Introduction
The identification of mutations in genes that
determine a high risk of developing hereditary colorectal cancer
(CRC) has allow to performed genetic testing to identify subjects
with strong predisposition to cancer (1). The hereditary CRC syndromes are
divided in polyposis, as FAP, MAP, PHTS (2–4), and
in non-polyposis, as Lynch syndrome (LS) (5). The LS is an autosomal dominant
syndrome; besides CRC, the phenotypic spectrum of LS includes other
primary tumors, as cancer of the stomach, endometrium, biliary and
pancreatic system, and urinary tract. The ‘Revised Amsterdam
Criteria’ represent the diagnostic guidelines designed to identify
LS families. Subsequently, Bethesda guidelines were developed to
improve identification of LS patients (5,6).
The LS is associated with germline mutations in one
of the mismatch repair (MMR) genes, including MutL homolog 1
(MLH1), MutS homolog 2 (MSH2), MSH6, PMS1
homolog 2, mismatch repair system component (PMS2), MLH3
and MSH3 (7–10). The loss of function of one MMR
protein prevents to repair's complex to work properly and this
determines a genetic instability known as microsatellite
instability (MSI) at somatic level (11). The mutations in MMR genes
are point mutations or large rearrangements (7,12,13).
Most of point mutations determine formation of truncated protein
through nonsense mutations, insertions or deletions; many often,
substitutions of a single nucleotide (as in missense and silent
mutations) may also create a truncated protein (14). Indeed, some exonic and intronic
variants create or disrupt splice sites and consequently, this
leads the creation of aberrant splicing mRNA transcripts (7). All these genetic variants in
MMR genes altering the protein function are considered to be
‘pathogenic’.
In this study, we have identified and characterized
a novel MSH2 gene splicing mutation in a LS young
patient.
Materials and methods
Patient history
Our proband is a 28-year-old male. He underwent
partial colon resection (proximal descending and transverse colon)
for a poorly differentiated adenocarcinoma in transverse colon
diagnosed at the age of 26 years. Subsequenty, at 27 years of age,
he developed sigmoid tubular adenomatous polyp, which was resected.
The proband's family history was also positive for colorectal and
extra-colonic cancer. A detailed pedigree is shown in Fig. 1.
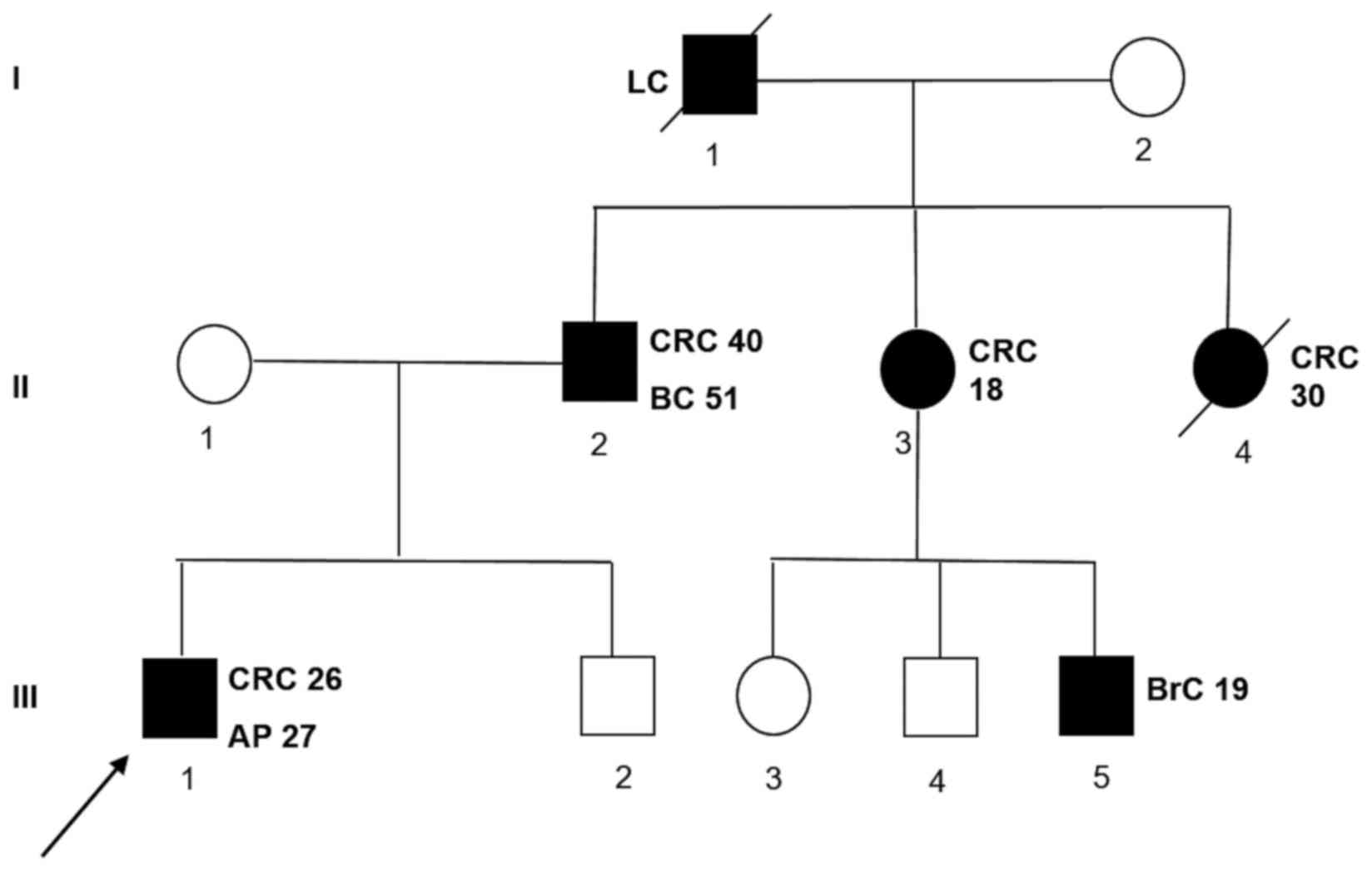 | Figure 1.Family pedigree of the patient with a
