin 1. To determine whether low dose of DEX
(10−8 M) induce autophagy in hFOB1.19 cells, we examined
the protein changes of LC3II and beclin 1 after cells were
incubated with DEX (10−8 M) for different times (0, 1,
3, 6, 12, and 24 h). Western blotting results showed that low dose
of DEX (10−8 M) induced autophagy in a time-dependent
manner. The level of LC3II and beclin 1 increased gradually
starting from the 3 h time-point and peaked at the 6 h time-point
(Fig. 3A and B). However, 3-MA
significantly inhibited the autophagy induced by DEX
(10−8 M) (Fig. 3C and
D).
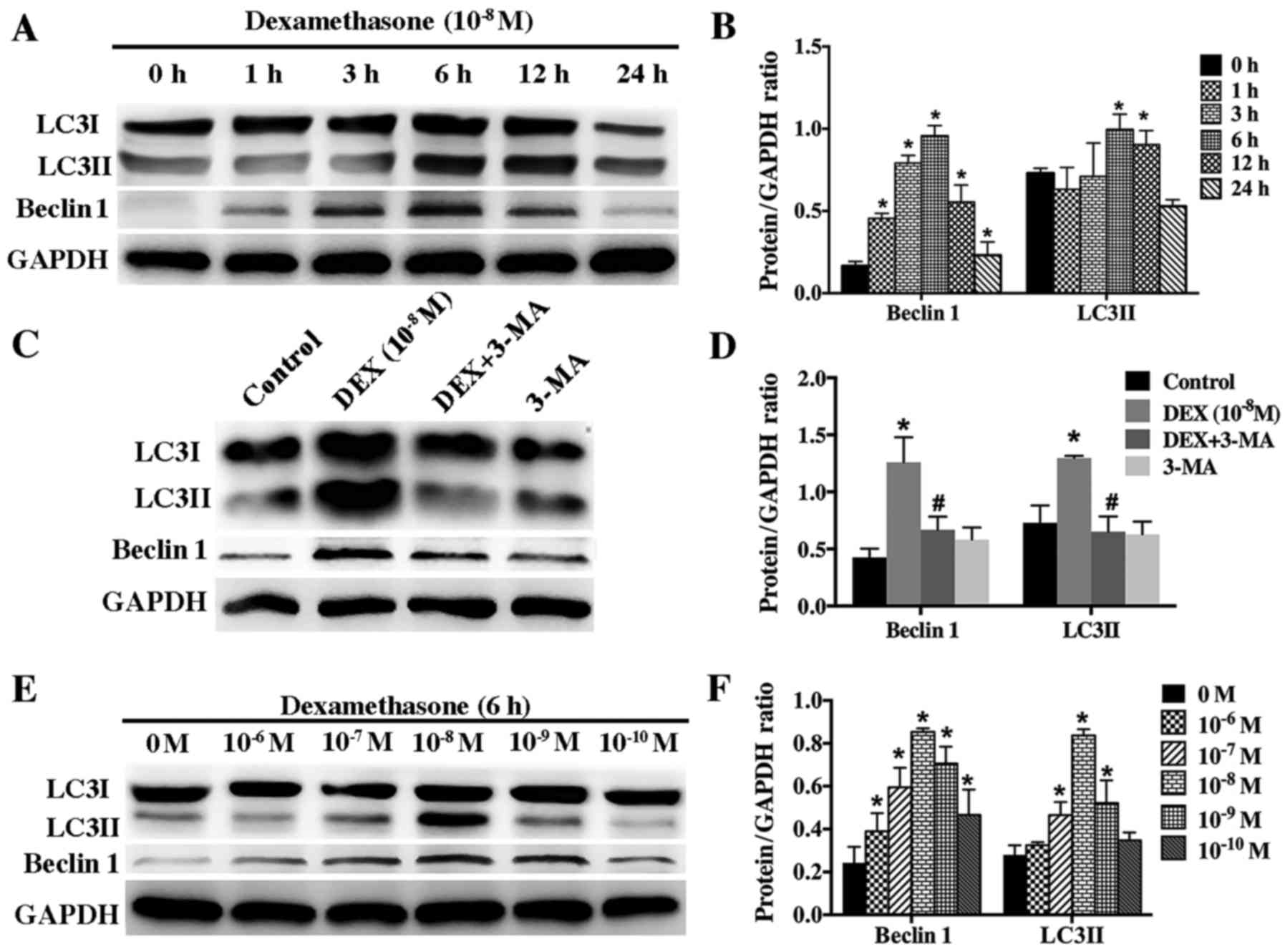 | Figure 3.Effect of DEX on the expression of
LC3II and beclin 1 in hFOB 1.19 cells. (A) Western blot analysis
showing the expression of beclin 1 and LC3 protein in hFOB 1.19
cells treated with DEX (10−8 M) for 0, 1, 3, 6, 12 or 24
h. (B) Quantitative analysis of beclin 1 and LC3II protein
expression. (C) Western blot analysis showing the expression of
beclin 1 and LC3 protein in hFOB 1.19 cells treated with DEX
(10−8 M) or DEX (10−8 M) + 3-MA (5 mM) for 6
h. (D) Quantitative analysis of beclin 1 and LC3II protein
expression. (E) Western blot analysis showing the expression of
beclin 1 and LC3 protein in hFOB 1.19 cells treated with DEX at
various concentration (0, 10−6, 10−7,
10−8, 10−9 and 10−10 M) for 6 h.
(F) Quantitative analysis of beclin 1 and LC3II protein expression.
