Introduction
Lung cancer, which accounts for more than one-tenth
of all cancer cases worldwide, is one of the most common types of
malignancies and perhaps, the first cause of cancer-related deaths
worldwide (1). In China, lung
cancer is currently the first most common cancer with more than
733,300 new cases and 610,200 deaths in 2015 (2). Based on histological
sub-classification, there are non-small cell lung cancer (NSCLC)
and SCLC with approximately 85% of lung cancer patients diagnosed
with NSCLC (3).
To date, surgical resection remains the only
treatment option for NSCLC (4,5).
However, with no specific symptoms associated with the early stage
of NCLSC and the lack of accurate and convenient diagnostic methods
for its early detection, most patients are diagnosed at the
advanced stage of this disease when surgical resection is no longer
feasible (6).
With the recent advances in the molecular biology,
molecule targeted therapy for NSCLC has equally made significant
progress. For example, approximately 10–20% of North American and
Western European patients and 30–50% of Asian patients with NSCLC
were shown to have mutation of EGFR gene (7). But, with the use of EGFR tyrosine
kinase inhibitors (TKIs), such as erlotinib and gefitinib, these
patients were found to response well (with a response rate of 10%,
and an estimated 2-month prolongation in median survival over
placebo) (8,9). It was also found that 3–7% of NSCLC
patients had an activated ALK gene, but treatment with crizotinib
(an ALK inhibitor) elevated their response rate from 10 to 55%
(compared to conventional chemotherapy), and increased their
6-month progression-free survival rate to 72% (8). Nevertheless, these situations can
only represent a cohort of NSCLC patients and due to drug
resistance these targeted therapy agents are often transient.
Moreover, even with the remarkable successes and
withdraw that have been obtained in the targeted therapy of NSCLC,
the precise molecular mechanisms of this disease are still far from
being fully understood. Thus, the need to find more potential
targets for more effective therapeutic strategies is very
paramount.
In order to better understand the DEGs influence on
molecular pathogenesis of NSCLC, in this study, we downloaded 4
NSCLC related mRNA datasets from Gene Expression Omnibus (GEO) and
identified the DEGs between the NSCLC patients and healthy control
samples. Subsequently, we performed Gene Ontology (GO) and
protein-protein interaction (PPI) networks analysis so as to study
and identify potential key DEGs for the diagnosis and treatment of
NSCLC. Further studies on these identified key DEGs can aid a
comprehensive understanding of the molecular development of NSCLC
and be explored as potential biomarkers for its diagnosis,
prognosis, and drug targets.
Materials and methods
Affymetrix microarray data
Series matrix files of the microarray data:
GSE21933, GSE33532, GSE44077 and GSE74706 were downloaded from the
public GEO database (http://www.ncbi.nlm.nih.gov/geo/), and the probe names
based on platforms were annotated to gene symbols according to
their corresponding new version of annotation files, such as
GPL6254, GPL570, GPL6244 and GPL13497. In each GSE file, we only
choose the NSCLC samples and their matched normal samples, and from
which we obtained 21 tumor samples and 21 normal samples for
GSE21933 (10), 80 tumor samples
and 20 normal samples for GSE33532 (11), 65 tumor samples and 65 normal
samples for GSE44077 (12) and 18
tumor samples and 18 normal samples for GSE74706 (13) to analyze downstream.
Data pre-processing and differential
expression analysis
The original array data of each series were analyzed
separately using R software (version 3.4.0) and R packages. In
brief: Firstly, background correction and quantile normalization
was performed on the raw data using the robust multi-array average
(RMA) algorithm in R affy package (14). The differentially expressed
proteins (DEGs) between the tumor and normal group samples were
then analyzed by the paired t-test based on the limma package in R.
Multiple testing was corrected by the Benjamini-Hochberg method to
obtain the adjusted P-value (15).
Finally, the Genes with adjusted P<0.05 and
|log2fold-change (FC)|>1 were considered to be
significant. To further enhance the reliability of the
bioinformatics analysis, the overlapping DEGs co-existed in all 4
GSE files were identified using FunRich software version 2.1.2
(http://www.funrich.org) (16).
GO enrichment analysis of DEGs
GO analysis is widely used to identify
characteristic biological processes (BP) for a large number of
genes in microarray analysis (17). DAVID (Database for Annotation,
Visualization and Integrated Discovery, https://david.ncifcrf.gov/), as a comprehensive set of
functional annotation tools (18),
was used to investigate enriched BP terms for all the overlapping
DEGs. P<0.05 and Count ≥5 were considered statistically
significant.
PPI network construction and hub gene
analysis
STRING (Search Tool for the Retrieval of Interacting
Genes, https://string-db.org/) database is a
useful online tool dedicated to evaluate the PPI interaction
information (19). STRING (version
10.5) was used to explore the interactive relationships of the
overlapping DEGs, and only the experimentally validated
interactions with a combined score >0.4 were selected as
significant. Then, the PPI network was built by Cytoscape software
(version 3.5.1) (20). The plug-in
cytoHubba in Cytoscape was used to screen the hub genes from the
PPI network and only DEGs with a degree score ≥19 was selected as
hub genes.