novel MutS homolog 2 gene mutation. The numbers next to each
diagnosis denote the age at onset. Symbols and abbreviations used
are denoted as follows: Arrow, index case (proband); symbols with
diagonal lines, succumbed; black symbol, CRC or tumors associated
with Lynch syndrome; CRC, colorectal cancer; LC, lung cancer; BC,
bladder cancer; BrC, brain cancer; AP, adenomatous polyp; squares,
males; circles, females. |
Sample from our patient was collected after being
granted authorisation from the Ethics Committee ‘Comitato etico per
le attività Biomediche-Carlo Romano’ of the University of Naples
Federico II (Naples, Italy; protocol no. 120/10). Once the
authorisation has been obtained the study has received ethical
approval, and the participant informed and written consent has been
obtained.
Furthermore, as negative controls we collected 100
healthy samples from Clinical Department of Laboratory Medicine of
our Hospital (Federico II of Naples).
Isolation of genomic DNA
The genomic DNA was extracted from peripheral blood
lymphocytes and from paraffin-embedded tumor tissue of our patient.
Total genomic DNA was extracted from 4 ml peripheral blood
lymphocytes using a BACC2 Nucleon kit (Amersham; GE Healthcare,
Chicago, IL, USA). For each paraffin block, five 20-µm-thick
sections were cut and collected in a 1.5-ml micro tube and 1 ml
xylene was added to each tube and kept at room temperature for 20
min to remove the paraffin completely, according to the protocol
described by Duraturo et al (7). Subsequently, the somatic DNA was
extracted using a BACC2 Nucleon kit.
Microsatellite analysis and V600E BRAF
mutation analysis
Microsatellite instability was tested on paired
samples from lymphocyte DNA and from paraffin-embedded tumor colon
tissues. The MSI status was evaluated using the CC-MSI kit
(AbAnalitica, Padova, Italy) and subsequent capillary
electrophoresis analysis using an ABI 3130 Prism (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA), as
previously described (7). For
V600E genotyping, genomic DNA extracted from paraffin-embedded
tumor tissue and blood lymphocytes were amplified using customized
primer pair, (15F-5′-TGCTTGCTCTGATAGGAAAATGAGA-3′- and
15R-5′-GGCCCTGAGATGCTGCTGAG-3′-), and sequenced in both the forward
and reverse directions using an ABI 3100 Genetic Analyser (Applied
Biosystems; Thermo Fisher Scientific, Inc.).