Values are presented as the mean ± standard deviation from three
independent experiments. *P<0.05 vs. control;
#P<0.05 vs. DEX (10−8 M) group. DEX,
dexamethasone; 3-MA, 3-methyladenine; LC3, light chain 3. |
To further investigate the relationship between DEX
and autophagy, we treated hFOB1.19 cells with gradient
concentration of DEX (10−6-10−10 M) for 6 h.
Our results showed that autophagy was activated most significantly
by 10−8 M DEX (Fig. 3E and
F). Higher and lower concentration of DEX could also induce
autophagy but at lower level, indicating that DEX modulated
autophagy in a dose-dependent manner.
The assessment of autolysosome presence and number
via transmission electron microscopy (TEM) is recognized as the
gold standard for monitoring autophagy intensity. To confirm the
role of low dose of DEX (10−8 M) in inducing autophagy,
we used TEM to observe and count autolysosomes. The quantity of
autolysosomes was significantly elevated in hFOB1.19 cells treated
with DEX (10−8 M) for 6 h compared with those in control
cells. Furthermore, large numbers of mitochondria that were
undergoing phagocytosis were observed. GCs have been reported to
increase intracellular oxidative stress by damaging mitochondria
(28). In response to this
oxidative stress, autophagy is upregulated to help to clear damaged
mitochondria with autophagosomes (29). The number of autolysosomes was
significantly lower in the group treated with 3-MA (5 mM) than that
in the corresponding group treated with DEX alone (Fig. 4A and B). This result confirmed that
autophagy protected osteoblasts against oxidative stress by
clearing damaged organelles (30).
In addition, cells with punctate aggregates (autolysosomes) was
defined as LC3-positive cells and they were observed via
fluorescence microscopy (Fig. 4C and
D). This result was consistent with the finding obtained via
TEM.
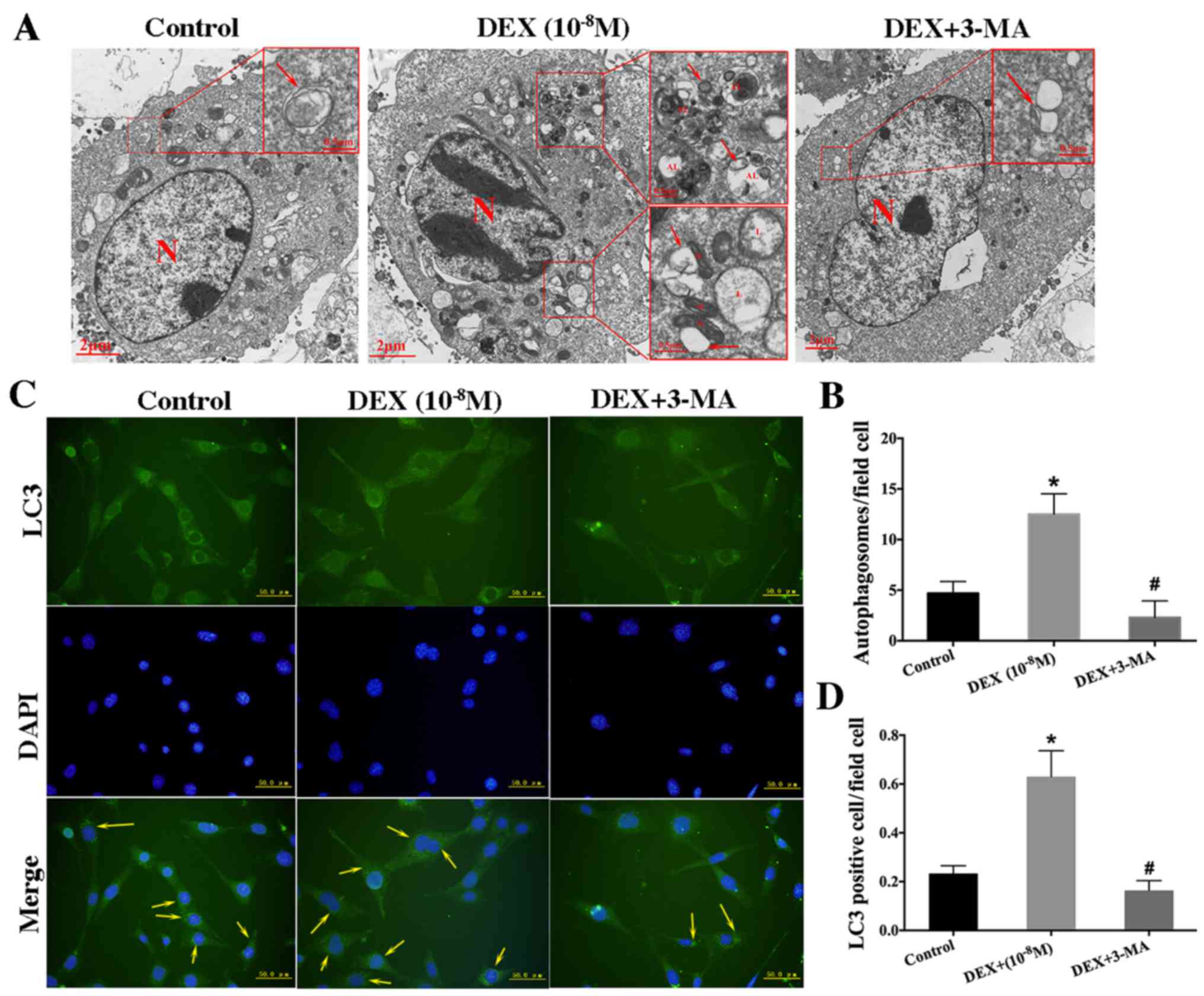 | Figure 4.Low-dose DEX (10−8
M)-induced autophagy in hFOB 1.19 cells. (A) Transmission electron
microscopy showing autophagosomes in hFOB 1.19 cells treated with
DEX (10−8 M) or DEX (10−8 M) + 3-MA (5 mM)
for 6 h. Arrows indicate autophagosomes. Scale bars, main images, 2
µm; enlarged areas, 0.5 µm. (B) Quantitation of autophagosomes. (C)
Fluorescence microscopy showing LC3-positive cells (cells with
visible green fluorescence, LC3 foci) and nuclei (blue
fluorescence) in hFOB 1.19 cells treated with DEX (10−8
M) or DEX (10−8 M) + 3-MA (5 mM) for 6 h. Arrows
indicate LC3-positive cells. Scale bars, 50 µm. (D) Quantitation of
LC3-positive cells. Values are presented as the mean ± standard
from three independent experiments. *P<0.05 vs. control;
#P<0.05 vs. DEX only treatment. AL,
autophagolysosome; N, nucleus; M, mitochondria; L, lysosome; DEX,
dexamethasone; 3-MA, 3-methyladenine; LC3, light chain 3. |
Autophagy was initiated in response to
the increase of ROS induced by low-dose DEX
After hFOB1.19 cells were exposed to various
concentration of DEX (10−4, 10−6, or
10−8 M) for 6 h, these cells' intracellular ROS level
was recorded by measuring DCF fluorescence intensity. The result
showed that intracellular ROS level increased (Fig. 5A and B) as DEX dose increased,
suggesting that DEX upregulated the ROS level in a dose-dependent
manner.