Association of hub genes and patient
prognosis
An online survival analysis tool Kaplan Meier
plotter (http://kmplot.com/analysis/)
(21), which includes both
clinical data and gene expression data of lung, breast, gastric and
ovarian cancers, was used to evaluate the prognostic significance
of each hub gene in NSCLC. According to the median expression value
of a gene, the patient samples were divided into high and low
expression groups. In this study, the analysis was carried out
under the default parameters, which is in brief, no subtypes
restriction, ‘univariate’ for Cox regression and ‘exclude biased
arrays’ for array quality control and in each survival plot, the
log rank P-value and hazard ratio (HR, 95% confidence intervals)
were calculated and displayed on the main plot.
Results
Identification of DEGs
In order to identify the DEGs, we downloaded 4
public RNA-seq datasets of NSCLC from the GEO database for our
analysis: GSE21933, GSE33532, GSE44077 and GSE74706. We defined
DEGs between tumor samples and matched normal samples according to
the threshold criterion (logFC >1 or logFC <-1 and adjusted
P<0.05), and we got 1437, 2127, 963 and 4147 DEGs in GSE21933,
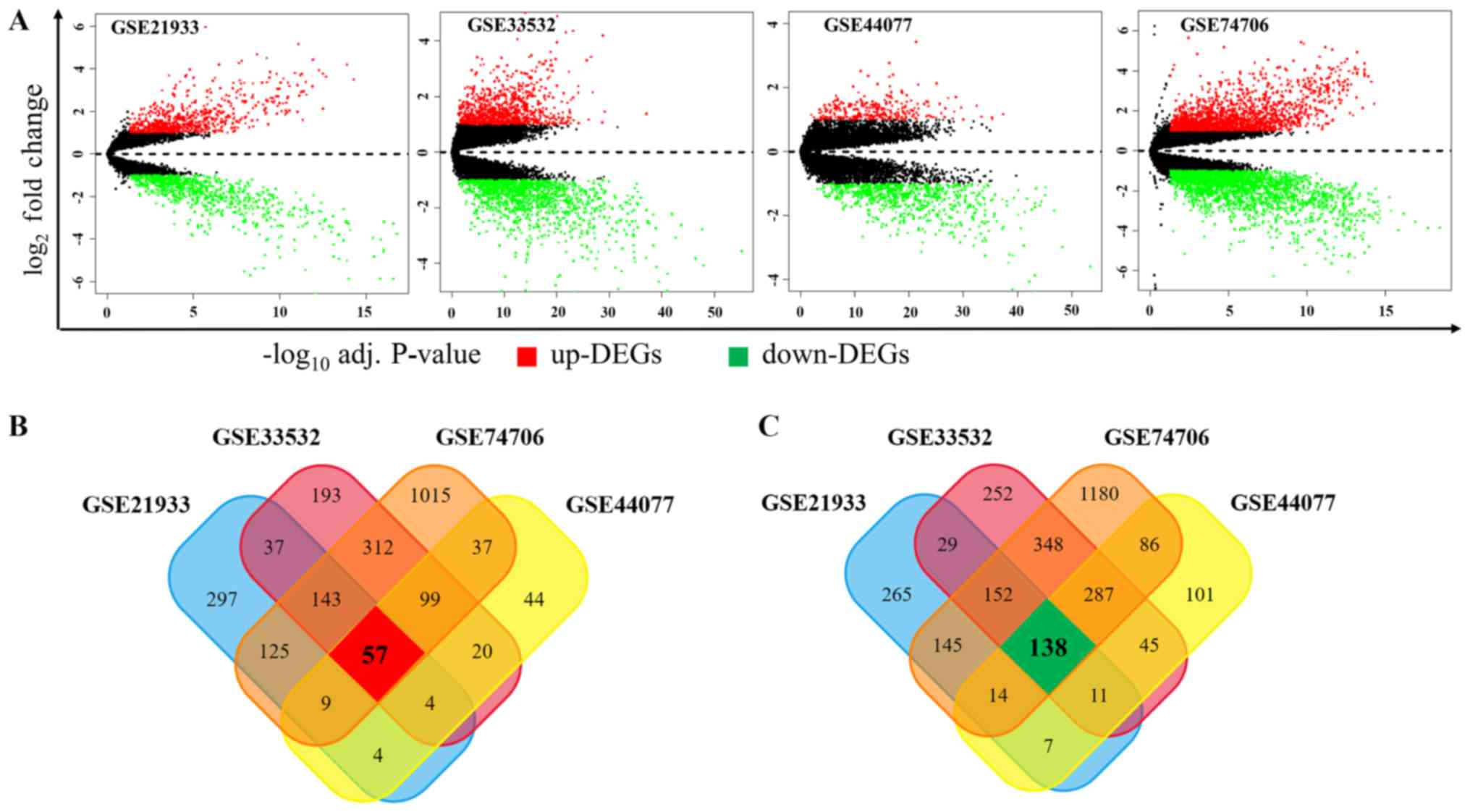
GSE33532, GSE44077 and GSE74706, respectively. As shown in Fig. 1A, among these DEGs, 676, 865, 274
and 1797 genes were upregulated while 761, 1262, 689, 2350 ones
were downregulated in GSE21933, GSE33532, GSE44077 and GSE74706,
respectively. In order to obtain the most reliable DEGs, we
isolated the DEGs presented in all four datasets and finally got a
total of 195 DEGs, consisting of 57 upregulated (Fig. 1B) and 138 downregulated DEGs
(Fig. 1C).
