Introduction
Choroideremia (OMIM: 303100) is an X-linked
recessive chorioretinal disorder characterized by progressive
degeneration of photoreceptors, retinal pigment epithelium (RPE)
and the choroid, with an estimated incidence estimated of 1 in
50,000 to 1 in 100,000 in men (1).
Affected hemizygous males usually develop progressive vision loss
beginning with night blindness in the first or second decade of
their lives, that progresses to reduced function of the peripheral
visual fields, and eventually complete blindness later in life
(1). Female carriers are often
asymptomatic or experience mild symptoms due to X-inactivation
(2). However, occasional female
carriers can also be fully affected by choroideremia (3).
The molecular basis of choroideremia involves
mutation in the CHM gene (MIM: 300390) on chromosome Xq21.2.
The CHM gene contains 15 exons spanning approximately 150 Kb
of genomic DNA that encodes a ubiquitously expressed protein, Rab
escort protein-1 (REP-1). Until recently, about 300 pathogenic
variations of the CHM gene have been identified in
choroideremia patients (LOVD Retinal and Hearing Impairment Genetic
Mutation Database; http://www.lovd.nl/CHM), including small deletions,
duplication, nonsense mutations, missense mutations, frameshifts,
splice site defects, substitution in the promoter, retrotransposon
insertion, and deletion of the entire CHM gene (4–6).
REP-1 is an essential component of the catalytic Rab
geranyl-geranyl transferase (GGTase) II complex, in which it is
involved in post-translational isoprenyl modification (7) and intracellular vesicular trafficking
(8). REP-1 loss-of-function
mutations can be compensated by REP-2 in most tissues; however,
REP-2 exhibits less effective geranylgeranylation of Rab27
(9), which plays an important and
unique role in regulating vesicular transport in RPE and
choriocapillaris cells (10,11).
Moreover, REP-1 is particularly crucial for the physical function
of RPE and photoreceptors (12,13).
In this study, we conducted a detailed clinical
investigation and targeted exome sequencing in a Chinese
choroideremia pedigree. A frameshift mutation c.280delA
(p.Thr94LeufsTer32) in CHM was detected in the male patients
as well as a female carrier. This mutation was absent in the
unaffected parents and 200 ethnicity-matched healthy controls. Our
findings indicate that this mutation might be an etiological factor
of the current pedigree, thus expanding the genetic variation
spectrum of choroideremia.
Subjects and methods
Subjects
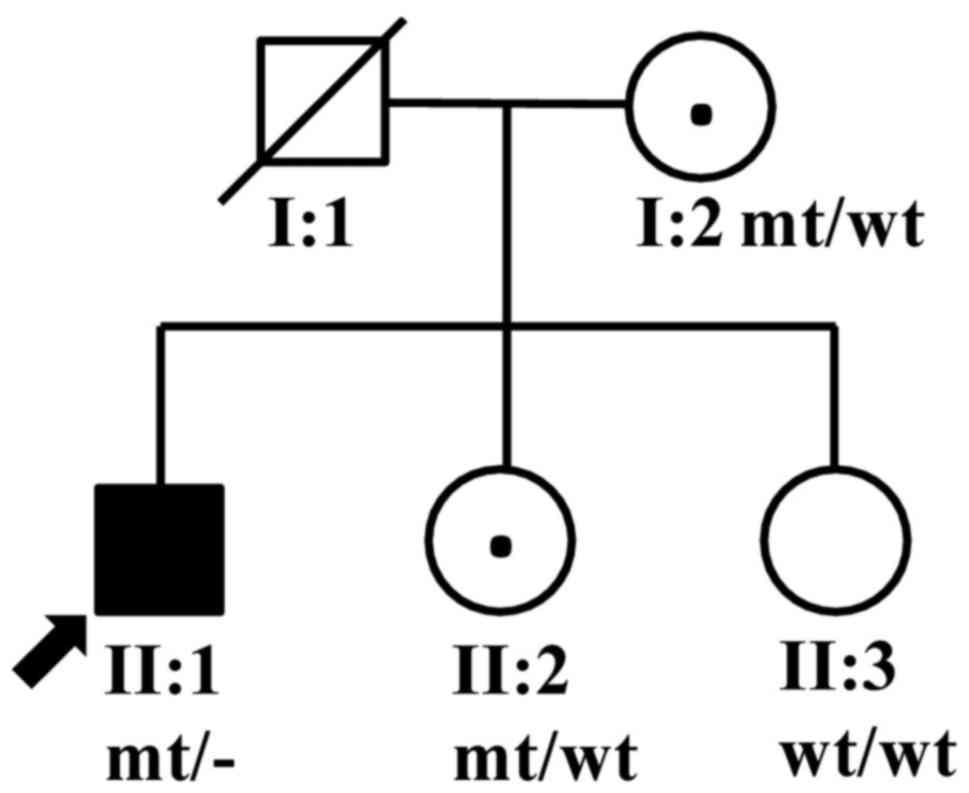
A choroideremia pedigree from Hunan Province, China,
consisting of five members (two males and three females) from two
generations (Fig. 1) was recruited
by the Second Xiangya Hospital, Central South University (Changsha,
China). Ocular examinations, including a slit lamp examination,
intraocular pressure (IOP) test, fundus photography (FP), best
corrected visual acuity (BCVA) test, A/B-scan ultrasonography,
ultrasound biomicroscopy (UBM), optical coherence tomography (OCT),
infrared reflectance (IR) imaging, fundus autofluorescence (FAF),
and fundus fluorescein angiography (FFA), were performed on these
patients. A group of 200 unrelated normal controls who were
ethnically matched with the patients were recruited mainly from the
hospital and as additional volunteers. This study was approved by
the Second Xiangya Hospital Ethics Committee. All of the pedigree
members and controls gave their written informed consent complying
with the Declaration of Helsinki principles (as revised in Brazil
2013).