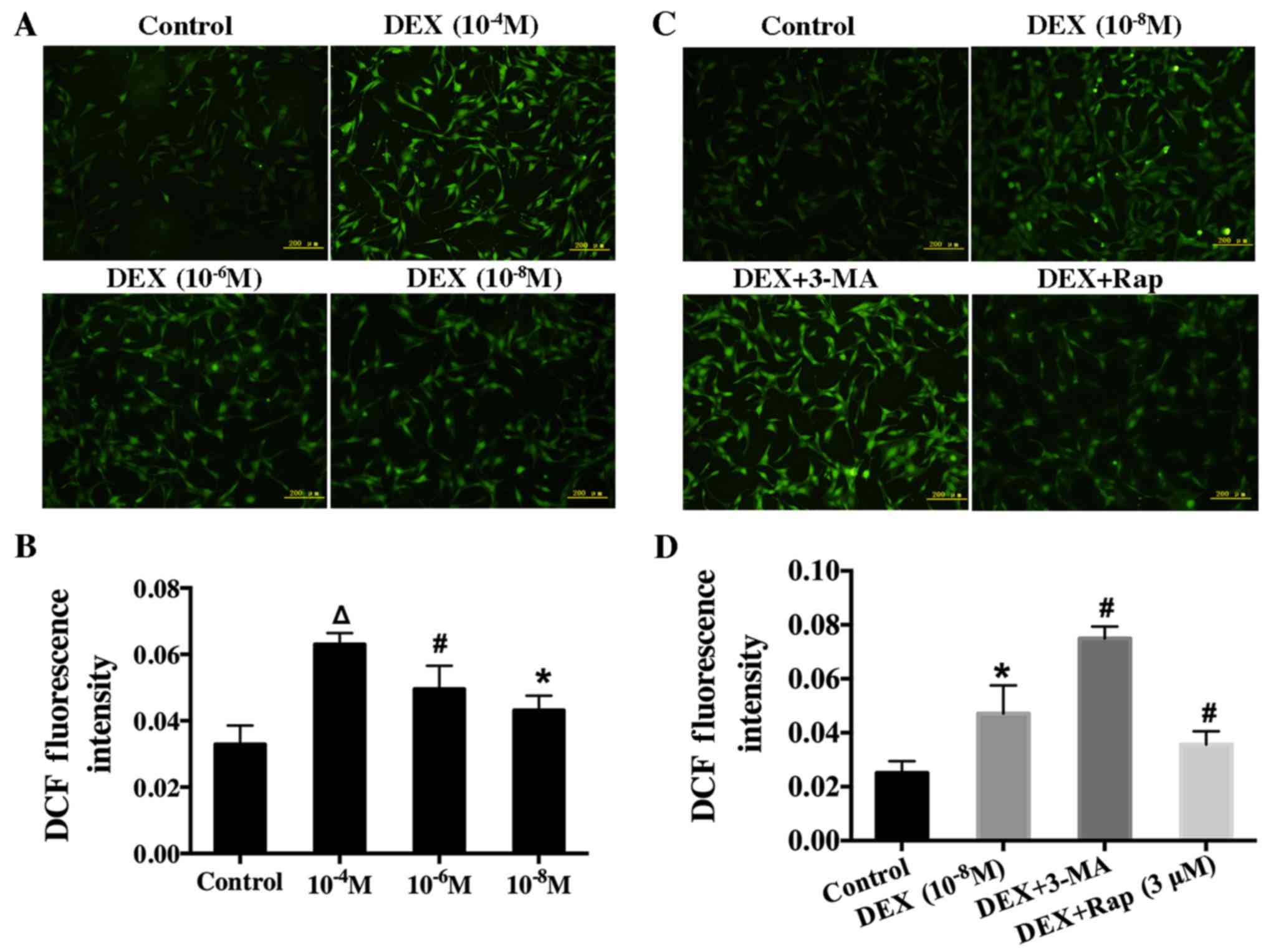 | Figure 5.Effect of DEX on intracellular ROS in
hFOB 1.19 cells and the effect of the autophagy modulator on the
induction of ROS in DEX-treated cells. (A) Cells were treated with
various concentration of DEX (0, 10−4, 10−6
or 10−8 M) for 6 h, and ROS levels were determined using
2′,7′-dichlorofluorescin diacetate staining. (B) Quantification
analysis of ROS. (C) Fluorescence microscopy showing intracellular
ROS level in hFOB 1.19 cells treated with DEX (10−8 M),
DEX (10−8 M) + 3-MA (5 mM) or DEX (10−8 M) +
Rap (3 µM) for 6 h. (D) Quantification analysis of ROS. Scale bars,
200 µm. Values are presented as the mean ± standard deviation from
three independent experiments. *P<0.05 vs. control;
#P<0.05 vs. DEX (10−8 M) treatment;
∆P<0.05 vs. DEX (10−6 M) treatment. DEX,
dexamethasone; ROS, reactive oxygen species; 3-MA, 3-methyladenine;
Rap, rapamycin; DCF, 2′,7′-dichlorofluorescin. |
To further investigate the relationship between ROS
and autophagy, we measured ROS level in cells after treatment with
Rap or 3-MA. In particular, hFOB1.19 cells were pretreated with Rap
(3 µM) or 3-MA (5 mM) for 1 h before incubation with DEX. Rap
significantly decreased intracellular ROS level. In contrast, ROS
level significantly increased when autophagy was suppressed with
3-MA (Fig. 5C and D). These
results indicated that autophagy could protect osteoblasts by
decreasing ROS level.