Mutation analysis: Amplification,
dHPLC and sequencing
All MLH1 (NM_000249) and MSH2 (NM_000251.2) exons
were amplified, including intron-exon boundaries, on DNA extracted
from blood lymphocytes of our patient, using customized primer
sets. Prior to dHPLC analysis, the polymerase chain reaction (PCR)
products were run on an 1–2% agarose gel to check for unspecific
amplicons. A Transgenomic Wave DNA Fragment Analysis System (3500
HT) was used to perform dHPLC analysis (Transgenomic Inc., Omaha,
NE, USA) using personal methods, available on request;
subsequently, genomic DNA was re-amplified and sequenced in both
the forward and reverse directions using an ABI 3100 Genetic
Analyser (Applied Biosystems; Thermo Fisher Scientific, Inc.).
In silico analysis
We analyzed the novel variant detected in this study
by the Human Splicing Finder (HSF) software (http://www.umd.be/HSF/) (15), a tool designed to predict the
effects of mutations on splicing signals or to identify splicing
motifs in human sequences, as previously described.
Reverse transcription (RT)-PCR MSH2
cDNA fragment
Total RNA was extracted from lymphocytes of our
patient and three normal controls using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) by standard procedure. cDNA was
synthesised using SuperScript II RT (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. PCR
amplification reactions of the cDNA fragment of 563 bp, including
the exons 1-2-3 of MSH2 gene was performed using customized primer
pair, (1F-5′-CTTCGTGCGCTTCTTTCAG-3′ and
1R-5′-TGTTTTACCCGGAGGAGAGA-3′). Amplified fragments were visualized
on 1% agarose gel and a 8% polyacrilamide gel. Each band was
excised from polyacrilamide gel and re-suspended only in 30 µl
water, over-night. Then, 1 µl was re-amplified and subsequently
sequenced using the same primer pair.
Results
First, we performed MSI analysis on our proband and
a MSI-H status was detected in DNA extracted from tumour tissues,