GO term enrichment analysis
In an attempt to find the potential biological
functions associated with these 195 DEGs, the online software DAVID
was used to identify the overrepresented GO categories. After GO
functional enrichment analysis, as shown in Table I, the upregulated DEGs were
significantly enriched in 18 biological process (BP) terms, which
includes mitosis, cell cycle, protein-DNA complex assembly,
microtubule-based process and cell proliferation. The downregulated
DEGs were also significantly enriched in 16 BP terms, such as
cellular response to hormone stimulus, angiogenesis, response to
drug and cell adhesion. All these terms are closely related to the
tumorigenesis and development.
 | Table I.Enriched Gene Ontology terms for
upregulated and downregulated differentially expressed genes. |
Table I.
Enriched Gene Ontology terms for
upregulated and downregulated differentially expressed genes.
| A, Upregulated |
|---|
|
|---|
| Term | Gene function | Count | P-value |
|---|
| GO:0007067 | Mitosis | 14 |
6.11×10‒13 |
| GO:0000280 | Nuclear
division | 14 |
6.11×10‒13 |
| GO:0000087 | M phase of mitotic
cell cycle | 14 |
7.70×10‒13 |
| GO:0048285 | Organelle
fission | 14 |
1.02×10‒12 |
| GO:0000279 | M phase | 15 |
6.02×10‒12 |
| GO:0022403 | Cell cycle
phase | 16 |
9.16×10‒12 |
| GO:0000278 | Mitotic cell
cycle | 15 |
2.91×10‒11 |
| GO:0022402 | Cell cycle
process | 16 |
7.21×10‒10 |
| GO:0007049 | Cell cycle | 16 |
5.28×10‒08 |
| GO:0051301 | Cell division | 11 |
8.09×10‒08 |
| GO:0065004 | Protein-DNA complex
assembly | 5 |
3.22×10‒04 |
| GO:0007018 | Microtubule-based
movement | 5 |
7.32×10‒04 |
| GO:0007017 | Microtubule-based
process | 6 |
2.14×10‒03 |
| GO:0034622 | Cellular
macromolecular complex assembly | 6 |
5.69×10‒03 |
| GO:0051726 | Regulation of cell
cycle | 6 |
6.72×10‒03 |
| GO:0034621 | Cellular
macromolecular complex subunit organization | 6 |
9.16×10‒03 |
| GO:0008283 | Cell
proliferation | 6 |
2.02×10‒02 |
|
| B,
Downregulated |
|
| Term | Gene function | Count | P-value |
|
| GO:0032870 | Cellular response
to hormone stimulus | 6 |
1.73×10‒05 |
| GO:0001525 | Angiogenesis | 10 |
3.41×10‒05 |
| GO:0050900 | Leukocyte
migration | 7 |
2.53×10‒04 |
| GO:0030336 | Negative regulation