To further investigate the role of ROS in low-dose
DEX-induced autophagy, hFOB1.19 cells were pretreated with catalase
(500 U/ml), an ROS inhibitor, for 1 h prior to treatment with DEX
(10−8 M). Western blot analysis showed that catalase
remarkably inhibited the autophagy induced by low-dose DEX
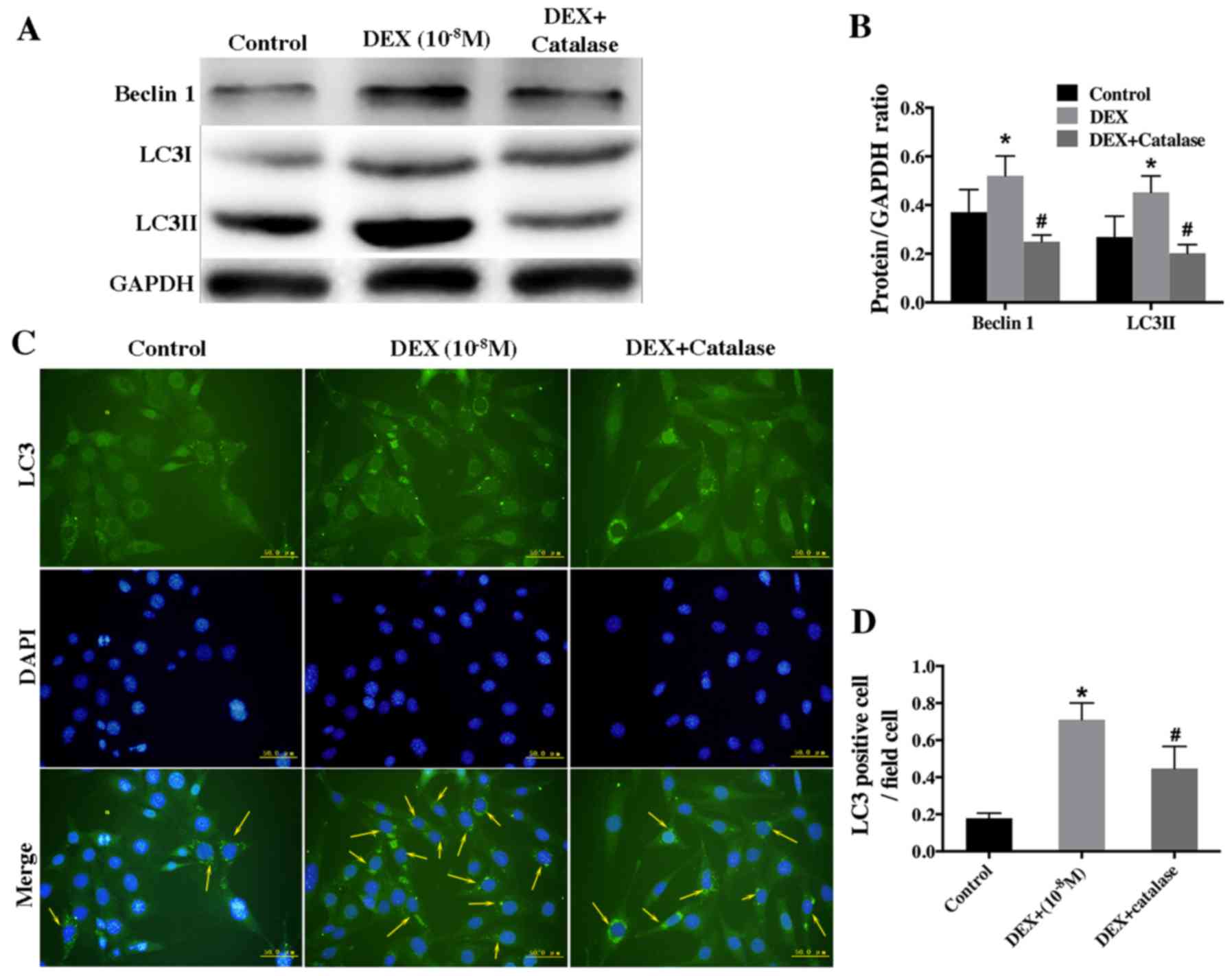
(Fig. 6A and B). Furthermore, the
result of immunofluorescence staining revealed that the rate of
LC3-positive cells was lower in the group treated with catalase and
DEX than that treated with DEX alone (Fig. 6C and D).