with instability of all nucleotide markers analyzed and complete
absence of BAT26 mononucleotide. No V600E mutation in BRAF
gene was identified. Subsequently, all MLH1 and MSH2
exons were analysed by DHPLC and the altered exonic fragments were
only sequenced. In this manner, we identified a novel mutation in
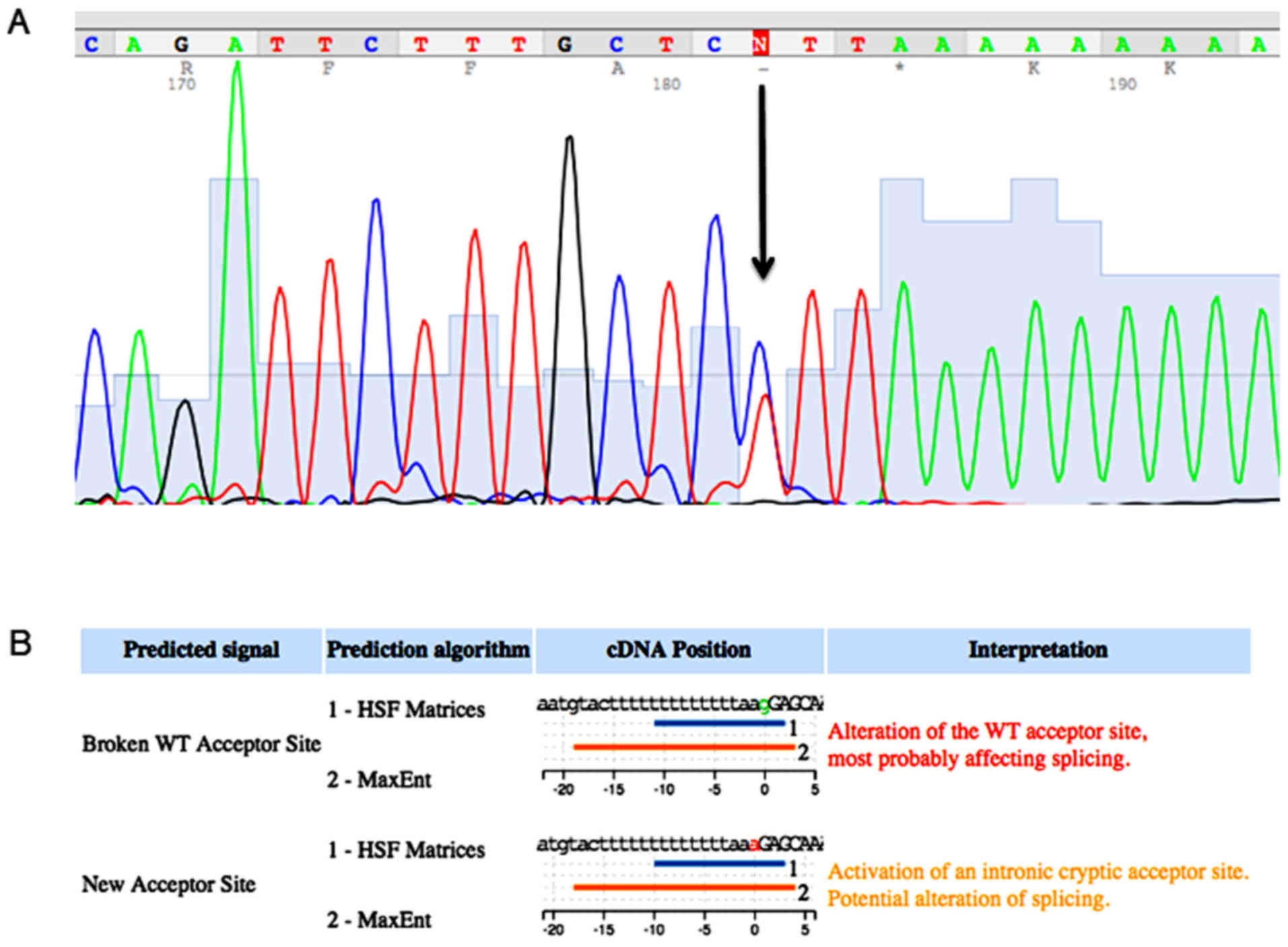
the MSH2 gene that determined a nucleotide substitution
(g>a) in the acceptor site, upstream at exon 2. The mutation
that is named c. 212-1g>a, has not been reported before in the
international database of InSiGHT-Group (http://www.insight-group.org/) and it was not detected
in 100 healthy controls analyzed, Fig.
2A. In silico analysis carried out using the HSF software
showed that this mutation altered the wild type acceptor site and
most probably it created a new acceptor site, Fig. 2B. The analysis by PCR of the
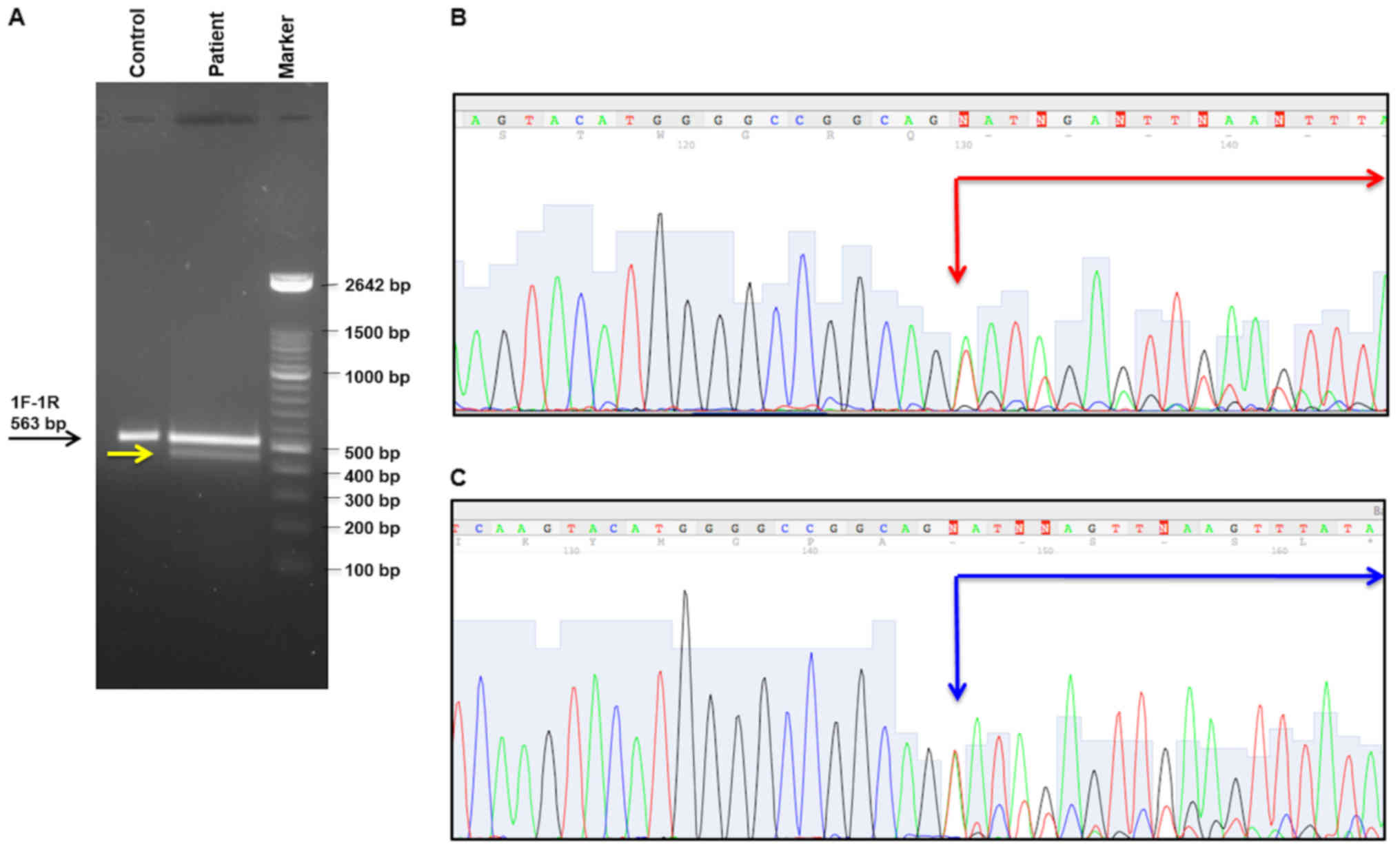
MSH2 cDNA fragment including the exons 1-2-3 of this gene
confirmed this computational data. In particular, abnormal band was
showed by this analysis, Fig. 3A.
Each amplification product visualized on the polyacrilamide 8% gel
has been extracted and sequenced, as described in Materials and
Methods. Two bands revealed two splicing isoforms of MSH2
mRNA determining a skipping of 80 and 85 bp, respectively
[(c.212_c.292) and (c.212_c.297)], Fig. 3B and C. Both two transcripts
determined a frameshift with premature stop codon and consequently,
the formation of a truncated protein (p.Gly71Valfs*2 and
p.Gly71Glufs*75, respectively). Therefore, this mutation is
considered as pathogenetic. No large rearrangements in MLH1
and MSH2 genes and no other point mutations in MLH1
and MSH6 genes were identified in our patient.
Discussion
Genetic testing of MMR genes has been widely
applied to aid the diagnosis of LS. So far, many pathogenetic
mutations were identified in these genes, in particular in