of cell migration | 6 |
6.18×10‒04 |
| GO:0042493 | Response to
drug | 9 |
1.62×10‒03 |
| GO:0007165 | Signal
transduction | 18 |
3.98×10‒03 |
| GO:0007155 | Cell adhesion | 10 |
5.99×10‒03 |
| GO:0016337 | Single organismal
cell-cell adhesion | 5 |
6.12×10‒03 |
| GO:0002576 | Platelet
degranulation | 5 |
6.56×10‒03 |
| GO:0006898 | Receptor-mediated
endocytosis | 6 |
1.12×10‒02 |
| GO:0043065 | Positive regulation
of apoptotic process | 7 |
2.15×10‒02 |
| GO:0018108 | Peptidyl-tyrosine
phosphorylation | 5 |
2.48×10‒02 |
| GO:0045893 | Positive regulation
of transcription, DNA-templated | 9 |
3.28×10‒02 |
| GO:0045087 | Innate immune
response | 8 |
3.61×10‒02 |
| GO:0006810 | Transport | 7 |
4.02×10‒02 |
Hub genes screening from the PPI
network
To further investigate the interaction between these
195 DEGs, the STRING database was used to analysis their PPI
networks, and the resulted PPI networks were constructed by
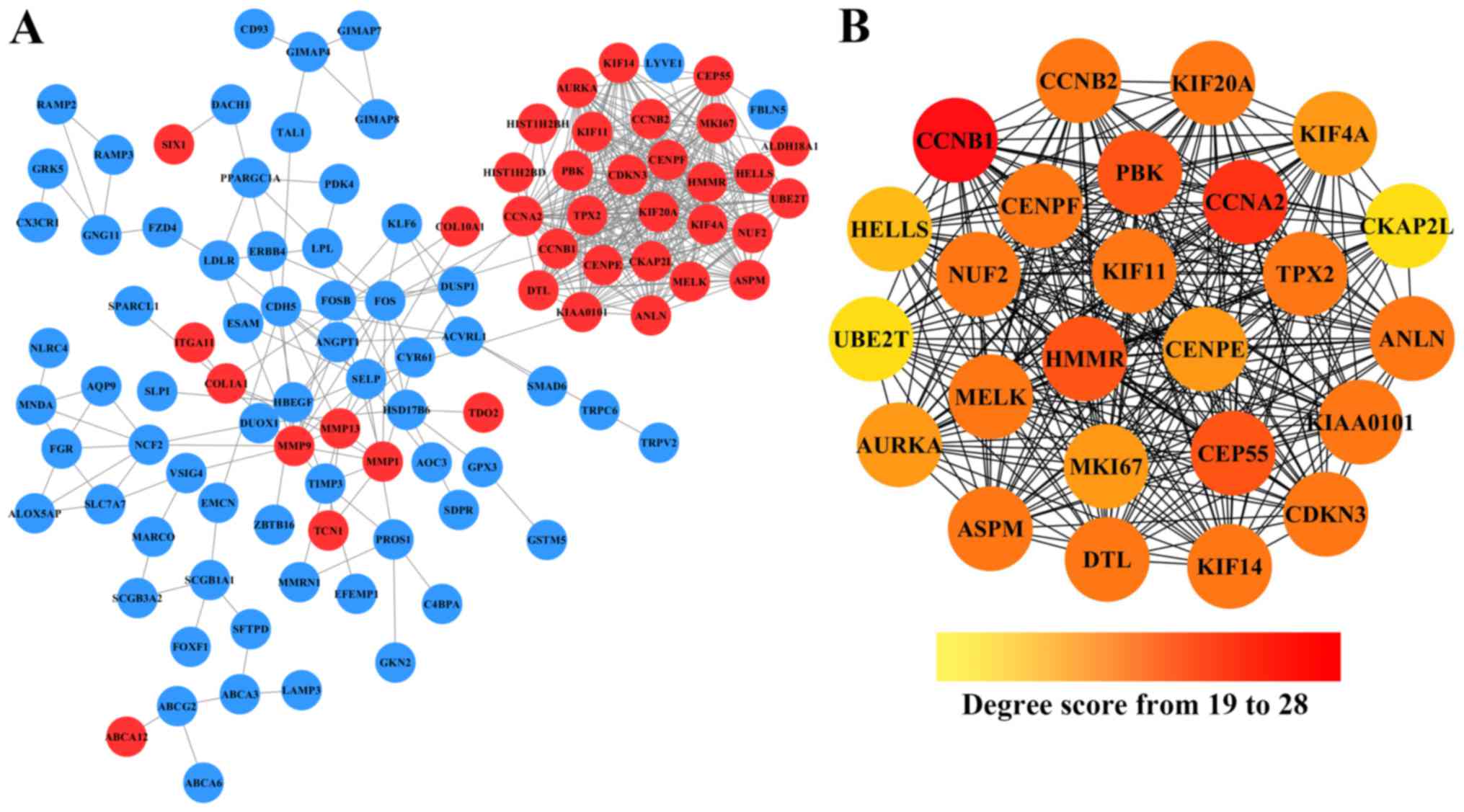
Cytoscape. As shown in Fig. 2A,
after removing the effect of free protein pairs, we obtained a
network with 38 upregulated DEGs, 66 downregulated DEGs and 424
edges.
In addition, in the PPI networks, genes that have
strong interactions with many other genes are called ‘hub genes’.
Due to their key positions in the PPI network, the hub genes are
potential drivers of the diseases status. In order to identify the
key tumorigenic genes of NSCLC, the cytoHubba plugin for Cytoscape
was used to screen the hub genes among all the DEGs. As shown in
Fig. 2B and Table II, we obtained 25 hub genes (the
nodes with red or orange colors) that exhibited especially high
degree scores (≥19). To our surprise, all these 25 hub genes were
upregulated DEGs. The genes G2/mitotic-specific cyclinB1 (CCNB1),
CyclinA2 (CCNA2), Centrosomal protein of 55 kDa (CEP55),
lymphokine-activated killer T-cell-originated protein kinase (PBK),
and hyaluronan mediated motility receptor (HMMR) were selected as
the top five hub genes with high connectivity degree. CCNB1, which
is also known as cyclin B1, was observed to exhibit the highest
degree of connectivity as shown in Table II.
| Table II.Statistical results of connectivity
degrees of the hub genes in the PPI network. |
Table II.