Targeted exome sequencing
DNA samples obtained from the proband II:1 and the
patient II:2 were used for targeted exome sequencing of the
CHM gene. Library construction was performed with the custom
NimbleGen SeqCap EZ System (Roche NimbleGen, Madison, WI, USA), and
90-cycles paired-end sequencing was performed on Illumina HiSeq2500
Analyzers (Illumina, Inc., San Diego, CA, USA). Illumina Pipeline
software (version 1.3.4) was used to perform base-calling and the
calculation of quality values for every base.
Reads were aligned to the human reference genome
hg19 using Burrows-Wheeler Aligner (BWA, http://bio-bwa.sourceforge.net/). Single nucleotide
variation (SNV) and insertion and deletion (indel) identification
was performed with SOAPsnp (http://soap.genomics.org.cn/soapsnp.html) and SAMtools
(http://samtools.sourceforge.net/). SNVs
and indels with read depth ≥8X and Phred score quality ≥30 were
reserved and annotated by Annotate Variation (ANNOVAR; http://annovar.openbioinformatics.org/en/latest/user-guide/download/).
Multiple sequence alignment was conducted through Clustal Omega
(https://www.ebi.ac.uk/Tools/msa/clustalo/) to compare
the amino acid sequences of CHM between multiple
species.
Sanger sequencing
PCR amplification and Sanger sequencing were used to
validate the presence of variation. The primer sequences used were
as follows: Forward primer, 5′-cat gtt tca cac tgc cca ct-3′, and
reverse primer, 5′-aaa ttc gga ggc gtt aag gt-3′. PCR was performed
in a 10-µl reaction mixture containing 5 µl 2X TaKaRa Taq™ HS
Perfect Mix (Takara Biotechnology Co., Ltd., Dalian, China), 30 ng
of each primer, and 30 ng of genomic DNA. The amplification
conditions consisted of denaturation at 94°C for 30 sec, followed
by 33 cycles of denaturation at 94°C for 5 sec, annealing at 60°C
for 20 sec and extension at 72°C for 20 sec. Final extension was
performed at 72°C for 7 min. Sanger sequencing was performed on ABI
3730 DNA Analyzer (Thermo Fisher Scientific, Inc., Waltham, MA,
USA).
X-chromosome inactivation (XCI)
assay
Human androgen receptor gene (NM_000044) and
ZNF261 gene (DXS6673E) were chosen to perform XCI analysis
on methylated HpaII and/or HhaI sites as described
previously (14–15). The primers sequences are as
follows: NM_000044 forward primer, 5′-GCTGTGAAGGTTGCTGTTCCTCAT-3′,
and reverse primer, 5′-TCCAGAATCTGTTCCAGAGCGTGC-3′; DXS6673E
forward primer, 5′-ATGCTAAGGACCATCCAGGA-3′, and reverse primer,
5′-GGAGTTTTCCTCCCTCACCA-3′. The two forward primers were labeled
with hexachlorofluorescein fluorescent dye. DNA (2 µg) was digested
with 20 units of HpaII or HhaI, respectively.
Reactions were performed in a 25-µl total volume including 2.5 µl
10X buffer. All incubations were performed for 12 h at 37°C and
then for 20 min at 65°C. For the PCR reaction, 100 ng DNA was added
to a 20-µl PCR reaction mixture containing 0.1 µl ABI Gold Taqase
(Applied Biosystems; Thermo Fisher Scientific, Inc.), 2 µl 10X PCR
Buffer, 2 µl 25 mM MgCl2, 0.4 µl 10 mM dNTP, 1 µl
forward primer, and 1 µl reverse primer. Samples were amplified
using a Bio-Rad T100 thermocycler (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) for 32 cycles (45 sec at 95°C, 30 sec at 60°C,
and 30 sec at 72°C), with initial denaturation at 95°C for 5 min.