MLH1 and MSH2.
In this study, we identified a novel germline
variant, MSH2 c.212-1g>a, which is a variant that affects
the correct mechanism of splicing. This variant is located in
acceptor splice site (−1) upstream of exon 2 in MSH2 and it
created two new splice acceptor site to 80 and 85 bases
respectively, downstream of the primary splice acceptor site. Thus,
this mutation didn't determine the skipping in-frame of exon 2 but
it promoted the formation of two aberrant splicing transcripts with
the loss of 80 and 85 bp, respectively. Both this splicing isoforms
determined a frameshift of MSH2 mRNA with consequent
premature stop codon, 2 and 75 codons after, respectively. The
formation of a premature stop codon located within and not at the
end of the transcript increases the probability that non-sense
mediated decay (NMD) might be involved in degrading aberrant
transcripts (16). Anyhow, two
aberrant transcripts likely formed a truncated protein thus not
functional. This result was concordant with the MSI-high status (it
showed instability of all repetitive sequences analyzed) and with
complete absence of BAT26 markers on DNA extracted from colon
cancer specimens of our patient. It is known that the variants
around the splicing donor and acceptor sites alter the correct
splicing mechanism (17).
According to the InSiGHT databases (http://www.insight-group.org/), the variants that fall
into the splice site, donor or acceptor (±1 or ±2) are classified
as ‘likely pathogenic’ variants. The most of these variants in
canonical splice site determines the skipping in-frame of relative
exon. Interestingly, the molecular characterization of our novel
variant, the c212-1g>a in MSH2 gene showed that it
created two aberrant splicing transcripts [(c.212_c.292) and
(c.212_c.297)] and that both determined the formation of protein
truncated (p.Gly71Valfs*2 and p.Gly71Glufs*75).