Taken together, the aforementioned results suggested
that a low dose of DEX induced autophagy by upregulating
intracellular ROS level.
ROS inhibitor suppressed the increased
viability of hFOB1.19 cells induced by low-dose DEX
Based on the previous results, we concluded that
low-dose DEX increased hFOB 1.19 cells viability by inducing
autophagy via intracellular ROS. To verify the key role of ROS in
the improvement of osteoblast viability induced by a low dose of
DEX, we firstly investigated cell viability via MTT analysis.
Significantly lower viability was observed in hFOB1.19 cells
co-treated with catalase (500 U/ml) and DEX (10-8 M), which was
consistent with 3-MA group (Fig.
7A). The same result was obtained in Annexin V-FITC/PI staining
analyses; in particular, more apoptotic cells were observed after
treatment with catalase and DEX than treatment with DEX alone
(Fig. 7B and C). All of our
results confirmed that low-dose DEX upregulated the viability of
hFOB1.19 cells by increasing intracellular ROS.
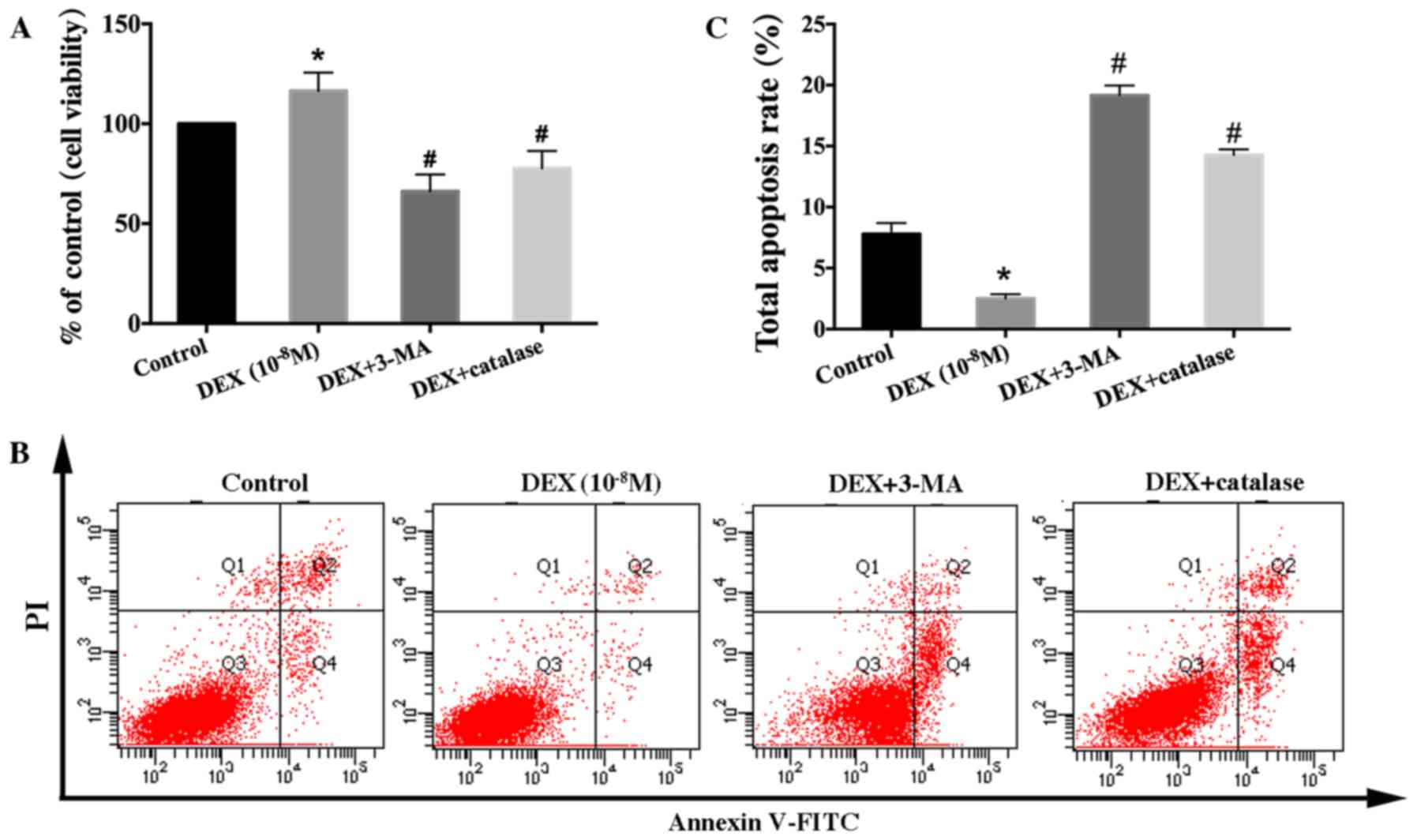 | Figure 7.Effect of a reactive oxygen species
inhibitor on the viability and apoptosis of low-dose DEX-treated
hFOB 1.19 cells. Cells were treated with DEX (10−8 M),
DEX (10−8 M) + 3-MA (5 mM), or DEX (10−8 M) +
catalase (500 U/ml) for 24 h. (A) Cell viability was estimated
using the MTT assay. (B) Cell apoptosis was estimated using Annexin
V-FITC/PI staining. (C) Quantification analysis of apoptotic cells.