The PCR products were separated by capillary electrophoresis on an
ABI 3100 DNA analyzer (Applied Biosystems; Thermo Fisher
Scientific, Inc.), and were analyzed with GeneMarker®
HID software (SoftGenetics, LLC., State College, PA, USA;
https://www.softgenetics.com/GeneMarkerHID.php).
Results
Clinical findings
In this family, the proband II:1 was a 46-year-old
man who reported having sudden ocular pain and decreased vision in
his left eye for one day before he came to our hospital. He also
complained of headache and nausea. The BCVA was hand motion in both
eyes. The Goldmann IOP of the right eye was 16 mmHg and of the left
eye was 42 mmHg. The slit-lamp examination identified a shallow
anterior chamber and peripheral chambers of <1/4 cornea
thickness (CT) in both eyes, and conjunctival congestion and
corneal edema in the left eye. The fundus examination results were
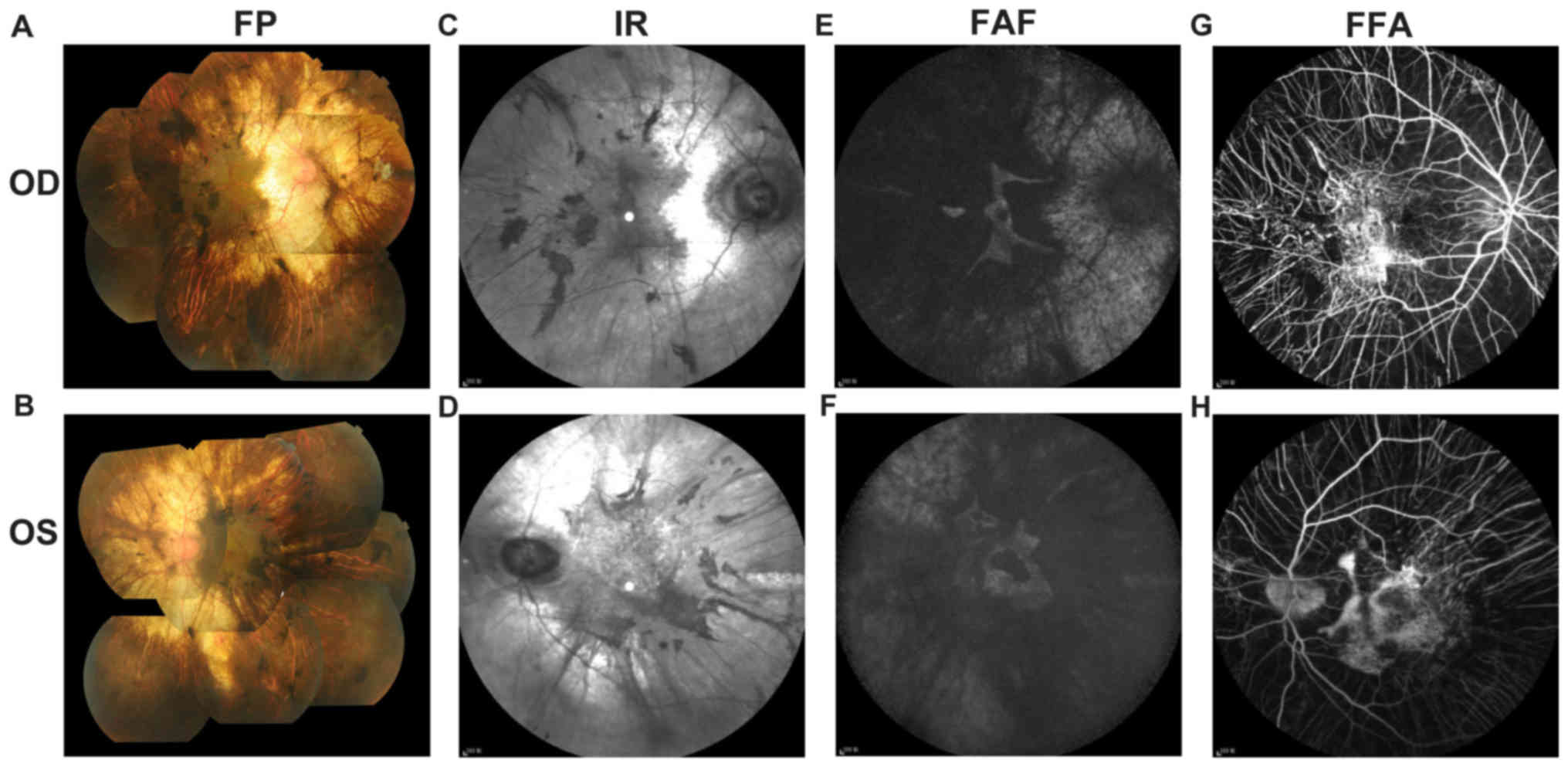
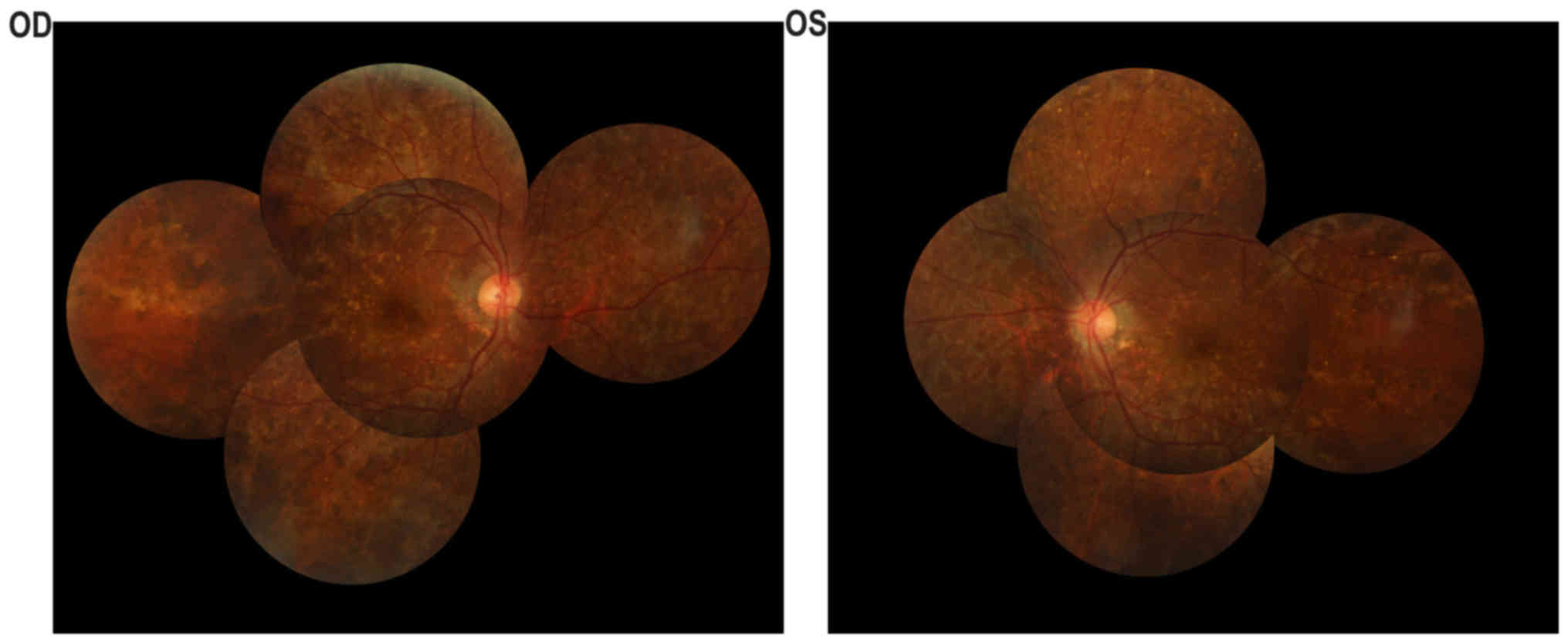
indicated in Fig. 2. Fundus
examination (Fig. 2A and B)