This novel MSH2 mutation was identified in a
28-year-old patient who developed an adenocarcinoma in transverse
colon diagnosed at the age of 26 years and, subsequently he
developed sigmoid tubular adenomatous polyp at 27 years of age. Our
patient's family history was also positive for colorectal and
extra-colonic cancer and met the ‘Revised Amsterdam Criteria’,
Fig. 1. In this family, there were
two cases of colon cancers early onset (II-3 and II-4 subjects) and
several cases of extracolic cancers. Indeed, the subject III-5
developed a brain cancer at 18 years of age, the proband's father
(II-1) has developed a bladder cancer beyond colon cancer and the
grandfather (I-1) a lung cancer. Unfortunately, we were not able to
performed genetic testing for other affected subjects of this
family due to their limited availability. However, this variant,
that is considered to be ‘pathogenic’ is likely responsible of LS
phenotype in this family.
It remains to be clarified whether MSH2
mutation alone induces the occurrence of LS-related cancers and
very early onset of these cancers, in this family. Literature
review indicated that genetic disorder and dietary and/or
environment factors had synergistic effect in promoting cancer
initiation in MSH2-defective individuals (17). For example, it was reported that
germline ablation of SMUG1 DNA glycosylase causes loss of
5-hydroxymethyluracil- and UNG-backup uracil-excision activities
and increases cancer predisposition of Ung-/-Msh2-/-
mice (18); as it has also been
shown that interaction between microbiota and dietary factors tends
to reduce the occurrence of CRC and other cancers in APC
(Min/+)MSH2(−/-) mice (19). Therefore, this suggested that other
genetic instabilities could increase the risk to develop LS-releted
cancers and to anticipate age onset MSH2-defective resultant
cancers.
In conclusion, in this study we have identified and
characterized a novel splicing mutation in MSH2 gene. In
particular, we investigated how one novel mutation into the
consensus splice site of MSH2 exon 2 leads the formation of
two aberrant transcripts, due to activation of new splice sites
inside of exon 2. Moreover, in the light of recent literature data
according to which molecular characterization of cancer-associated
mutations can provide valuable information on disease prognosis and
patient response to therapy (20)
this study reaffirms the importance to identify pathogenic
mutations in LS families to facilitate pre-symptomatic diagnosis
and to improve therapeutic pathway in order to promote a
personalized medicine.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Campania
Regional Authority [granted to CEINGE, Biotecnologie Avanzate
(Naples, Italy; 2010–2012 POR Campania, grant no.
fSe2007-2013)].
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FD and PI conceived and designed the experiments. RL
performed the experiments. FD and MDR analyzed the data. FD and MDR
interpreted the data, and FD wrote the first draft of the
manuscript. FD and RL agreed with the results and conclusions of
the manuscript. FD developed the structure and arguments for the
paper. RL, MDR and PI made critical revisions. All authors reviewed
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of ‘Comitato etico per le attività Biomediche-Carlo
Romano’ of the University of Naples Federico II (Naples, Italy;
protocol no. 120/10), and written informed consent was obtained
from all participants.
Consent for publication
Written informed consent was obtained from all
participants.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
De Rosa M, Pace U, Rega D, Costabile V,
Duraturo F, Izzo P and Delrio P: Genetics, diagnosis and management
of colorectal cancer (Review). Oncol Rep. 34:1087–1096. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dodaro C, Grifasi C, Florio J, Santangelo
ML, Duraturo F, De Rosa M, Izzo P and Renda A: The role of mutation
analysis of the APC gene in the management of FAP patients. A
controversial issue. Ann Ital Chir. 87:321–325. 2016.PubMed/NCBI
|
|
3
|
De Rosa M, Galatola M, Borriello S,
Duraturo F, Masone S and Izzo P: Implication of adenomatous
polyposis coli and MUTYH mutations in familial colorectal
polyposis. Dis Colon Rectum. 52:268–274. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Paparo L, Rossi GB, Delrio P, Rega D,
Duraturo F, Liccardo R, Debellis M, Izzo P and De Rosa M:
Differential expression of PTEN gene correlates with phenotypic
heterogeneity in three cases of patients showing clinical
manifestations of PTEN hamartoma tumour syndrome. Hered Cancer Clin
Pract. 11:82013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liccardo R, De Rosa M, Izzo P and Duraturo
F: Novel implications in molecular diagnosis of lynch syndrome.