Statistical results of connectivity
degrees of the hub genes in the PPI network.
| Gene | Degree | Gene | Degree |
|---|
| CCNB1 | 28 | KIF11 | 24 |
| CCNA2 | 27 | KIF20A | 24 |
| CEP55 | 25 | CCNB2 | 24 |
| PBK | 25 | CENPF | 24 |
| HMMR | 25 | TPX2 | 24 |
| DTL | 24 | MKI67 | 23 |
| KIAA0101 | 24 | KIF4A | 23 |
| MELK | 24 | CENPE | 23 |
| ANLN | 24 | AURKA | 23 |
| KIF14 | 24 | HELLS | 21 |
| CDKN3 | 24 | CKAP2L | 19 |
| ASPM | 24 | UBE2T | 19 |
| NUF2 | 24 |
|
|
Survival analysis of each hub genes
using online tool Kaplan-Meier Plotter
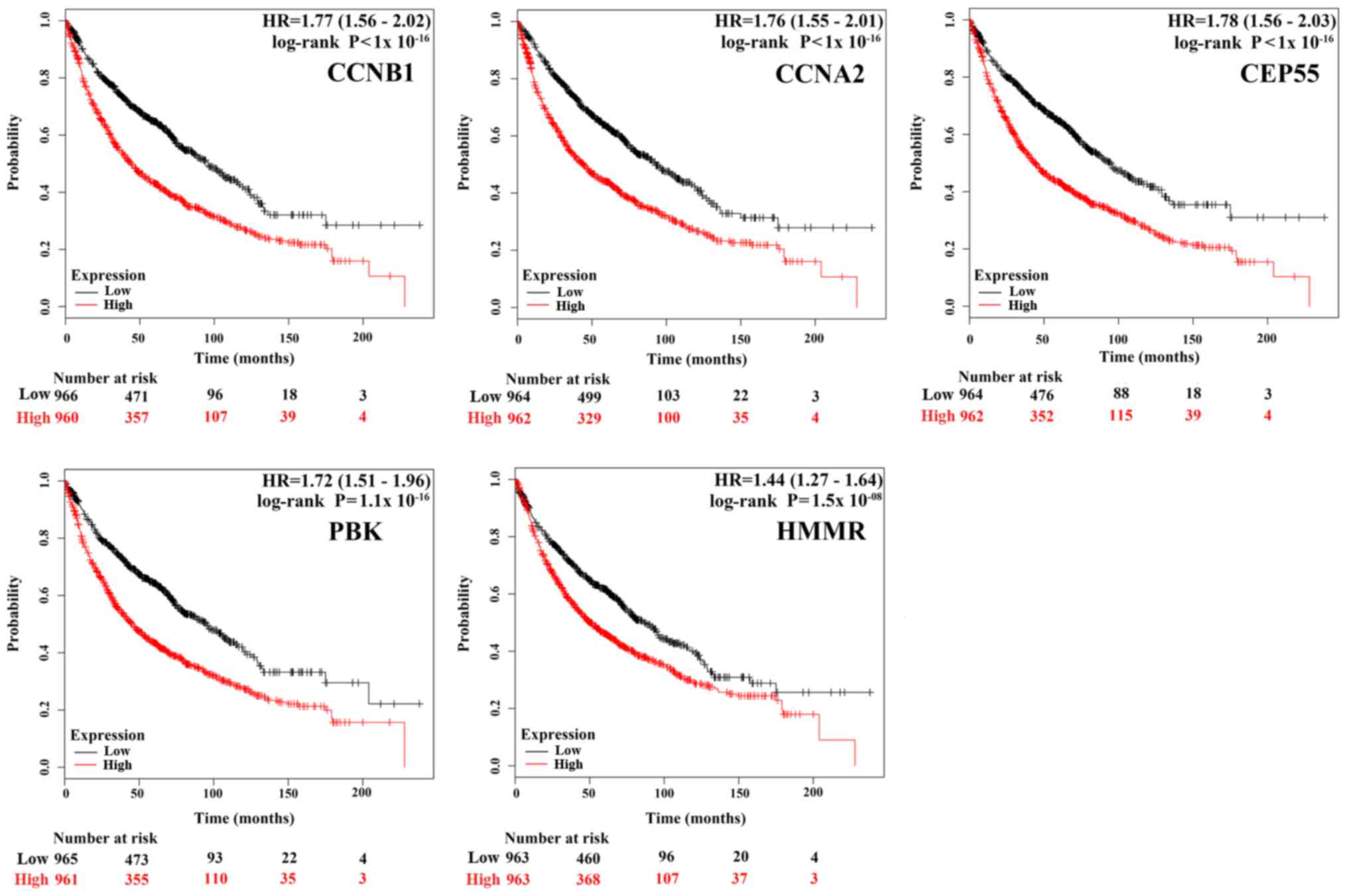
To gain insight into the associations between the
hub genes and NSCLC, we did a survival analysis through an online
tool Kaplan Meier Plotter, which contains a large number of
microarray datasets of lung cancer. As shown in Fig. 3, the survival time of the top five
hub genes was significantly separated between the high expression
groups and low expression groups in NSCLC patients (P<0.05),
with the low expression groups of these 5 hub genes exhibiting a
good prognostic effect in NSCLC. Moreover, the remaining 20 hub
genes also showed the same trend (data not shown). These imply that
the expression levels of the 25 hub genes are significantly
associated with clinical prognosis of NSCLC and they may play
important roles in the progression of NSCLC.
Discussion
The tumorigenesis, progression and metastasis of
lung cancer, like in any other cancer is considered as a very
complex process as it involves aberrations of multiple genes and
cellular pathways (22). Some DEGs
that have multi-interactions with other DEGs are probably core
functional genes in promoting the carcinogenesis (23,24).