identified a cup-to-disc ratio of 0.4 in the right eye and 0.8 in
the left eye, pink optic nerves in both eyes, vessel narrowing in
both eyes, dimming of the color and lustre of the macular in both
eyes, severe chorioretinal atrophy with exposure of the sclera in
both eyes, and pigment proliferation in the posterior polar and
peripheral fundus in both eyes. The A-scan showed that the axial
lengths of the right and left eye were 23.11 and 23.10 mm,
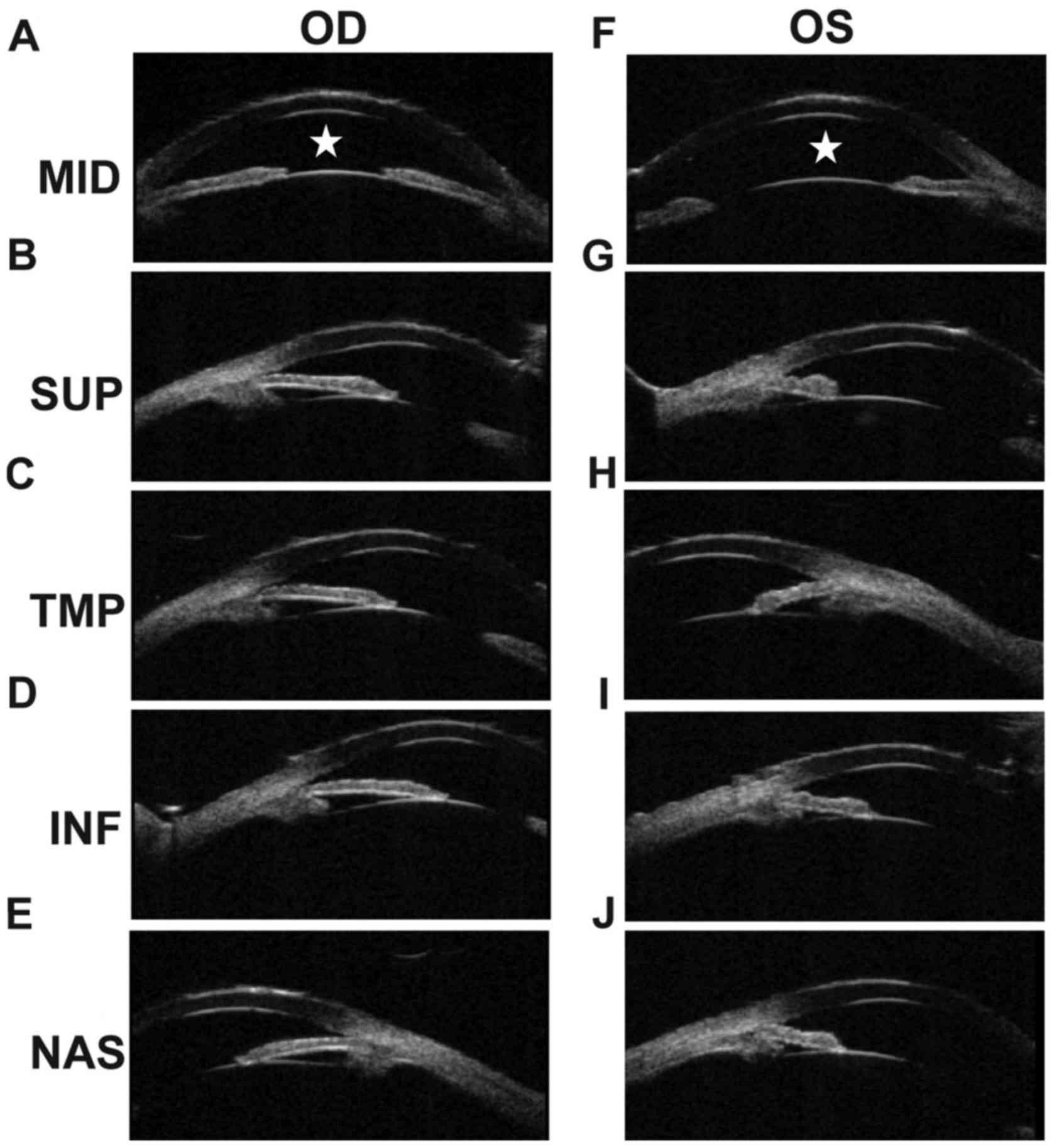
respectively. The B-scan showed vitreous opacity in both eyes. UBM
of the right eye showed that the anterior chamber depth (ACD) was
shallow (Fig. 3A), and also that
the anterior chamber angles were narrow at the superior side, the
temporal side, the inferior side and the nasal side (Fig. 3B-E). The ACD of the left eye was
shallow (Fig. 3F), and was closed
at the superior side, the temporal side, and the inferior side
(Fig. 3G-I). The angles of the
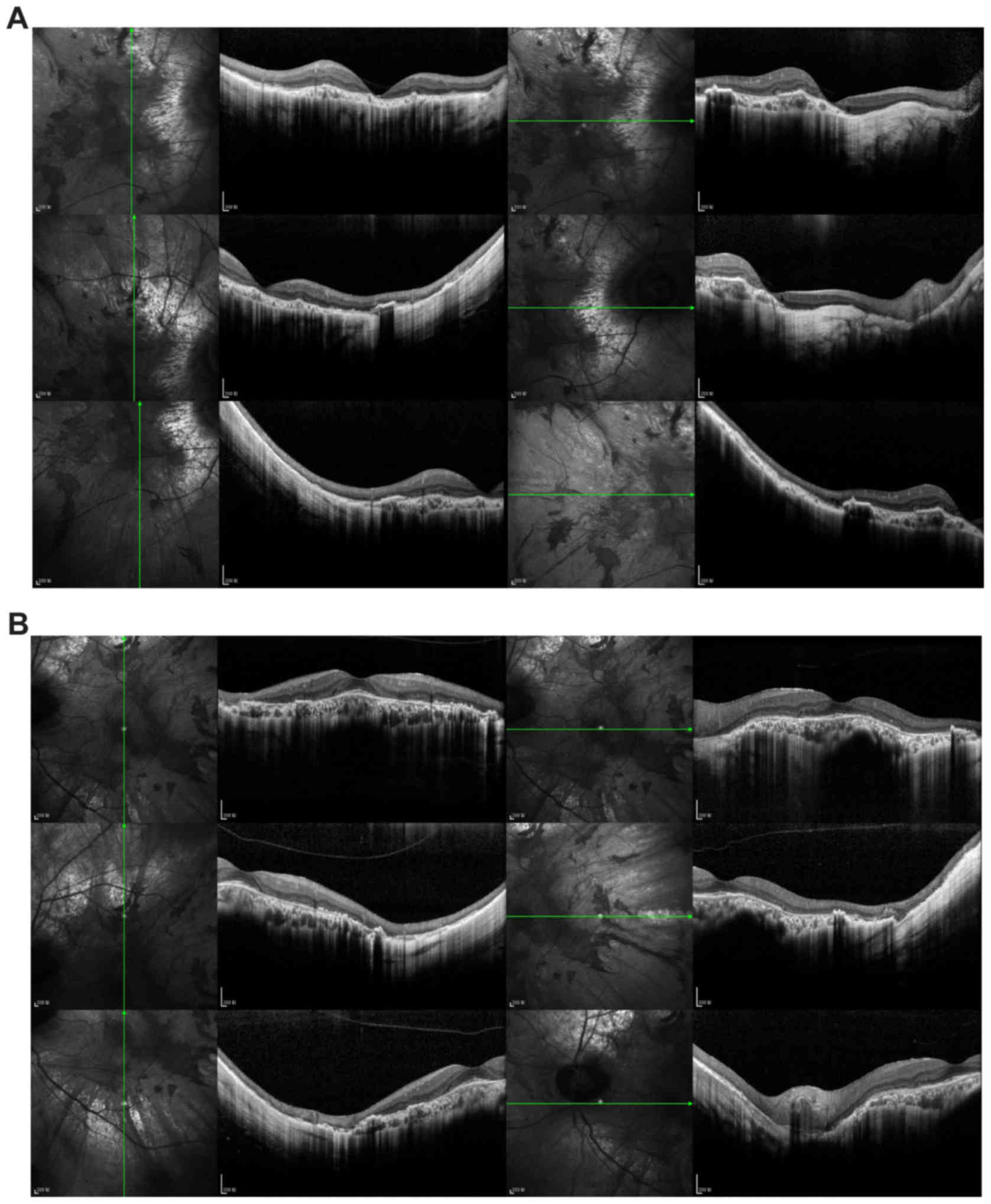