Viable cells, early apoptotic cells, late apoptotic cells and
necrotic cells appear in the bottom left quadrant (Q3), bottom
right quadrant (Q4), top right quadrant (Q2) and top left quadrant
(Q1), respectively. The apoptotic rate was determined as the
percentage of Q2 + Q4. Values are expressed as the mean ± standard
deviation from three independent experiments. *P<0.05 vs.
control group; #P<0.05 vs. DEX (10−8 M)
treatment. FITC, fluorescein isothiocyanate; PI, propidium iodide;
DEX, dexamethasone; 3-MA, 3-methyladenine. |
Discussion
Many studies have suggested that GCs contributed to
the development and progression of GIOP by repressing viability and
promoting apoptosis among osteoblasts (2,6).
However, there is controversy regarding the effect of GCs on
osteoblasts in vivo and in vitro. Endogenous GCs,
which are secreted by the adrenal cortex, are essential for bone
development (31,32). Moreover, investigations have found
that in vitro, DEX can accelerate the osteogenic activity of
osteoblasts (33) and the
proliferation of bone marrow stromal cells (34), which are precursors and source of
osteoblasts. Our results showed that DEX exerted a biphasic effect
on the viability of hFOB1.19 cells: High-dose DEX (≥10−6
M) continuously inhibited cell viability and markedly accelerated
apoptosis, whereas lower-dose DEX (10−8 M) increased
cell viability in the early stage, implying that both dose and
treatment time contributed to the effect of GCs on osteoblast
viability. This result is similar to the finding obtained by Jia
et al (8) and Shi et
al (9), who claimed that the
effect of GCs on cell viability was determined by GCs dose. In
fact, the treatment dose of GCs used for autoimmune and
inflammatory diseases is higher than the physiological level of
these compounds, and treatment duration is often months or even
years. This observation may partially resolve the controversy
regarding the opposite effect of GCs on osteoblast viability.
As we know, basal autophagy occurs at low level in
all cells and is considered as a quality control mechanism of
protein and organelle, maintaining normal cellular homeostasis
(12). When encountering some
stresses, autophagy is usually upregulated (35). If autophagy is excessively
upregulated, autophagosomes will devour the cellular proteins and
organelles, and cause cells apoptosis. Hence, autophagy is also
regarded as type II form of programmed cell death (36). However, under some moderate
stimulus conditions, autophagy can be moderately induced to promote
cell survival and avert apoptosis by eliminating stress inducers
(37). Autophagy ensures the
delivery of metabolic substrates to cells in order to fulfill their
energy demand during stress, thus supports cell growth and survival
(38,39). This function is critical for the
survival of some terminally differentiated cells, such as
osteoblasts and osteoclasts (40).
Many recent studies have demonstrated the protective effect of
autophagy with respect to facilitating osteoblast viability under
various stress (16,17,41).
Moreover, the involvement of GC-induced autophagy in modulating the
viability of various cells has been well demonstrated (42–44).
However, the role of autophagy in affecting the
viability of osteoblasts exposed to low dose of GCs has rarely been
reported. Based on prior studies and our results, we hypothesize
that the increased viability of osteoblasts exposed to low-dose DEX
may be associated with autophagy. Our results indicated that
treatment with 3-MA markedly inhibited the increased osteoblast
activity induced by a low dose of DEX (10−8 M) and
significantly increased the apoptosis rate; both effects verified
our hypothesis. In conclusion, all of the aforementioned results
indicated that a low dose of GCs increased osteoblast viability by
inducing autophagy at early stage.
DEX, as an inducer of oxidative stress, can lead to
mitochondrial dysfunction and upregulate oxidative stress in cells
(21,45). Therefore, it could be observed that
intracellular ROS level was consistently upregulated as DEX dose
increased. To respond to this oxidative stress, autophagy was
upregulated to protect osteoblasts from apoptosis due to DEX
exposure; this phenomenon could be observed by detecting
autophagy-related proteins and autophagosomes in osteoblasts
incubated with low-dose DEX. If the DEX dose and the corresponding
oxidative stress are low and the treatment time is short, this
attempt to protect osteoblasts will be successful. However, as DEX
dose and treatment time increase, the antioxidant capacity of
autophagy will be overwhelmed, and the ability of protecting
against stress will be reduced, potentially leading to osteoblast
apoptosis. Our study demonstrated that osteoblast viability was
strictly determined by GCs dose. A low dose of GCs activated
autophagy to protect cells from apoptosis, whereas a high dose of
GCs induced apoptosis. This finding was consistent with the results
of an in vivo investigation by Jia et al (8), who reported that autophagic gene
expression and osteocyte autophagy increased when mice were treated
with a low dose of GCs but the expression of apoptotic genes
increased when mice were treated with higher dose of GCs.
The generation of ROS is known to be a normal
physiological activity of cellular metabolism (46). However, researches on the
pathological mechanism of GCs have demonstrated that excessive and
long-term treatment with GCs caused the excessive accumulation of
intracellular ROS by dysregulating mitochondria function, leading
to the upregulation of oxidative stress (21). Similarly, our results showed that
ROS level was increased in hFOB1.19 cells treated with DEX.
Moreover, the higher DEX dose, the higher ROS level. The excessive
accumulation of ROS might disturb cellular homeostasis, resulting
in cell injury and death (47,48).
Both in vitro and in vivo studies have demonstrated
that excessive ROS and oxidative stress reduce bone formation by
inhibiting the differentiation and viability of osteoblasts
(45,49). Therefore, we concluded that the
reduced viability of osteoblasts treated with high-dose DEX was
associated with excessive intracellular ROS.
However, studies have showed that physiological ROS
acted as intracellular signalling molecule, and moderate ROS was
thought to modulate cell viability by upregulating autophagy
(17,46,50).
Based on our investigation and works by other researchers, we
hypothesize that low-dose DEX increased osteoblast activity by
inducing autophagy via intracellular ROS. To verify this
hypothesis, we treated osteoblasts with a low dose of DEX
(10−8 M) combined with or without the ROS scavenger
catalase. The results showed that the increased cell activity and
autophagy induced by a low dose of DEX was significantly inhibited
by catalase. In addition, increased autophagy can decrease or
eliminate intracellular ROS (51).