Gastroenterol Res Pract. 2017:25950982017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Giardiello FM, Allen JI, Axilbund JE,
Boland CR, Burke CA, Burt RW, Church JM, Dominitz JA, Johnson DA,
Kaltenbach T, et al: Guidelines on genetic evaluation and
management of Lynch syndrome: A consensus statement by the US
multi-society task force on colorectal cancer. Gastroenterology.
147:502–526. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Duraturo F, Liccardo R, Cavallo A, De Rosa
M, Rossi GB and Izzo P: Multivariate analysis as a method for
evaluating the pathogenicity of novel genetic MLH1 variants in
patients with colorectal cancer and microsatellite instability. Int
J Mol Med. 36:511–517. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Duraturo F, Liccardo R, Cavallo A, De Rosa
M, Grosso M and Izzo P: Association of low-risk MSH3 and MSH2
variant alleles with Lynch syndrome: Probability of synergistic
effects. Int J Cancer. 129:1643–1650. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liccardo R, De Rosa M, Rossi GB,
Carlomagno N, Izzo P and Duraturo F: Incomplete segregation of MSH6
frameshift variants with phenotype of lynch syndrome. Int J Mol
Sci. 18:pii: E999. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duraturo F, Liccardo R and Izzo P:
Coexistence of MLH3 germline variants in colon cancer patients
belonging to families with Lynch syndrome-associated brain tumors.
J Neurooncol. 129:577–578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gelsomino F, Barbolini M, Spallanzani A,
Pugliese G and Cascinu S: The evolving role of microsatellite
instability in colorectal cancer: A review. Cancer Treat Rev.
51:19–26. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Duraturo F, Cavallo A, Liccardo R, Cudia
B, De Rosa M, Diana G and Izzo P: Contribution of large genomic
rearrangements in Italian Lynch syndrome patients: Characterization
of a novel alu-mediated deletion. Biomed Res Int. 2013:2198972013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cudia B, Liccardo R, Di Carlo G, Damiano
G, Ignazio A, Monte L, Izzo P and Duraturo F: Clinical and
anamnestic evaluation rôle for the diagnosis and treatment of
families affected by lynch syndrome. Case report and review of the
literature. Eur J Oncol. 19:265–271. 2014.
|
|
14
|
Van der Klift HM, Jansen AM, van der
Steenstraten N, Bik EC, Tops CM, Devilee P and Wijnen JT: Splicing
analysis for exonic and intronic mismatch repair gene variants
associated with Lynch syndrome confirms high concordance between
minigene assays and patient RNA analyses. Mol Genet Genomic Med.
3:327–345. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Desmet FO, Hamroun D, Lalande M,
Collod-Béroud G, Claustres M and Béroud C: Human splicing finder:
An online bioinformatics tool to predict splicing signals. Nucleic
Acids Res. 37:e672009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
De Rosa M, Morelli G, Cesaro E, Duraturo
F, Turano M, Rossi GB, Delrio P and Izzo P: Alternative splicing
and nonsense-mediated mRNA decay in the regulation of a new
adenomatous polyposis coli transcript. Gene. 395:8–14. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hu H, Li H, Jiao F, Han T, Zhuo M, Cui J,
Li Y and Wang L: Association of a novel point mutation in MSH2 gene
with familial multiple primary cancers. J Hematol Oncol.
10:1582017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kemmerich K, Dingler FA, Rada C and
Neuberger MS: Germline ablation of SMUG1 DNA glycosylase causes
loss of 5-hydroxymethyluracil- and UNG-backup uracil-excision
activities and increases cancer predisposition of Ung-/-Msh2-/-
mice. Nucleic Acids Res. 40:6016–6025. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Belcheva A, Irrazabal T, Robertson SJ,
Streutker C, Maughan H, Rubino S, Moriyama EH, Copeland JK,
Surendra A, Kumar S, et al: Gut microbial metabolism drives
transformation of MSH2-deficient colon epithelial cells. Cell.
158:288–299. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
De Rosa M, Rega D, Costabile V, Duraturo
F, Niglio A, Izzo P, Pace U and Delrio P: The biological complexity
of colorectal cancer: Insights into biomarkers for early detection
and personalized care. Therap Adv Gastroenterol. 9:861–886. 2016.
View Article : Google Scholar : PubMed/NCBI
|