left eye were narrow at the nasal side (Fig. 3J). OCT imaging (Fig. 4) of both eyes indicated widespread
disappearance of the choroid and the outer retinal layer, which was
accompanied with pigment hyperplasia, though a small part of the
choroid was retained in the central macula. In the left fovea, the
outer nuclear layer, external limiting membrane, and photoreceptor
layer were still present. IR imaging (Fig. 2C and D) demonstrated
hyperreflective lesions corresponding to the area of serious
chorioretinal atrophy and hyporeflective lesions corresponding to
the area of pigment proliferation. FAF (Fig. 2E and F) of both eyes showed an
isolated area of hyperautofluorescence in the foveal area. Part of
the transparent sclera was also observed as hyperreflective in FAF
image. FFA (Fig. 2G and H) of both
eyes showed an absence of the choriocapillaris in the regions of
pigment accumulation. The retinal vessels were normal in FFA.
According to the above information, the patient's clinical
diagnosis was choroideremia in both eyes and angle-closure glaucoma
(preclinical stage in the right eye, acute attack stage in the left
eye).
The proband's sister, II:2, did not report any
vision problems, and the anterior segments of both of her eyes were
normal. However, the fundus examination identified darkened color
and lustre of the retina, and a crystal-like appearance in the
retina of both eyes (Fig. 5). The
proband's parents, I:1 and I:2, showed no clinical
manifestation.
Targeted exome sequencing and sanger
validation
We selected the proband II:1 and the patient II:2
for targeted exome sequencing, which was designed to isolate a
1,962-bp DNA region of the CHM gene. In our sequencing
results, 96.00 and 95.82% of the qualified bases for II:1 and II:2
were mapped to the targeted sequence, with mean read depths of
approximately 210.32- and 208.61-fold for II:1 and II:2 (Table I). The exon coverage rates were
99.75 and 99.26% for the patient and female carrier samples,
respectively, which was sufficient to pass our thresholds for
calling SNVs and indels (Table I).