In our study, the upregulation of autophagy by Rap caused the
reduction of intracellular ROS in osteoblasts treated with low-dose
DEX, whereas pretreatment with 3-MA produced the opposite result.
In summary, our data implied that the low-dose GCs induced
autophagy via intracellular ROS and autophagy protected osteoblasts
from apoptosis by reducing ROS level.
In conclusion, the present study demonstrated that
GCs modulated osteoblast viability in a dose-dependent manner.
High-dose of GCs induced osteoblast apoptosis, whereas a lower dose
of GCs enhanced osteoblast viability by inducing autophagy via
intracellular ROS. Our findings suggested that the modulation of
osteoblast autophagy could be a novel approach for the prevention
and treatment of GIOP. Next, further studies should be performed to
explore the underlying mechanism of low-dose GC-induced autophagy
via ROS.
References
|
1
|
Kenanidis E, Potoupnis ME, Kakoulidis P,
Leonidou A, Sakellariou GT, Sayegh FE and Tsiridis E: Management of
glucocorticoid-induced osteoporosis: Clinical data in relation to
disease demographics, bone mineral density and fracture risk.
Expert Opin Drug Saf. 14:1035–1053. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Frenkel B, White W and Tuckermann J:
Glucocorticoid-induced osteoporosis. Adv Exp Med Biol. 872:179–215.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dalle Carbonare L, Arlot ME, Chavassieux
PM, Roux JP, Portero NR and Meunier PJ: Comparison of trabecular
bone microarchitecture and remodeling in glucocorticoid-induced and
postmenopausal osteoporosis. J Bone Miner Res. 16:97–103. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Van Staa TP, Laan RF, Barton IP, Cohen S,
Reid DM and Cooper C: Bone density threshold and other predictors
of vertebral fracture in patients receiving oral glucocorticoid
therapy. Arthritis Rheum. 48:3224–3229. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Capulli M, Paone R and Rucci N: Osteoblast
and osteocyte: Games without frontiers. Arch Biochem Biophys.
561:3–12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moriishi T and Komori T: Glucocorticoid
and bone. The inhibition of osteoblast differentiation and
induction of osteocyte apoptosis through the regulation of Bcl-2 by
glucocorticoids. Clin Calcium. 24:1329–1336. 2014.(In
Japanese).
|
|
7
|
Komori T: Glucocorticoid signaling and
bone biology. Horm Metab Res. 48:755–763. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jia J, Yao W, Guan M, Dai W, Shahnazari M,
Kar R, Bonewald L, Jiang JX and Lane NE: Glucocorticoid dose
determines osteocyte cell fate. FASEB J. 25:3366–3376. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shi J, Wang L, Zhang H, Jie Q, Li X, Shi
Q, Huang Q, Gao B, Han Y, Guo K, et al: Glucocorticoids:
Dose-related effects on osteoclast formation and function via
reactive oxygen species and autophagy. Bone. 79:222–232. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Martinez-Lopez N, Athonvarangkul D and
Singh R: Autophagy and aging. Adv Exp Med Biol. 847:73–87. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang JY, Yao WX, Wang Y, Fan YL and Wu JB:
Network analysis reveals crosstalk between autophagy genes and
disease genes. Sci Rep. 7:443912017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee JY, Koga H, Kawaguchi Y, Tang W, Wong
E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, et al: HDAC6
controls autophagosome maturation essential for ubiquitin-selective
quality-control autophagy. EMBO J. 29:969–980. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sid B, Verrax J and Calderon PB: Role of
AMPK activation in oxidative cell damage: Implications for
alcohol-induced liver disease. Biochem Pharmacol. 86:200–209. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kouroku Y, Fujita E, Tanida I, Ueno T,
Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E and Momoi T: ER
stress (PERK/eIF2alpha phosphorylation) mediates the
polyglutamine-induced LC3 conversion, an essential step for
autophagy formation. Cell Death Differ. 14:230–239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gu X, Han D, Chen W, Zhang L, Lin Q, Gao
J, Fanning S and Han B: SIRT1-mediated FoxOs pathways protect
against apoptosis by promoting autophagy in osteoblast-like
MC3T3-E1 cells exposed to sodium fluoride. Oncotarget.
7:65218–65230. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang L, Meng H and Yang M: Autophagy
protects osteoblasts from advanced glycation end products-induced
apoptosis through intracellular reactive oxygen species. J Mol
Endocrinol. 56:291–300. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tanida I, Ueno T and Kominami E: LC3 and
autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fu LL, Cheng Y and Liu B: Beclin-1:
Autophagic regulator and therapeutic target in cancer. Int J
Biochem Cell Biol. 45:921–924. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Grishko V, Xu M, Ho R, Mates A, Watson S,
Kim JT, Wilson GL and Pearsall AW IV: Effects of hyaluronic acid on
mitochondrial function and mitochondria-driven apoptosis following
oxidative stress in human chondrocytes. J Biol Chem. 284:9132–9139.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ong SL, Zhang Y and Whitworth JA: Reactive
oxygen species and glucocorticoid-induced hypertension. Clin Exp
Pharmacol Physiol. 35:477–482. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Henrotin Y, Kurz B and Aigner T: Oxygen
and reactive oxygen species in cartilage degradation: Friends or
foes? Osteoarthritis Cartilage. 13:643–654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghavami S, Shojaei S, Yeganeh B, Ande SR,
Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam
AR, Kashani HH, et al: Autophagy and apoptosis dysfunction in
neurodegenerative disorders. Prog Neurobiol. 112:24–49. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Almeida M, Han L, Martin-Millan M, Plotkin
LI, Stewart SA, Roberson PK, Kousteni S, O'Brien CA, Bellido T,
Parfitt AM, et al: Skeletal involution by age-associated oxidative
stress and its acceleration by loss of sex steroids. J Biol Chem.