To improve diagnosis and treatment of NSCLC, it is vitally
important to find these abnormal genes and understand their roles
in the molecular mechanism of NSCLC. With the development of
microarray and high throughput technologies, we have been able to
detect the cancer etiology by examining aberrations in whole-genome
level as these technologies have been widely used to predict the
potential therapeutic targets for cancers (25).
In the present study, using the gene expression
profiles of GSE21933, GSE33532, GSE44077 and GSE74706 to screen the
co-expressed DEGs between NSCLC and normal samples, a total of 195
DEGs, consisting of 57 upregulated and 138 downregulated DEGs were
obtained. The observation that, upregulated DEGs were enriched in
BP terms related to mitosis, cell cycle and cell proliferation is
consistent with previous knowledge that the functional deficits of
cell cycle and cell proliferation regulators are the main causes of
tumorigenesis and progression (26). The downregulated DEGs were enriched
in BP terms such as angiogenesis and cell adhesion. Angiogenesis is
an essential biological process for tumor growth and metastasis in
that, its blockage in tumor tissue has been recognized as a
charming strategy to inhibit tumor growth (27). According to Cavallaro and
Christofori (28), changes in the
expression of cell adhesion molecules could affect the adhesive
repertoire of a cell, signal transduction status of cells, the
interactions between cells and their environment, and play a
crucial role in tumor progression, invasion and metastasis. Our
results suggested that these up and downregulated DEGs are involved
in these BP and may play important role in the progression of
NSCLC.
Based on the PPI network, we identified a series of
hub genes and among the list of the top five hub genes were CCNB1,
CCNA2, CEP55, PBK and HMMR, respectively. CCNB1, as a key
regulatory protein involved in mitosis, is essential for G2/M
transition during the cell cycle (29). Yuan et al (30). reported that its depletion
inhibited proliferation and induced apoptosis in human tumor cells.
The work of Soria et al (31). established the expression of CCNB1
in a significant fraction but not all types of NSCLC. They showed
that, different subtypes of NSCLC are not only biologically
different but also showed variation in their CCNB1 expressions. Of
all the histological subtypes examined, overexpression of CCNB1 was
more frequently observed in the squamous cell carcinoma (SCC)
subtypes. This overexpression was also reported to affects patient
survival time and might be an adverse prognostic marker for
SCC-subtype NSCLC patients (31).
While our work establishes CCNB1 as a top hub gene in NSCLC in
agreement with these reports, it did not specify the subtypes,
which could be a possible limitation to our analysis. The second
hub gene CCNA2, encoding cyclins controls both the G1/S and the
G2/M transition of the cell cycle (32). Its protein expression was found to
be elevated in variety of tumors, including breast, liver, prostate
and lung cancers, and appears to be a prognostic marker in
prediction of survival or early relapse (33). The third hub gene CEP55 (also known
as c10orf3 and FLJ10540), a pivotal component of cytokinesis, is
associated with the PI3K/AKT pathway activation via an interaction
with the catalytic subunit of PI3K (34). CEP55 overexpression is highly
correlated with carcinogenesis across multiple cancer types
including lung, breast, liver, and colon cancers and has been
identified as a member of several prognostic ‘gene signatures’ for
cancer (35). Tao et al
(36). has reported that ectopic
expression of CEP55 induces tumorigenesis in nude mice, while its
knockdown in gastric cancer cells suppressed cellular proliferation
by inducing G2/M phase arrest, indicating its potential as an
anti-tumor target. PBK also known as TOPK, was found to play an
important role in tumor formation and progression. Its high
expression level was detectable in the majority of lung cancer
tissues and cell lines, but not in normal tissues (37–40).
Overexpression of PBK was reported to promote a PI3K/AKT-dependent
cell migration (5). Lei et
al (40). suggested that PBK
expression was positively correlated with Ki67 and p53 expression,
and could be used as an independent prognostic factor in NSCLC.
Another hub gene which we found to be hyper-expressed in NSCLC is
HMMR, a multifunctional oncogene. Its high expression level, which
was also found in breast cancer, is associated with poor disease
outcome (41). Recent studies have
deciphered that HMMR could regulate cell proliferation, survival,
and migration via forming a complex with CD44 (a well-known cancer
stem cell marker) and hyaluronan (a key component of the
microenvironment of most malignant tumors) (42). In this study, HMMR showed a strong
interaction with each of the top five hub genes, which indicates
their joint functions in NSCLC, although further investigations are
still needed to clarify underlying biological associations between
HMMR and NSCLC. All these reports indicate that CCNB1, CCNA2,
CEP55, PBK, and HMMR are involved in the pathogenesis of malignant
tumors mainly by affecting cell cycle and mitosis. This is
consistent with our findings. Furthermore, the Kaplan Meier Plotter
survival analysis from our study demonstrated that mRNA expression
levels of these five hub genes, as well as the remaining 20 hub
genes, were significantly associated with clinical prognosis of
lung cancer. While these could imply their roles in the progression
of NSCLC thereby making them a potential target for its diagnosis
and treatment, further experimental verification of the clinical
significance of each hub gene and its subtypes is still
necessary.