After alignment and variation calling, a total of 12 variations in
the CHM gene were identified among the two family members,
including 2 exonic variations, 9 intronic variations, and 1 in the
3′-UTR (Table II). However, only
1 frameshift variation, c.280delA (p.Thr94LeufsTer32), in
CHM was found in II:1 and II:2, that had not been reported
either in the dbSNP database or in the 1,000 Genomes database,
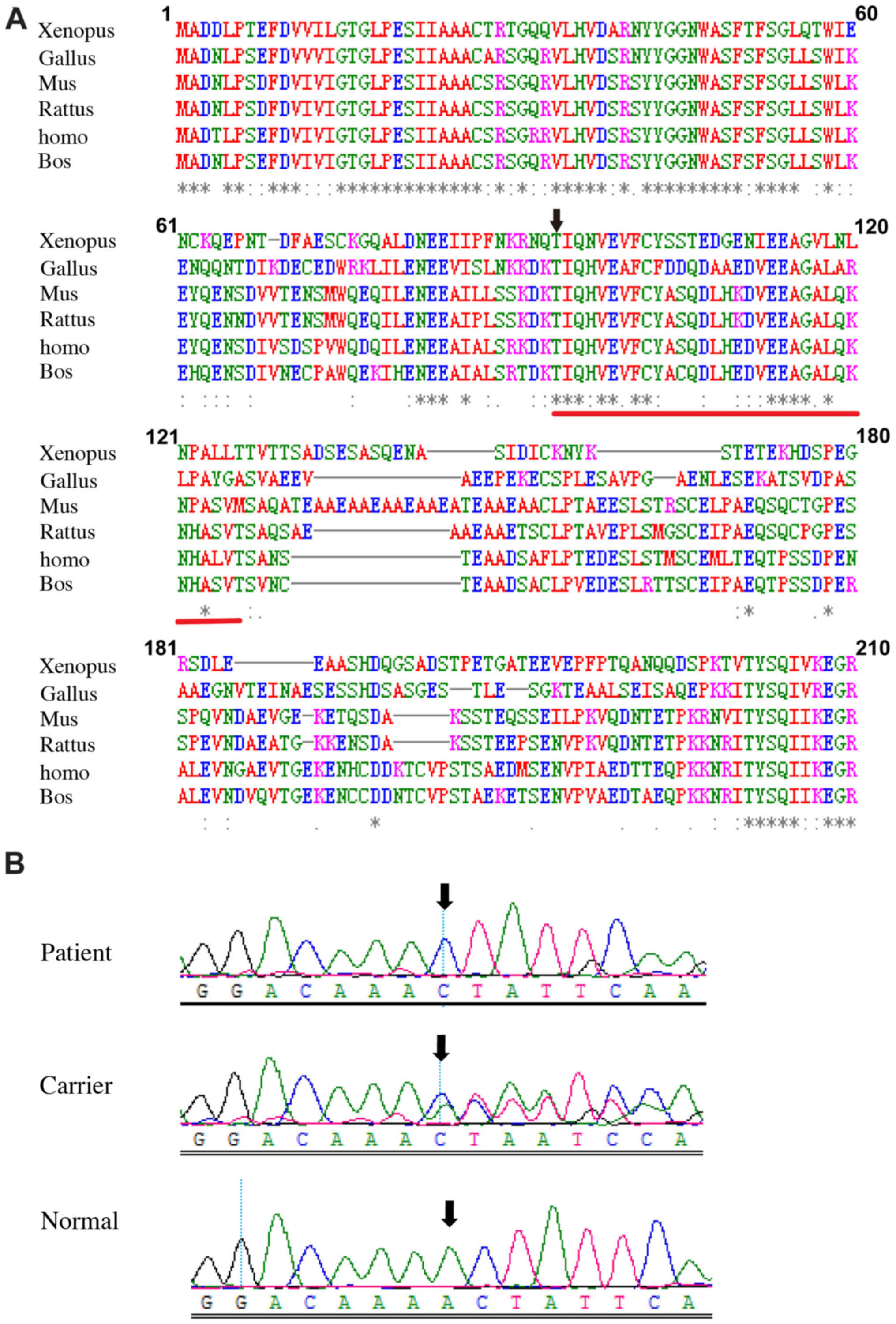
which was predicted to produce a truncated REP-1 protein. Comparing
the amino acid sequences of CHM between multiple species
demonstrated asterisks and codons among the amino acids 94 to 126
(Fig. 6A), which indicated
conserved amino acids on these sites.
 | Table I.Summary of the targeted exome
sequencing results. |
Table I.
Summary of the targeted exome
sequencing results.
| Sample | Targeted gene | Targeted region
(bp) | Targeted region
mapped (%) | Targeted exon
coverage (%) | Mean depth (x) | Mean depth >30×
(%) |
|---|
| II-1 | CHM | 1962 | 96.00 | 99.75 | 210.32 | 98.45 |
| II-2 |
|
| 95.82 | 99.26 | 208.61 | 95.87 |
| Table II.Variations detected in CHM gene
(NM_000325) in the pedigree by targeted exome sequencing. |
Table II.