282:27285–27297. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wauquier F, Leotoing L, Coxam V, Guicheux
J and Wittrant Y: Oxidative stress in bone remodelling and disease.
Trends Mol Med. 15:468–477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Harris SA, Enger RJ, Riggs BL and
Spelsberg TC: Development and characterization of a conditionally
immortalized human fetal osteoblastic cell line. J Bone Miner Res.
10:178–186. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brand MD, Affourtit C, Esteves TC, Green
K, Lambert AJ, Miwa S, Pakay JL and Parker N: Mitochondrial
superoxide: Production, biological effects, and activation of
uncoupling proteins. Free Radic Biol Med. 37:755–767. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wohlgemuth SE, Calvani R and Marzetti E:
The interplay between autophagy and mitochondrial dysfunction in
oxidative stress-induced cardiac aging and pathology. J Mol Cell
Cardiol. 71:62–70. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kalak R, Zhou H, Street J, Day RE,
Modzelewski JR, Spies CM, Liu PY, Li G, Dunstan CR and Seibel MJ:
Endogenous glucocorticoid signalling in osteoblasts is necessary to
maintain normal bone structure in mice. Bone. 45:61–67. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou H, Mak W, Kalak R, Street J, Fong-Yee
C, Zheng Y, Dunstan CR and Seibel MJ: Glucocorticoid-dependent Wnt
signaling by mature osteoblasts is a key regulator of cranial
skeletal development in mice. Development. 136:427–436. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ishida Y, Tertinegg I and Heersche JN:
Progesterone and dexamethasone stimulate proliferation and
differentiation of osteoprogenitors and progenitors for adipocytes
and macrophages in cell populations derived from adult rat
vertebrae. J Bone Miner Res. 11:921–930. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Atmani H, Chappard D and Basle MF:
Proliferation and differentiation of osteoblasts and adipocytes in
rat bone marrow stromal cell cultures: Effects of dexamethasone and
calcitriol. J Cell Biochem. 89:364–372. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang F, Jia J and Rodrigues B: Autophagy,
metabolic disease, and pathogenesis of heart dysfunction. Can J
Cardiol. 33:850–859. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Booth LA, Tavallai S, Hamed HA,
Cruickshanks N and Dent P: The role of cell signalling in the
crosstalk between autophagy and apoptosis. Cell Signal. 26:549–555.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bhutia SK, Kegelman TP, Das SK, Azab B, Su
ZZ, Lee SG, Sarkar D and Fisher PB: Astrocyte elevated gene-1
induces protective autophagy. Proc Natl Acad Sci USA. 107:pp.
22243–22248. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bhutia SK, Mukhopadhyay S, Sinha N, Das
DN, Panda PK, Patra SK, Maiti TK, Mandal M, Dent P, Wang XY, et al:
Autophagy: Cancer's friend or foe? Adv Cancer Res. 118:61–95. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Manolagas SC and Parfitt AM: What old
means to bone. Trends Endocrinol Metab. 21:369–374. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lv XH, Zhao DH, Cai SZ, Luo SY, You T, Xu
BL and Chen K: Autophagy plays a protective role in cell death of
osteoblasts exposure to lead chloride. Toxicol Lett. 239:131–140.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang L, Fan J, Lin YS, Guo YS, Gao B, Shi
QY, Wei BY, Chen L, Yang L, Liu J and Luo ZJ: Glucocorticoids
induce autophagy in rat bone marrow mesenchymal stem cells. Mol Med
Rep. 11:2711–2716. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao Y, Zuo Y, Huo HJ, Xiao YL, Yang XJ
and Xin DQ: Glucocorticoid induced autophagy in N1511 chondrocyte
cells. Eur Rev Med Pharmacol Sci. 18:3573–3579. 2014.PubMed/NCBI
|
|
44
|
Grander D, Kharaziha P, Laane E,
Pokrovskaja K and Panaretakis T: Autophagy as the main means of
cytotoxicity by glucocorticoids in hematological malignancies.
Autophagy. 5:1198–1200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Almeida M, Han L, Ambrogini E, Weinstein
RS and Manolagas SC: Glucocorticoids and tumor necrosis factor
alpha increase oxidative stress and suppress Wnt protein signaling
in osteoblasts. J Biol Chem. 286:44326–44335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kiffin R, Bandyopadhyay U and Cuervo AM:
Oxidative stress and autophagy. Antioxid Redox Signal. 8:152–162.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhen YF, Wang GD, Zhu LQ, Tan SP, Zhang
FY, Zhou XZ and Wang XD: P53 dependent mitochondrial permeability
transition pore opening is required for dexamethasone-induced death
of osteoblasts. J Cell Physiol. 229:1475–1483. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Manolagas SC: From estrogen-centric to
aging and oxidative stress: A revised perspective of the
pathogenesis of osteoporosis. Endocr Rev. 31:266–300. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Schroder K: NADPH oxidases in bone
homeostasis and osteoporosis. Cell Mol Life Sci. 72:25–38. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Suzuki M, Bandoski C and Bartlett JD:
Fluoride induces oxidative damage and SIRT1/autophagy through
ROS-mediated JNK signaling. Free Radic Biol Med. 89:369–378. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Scherz-Shouval R and Elazar Z: ROS,
mitochondria and the regulation of autophagy. Trends Cell Biol.
17:422–427. 2007. View Article : Google Scholar : PubMed/NCBI
|