In summary, using the bioinformatics, we have
provided information on the DEGs and hub genes that may be involved
in NSCLC. This analysis is, however, limited in that it does not
the capture the expression of these genes in the different NSCLC
subtypes. Another limitation of this work is that, in reducing the
number of false positive DEGs, we obtained co-expressed DEGs among
4 different NSCLC associated microarray datasets. By this, many
likely important genes might have been lost. While our work showed
the benefit and usefulness of microarray analysis in bringing out
the DEGs and hub genes that can be potential diagnostic and
treatment targets of NSCLC, the need for improved analysis and
prospective clinical studies is still imperative. Albeit, this
study can contribute to the overall understanding of the underlying
molecular mechanisms of NSCLC and serve as guide to subsequent
experimental studies.
Acknowledgements
Not applicable.
Funding
This study was financially supported by Fundamental
Research Funds for the Central Universities (grant no.
XDJK2017D143), Fundamental Research Program of Hunan University of
Medicine (grant no. 2014KY03) and Science and Technology Plan
Projects of Huaihua (2014–17). Funding was also received from
Chongqing Science and Technology Commission (grant no.
cstc2015jcyjAoo61), Chongqing Educational Ministry (grant no.
Kj1500637) and The National Key Research and Development Program of
China for Traditional Chinese Medicine Modernization (grant nos.
2017YFC1702600 and 2017YFC1702605).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YBX and XGL conceived and designed the study. YBX,
MF, HYR and XH performed the data analysis. YBX and HYR wrote the
paper. YBX, XGL, MF, HYR and XH reviewed and edited the manuscript.
All authors read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Semenova EA, Nagel R and Berns A: Origins,
genetic landscape, and emerging therapies of small cell lung
cancer. Genes Dev. 29:1447–1462. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Danesi R, Pasqualetti G, Giovannetti E,
Crea F, Altavilla G, Del Tacca M and Rosell R: Pharmacogenomics in
non-small-cell lung cancer chemotherapy. Adv Drug Deliv Rev.
61:408–417. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shih MC, Chen JY, Wu YC, Jan YH, Yang BM,
Lu PJ, Cheng HC, Huang MS, Yang CJ, Hsiao M and Lai JM: TOPK/PBK
promotes cell migration via modulation of the PI3K/PTEN/AKT pathway
and is associated with poor prognosis in lung cancer. Oncogene.
31:2389–2400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Keith RL and Miller YE: Lung cancer
chemoprevention: Current status and future prospects. Nat Rev Clin
Oncol. 10:334–343. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oxnard GR, Lo PC, Nishino M, Dahlberg SE,
Lindeman NI, Butaney M, Jackman DM, Johnson BE and Jänne PA:
Natural history and molecular characteristics of lung cancers
harboring EGFR exon 20 insertions. J Thorac Oncol. 8:179–184. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gerber DE and Minna JD: ALK inhibition for
non-small cell lung cancer: From discovery to therapy in record
time. Cancer cell. 18:548–551. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Janku F, Stewart DJ and Kurzrock R:
Targeted therapy in non-small-cell lung cancer-is it becoming a
reality? Nat Rev Clin Oncol. 7:401–414. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lo FY, Chang JW, Chang IS, Chen YJ, Hsu
HS, Huang SF, Tsai FY, Jiang SS, Kanteti R, Nandi S, et al: The
database of chromosome imbalance regions and genes resided in lung
cancer from Asian and Caucasian identified by array-comparative
genomic hybridization. BMC Cancer. 12:2352012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meister M, Belousov A, Xu EC, Schnabel PA,
Warth A, Hoffmann H, Dienemann H, Riedlinger J, Bodenmueller H,
Zolg W, et al: Intra-tumor heterogeneity of gene expression
profiles in early stage. Non-small cell lung cancer. 2014.
|
|
12
|
Kadara H, Fujimoto J, Yoo SY, Maki Y,
Gower AC, Kabbout M, Garcia MM, Chow CW, Chu Z, Mendoza G, et al:
Transcriptomic architecture of the adjacent airway field
cancerization in non-small cell lung cancer. J Natl Cancer Inst.
106:dju0042014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marwitz S, Depner S, Dvornikov D, Merkle
R, Szczygieł M, Müller-Decker K, Lucarelli P, Wäsch M, Mairbäurl H,
Rabe KF, et al: Downregulation of the TGFβ Pseudoreceptor BAMBI in
non-small cell lung cancer enhances TGFβ signaling and invasion.
Cancer Res. 76:3785–3801. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J Royal Statis Soc. 57:289–300. 1995.
|
|
16
|
Pathan M, Keerthikumar S, Ang CS, Gangoda
L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim
A, et al: FunRich: An open access standalone functional enrichment
and interaction network analysis tool. Proteomics. 15:2597–2601.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gene Ontology Consortium: The gene
ontology (GO) project in 2006. Nucleic Acids Res. 34:(Database
Issue). D322–D326. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Szklarczyk D, Morris JH, Cook H, Kuhn M,
Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al:
The STRING database in 2017: Quality-controlled protein-protein
association networks, made broadly accessible. Nucleic Acids Res.
45(D1): D362–D368. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Szász AM, Lánczky A, Nagy Á, Förster S,
Hark K, Green JE, Boussioutas A, Busuttil R, Szabó A and Győrffy B:
Cross-validation of survival associated biomarkers in gastric
cancer using transcriptomic data of 1,065 patients. Oncotarget.
7:49322–49333. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Saito M, Shiraishi K, Kunitoh H,
Takenoshita S, Yokota J and Kohno T: Gene aberrations for precision
medicine against lung adenocarcinoma. Cancer Sci. 107:713–720.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cardarella S and Johnson BE: The impact of
genomic changes on treatment of lung cancer. Am J Respir Crit Care
Med. 188:770–775. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liang B, Li C and Zhao J: Identification
of key pathways and genes in colorectal cancer using bioinformatics
analysis. Med Oncol. 33:1112016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Feng X, Ofstad W and Hawkins D:
Antiangiogenesis therapy: A new strategy for cancer treatment. US
Pharm. 35 Oncology Suppl:S4–S9. 2010.
|
|
28
|
Cavallaro U and Christofori G: Cell
adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev
Cancer. 4:118–132. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Petri ET, Errico A, Escobedo L, Hunt T and
Basavappa R: The crystal structure of human cyclin B. Cell Cycle.
6:1342–1349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yuan J, Yan R, Krämer A, Eckerdt F, Roller
M, Kaufmann M and Strebhardt K: Cyclin B1 depletion inhibits
proliferation and induces apoptosis in human tumor cells. Oncogene.
23:5843–5852. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Soria JC, Jang SJ, Khuri FR, Hassan K, Liu
D, Hong WK and Mao L: Overexpression of cyclin B1 in early-stage
non-small cell lung cancer and its clinical implication. Cancer
Res. 60:4000–4004. 2000.PubMed/NCBI
|
|
32
|
Pagano M, Pepperkok R, Verde F, Ansorge W
and Draetta G: Cyclin A is required at two points in the human cell
cycle. EMBO J. 11:961–971. 1992.PubMed/NCBI
|
|
33
|
Yam CH, Fung TK and Poon RY: Cyclin A in
cell cycle control and cancer. Cell Mol Life Sci. 59:1317–1326.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen CH, Lu PJ, Chen YC, Fu SL, Wu KJ,
Tsou AP, Lee YC, Lin TC, Hsu SL, Lin WJ, et al: FLJ10540-elicited
cell transformation is through the activation of PI3-kinase/AKT
pathway. Oncogene. 26:4272–4283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jeffery J, Sinha D, Srihari S, Kalimutho M
and Khanna KK: Beyond cytokinesis: The emerging roles of CEP55 in
tumorigenesis. Oncogene. 35:683–690. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tao J, Zhi X, Tian Y, Li Z, Zhu Y, Wang W,
Xie K, Tang J, Zhang X, Wang L and Xu Z: CEP55 contributes to human
gastric carcinoma by regulating cell proliferation. Tumour Biol.
35:4389–4399. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu F, Zykova TA, Kang BS, Wang Z, Ebeling
MC, Abe Y, Ma WY, Bode AM and Dong Z: Bidirectional signals
transduced by TOPK-ERK interaction increase tumorigenesis of HCT116
colorectal cancer cells. Gastroenterology. 133:219–231. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zykova TA, Zhu F, Lu C, Higgins L, Tatsumi
Y, Abe Y, Bode AM and Dong Z: Lymphokine-activated killer
T-cell-originated protein kinase phosphorylation of histone H2AX
prevents arsenite-induced apoptosis in RPMI7951 melanoma cells.
Clin Cancer Res. 12:6884–6893. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zlobec I, Molinari F, Kovac M, Bihl MP,
Altermatt HJ, Diebold J, Frick H, Germer M, Horcic M, Montani M, et
al: Prognostic and predictive value of TOPK stratified by KRAS and
BRAF gene alterations in sporadic, hereditary and metastatic
colorectal cancer patients. Br J Cancer. 102:151–161. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lei B, Liu S, Qi W, Zhao Y, Li Y, Lin N,
Xu X, Zhi C, Mei J, Yan Z, et al: PBK/TOPK expression in
non-small-cell lung cancer: Its correlation and prognostic
significance with Ki67 and p53 expression. Histopathology.
63:696–703. 2013.PubMed/NCBI
|
|
41
|
Pujana MA, Han JD, Starita LM, Stevens KN,
Tewari M, Ahn JS, Rennert G, Moreno V, Kirchhoff T, Gold B, et al:
Network modeling links breast cancer susceptibility and centrosome
dysfunction. Nat Genet. 39:1338–1349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hamilton SR, Fard SF, Paiwand FF, Tolg C,
Veiseh M, Wang C, McCarthy JB, Bissell MJ, Koropatnick J and Turley
EA: The hyaluronan receptors CD44 and Rhamm (CD168) form complexes
with ERK1,2 that sustain high basal motility in breast cancer
cells. J Biol Chem. 282:16667–16680. 2007. View Article : Google Scholar : PubMed/NCBI
|