Variations detected in CHM gene
(NM_000325) in the pedigree by targeted exome sequencing.
| Position | SNP no. | Variation type | Location | Nucleotide
change | Amino acid
change | Allele frequency in
1,000 genome | Affected member |
|---|
| chrX:85281760 | rs1015148 | Hemi, Het | Intron 2 | c.116+80C>T | None | 0.2024 | II-1, II-2 |
| chrX:85281760 | rs5923428 | Homo | Intron 2 | c.116+735A>G | None | 0.065 | II-2 |
| chrX:85261183 | rs62607865 | Homo | Intron 2 |
c.116+21312C>A | None | 0.0137 | II-2 |
| chrX:85236825 |
| Het | Intron 2 | c.117-12delT | None | 0 | II-2 |
| chrX:85219021 | rs10217950 | Het | Exon 5 | c.351A>G | p.A117A | 0.1401 | II-2 |
| chrX:85233805 |
| Hemi, Het | Exon 4 | c.280 delA | p.T94LfsTer32 | 0 | II-1, II-2 |
| chrX:85215758 | rs7880234 | Homo | Intron 5 |
c.703-1776G>A | None | 0.0183 | II-2 |
| chrX:85212717 |
| Het | Intron 7 |
c.940+143C>A | None | 0 | II-1 |
|
chrX:85166221~85166222 |
| Het | Intron 9 |
c.1244+44_+45insATAT | None | 0 | II-2 |
| chrX:85155596 |
| Hemi | Intron 11 |
c.1413+55G>T | None | 0 | II-1 |
| chrX:85145586 |
| Homo | Intron 12 | c.1510+3607
A>G | None | 0 | II-2 |
| chrX:85119508 |
| Hemi | 3-UTR | c.*127T>C | None | 0 | II-1 |
We used Sanger sequencing to validate the c.280delA
variation and analyze the co-segregating status of the variant in
all family members. Hemizygous c.280delA variation was found in
II:1, and heterozygous c.280delA variations were found in I:2 and
II:2, respectively, while the variation was not detected in II:3
(Fig. 6B). The variation was
completely cosegregated with the phenotype in all family members
except for carrier II:2. In addition, the variation was not
identified in the 200 unrelated normal controls. Thus, our results
indicate that the variation c.280delA (p.Thr94LeufsTer32) in the
CHM gene is a genetic etiological factor of the current
choroideremia pedigree. Our phenotype and variation data in this
study have been submitted to ClinVar (accession no.
SCV000600017).
XCI
Given that the carrier II:2 exhibited
disease-related phenotypes, we determined the XCI profile of the
female family members. The DNA of the carrier II:2 and normal
female I:2 showed 48 and 43% inactivated X chromosome,
respectively, indicating that both the female carrier and normal
female had a random XCI pattern. Thus, no correlation between
phenotypic status and X-inactivation pattern was established in
these females.
Discussion
Choroideremia is an X-linked, recessive,
chorioretinal degenerative disease characterized by progressive
centripetal loss of the photoreceptor, retinal RPE and choroid
layers. Our initial patient (II:1) revealed exhibited choroideremia
with acute angle-closure glaucoma. Subsequently, in our female
carrier II:2, we identified darkening of the color and lustre of
the retina, along with a crystal-like appearance of the retina was
observed, which was likely due to hypopigmentation of the RPE.
These clinical manifestations were similar to phenotypes reported
in other studies (16,17) with the exception of the
presentation of acute angle-closure glaucoma. II:2 also exhibited
narrow or closed anterior chamber angles in both eyes, which may
lead to high IOP and acute angle-closure glaucoma in the
future.
A novel frameshift variant, c.280delA
(p.Thr94LeufsTer32), was identified in the CHM gene through
targeted exome sequencing in our pedigree, and was validated by
Sanger sequencing. The variant was detected in the mother I:2, and
in the offspring II:1 and II:2, and thus the proband and the female
carrier inherited the variant from their carrier mother rather than
acquiring it from a de novo germline mutation. As with
phenotyped carriers reported in other studies (17,18),
our variant was completely cosegregated with the phenotypes.
Moreover, our data revealed that both a normal female and a carrier
had random XCI patterns, which suggested that there is no
preferential XCI of the mutant X chromosome allele. This result is
consistent with a previous study of XCI detection in 13
heterozygous females from two families (19).
Other previous studies have suggested that the
molecular basis of choroideremia may involve selective
underprenylation of Rab protein due to the absence of REP-1
(20). Our frameshift mutation was
predicted to create a premature stop codon and to thus lead to a
lack of full-length REP-1. In turn, this would likely lead to a
deficiency of GGTase function, with the insufficient transfer of
geranylgeranylpyrophosphate groups onto Rab proteins potentially
resulting in abnormal protein function, preventing their
participation in intracellular vesicular transport (21). This would ultimately cause several
ocular defects as observed in our patients.
In conclusion, our results suggest that c.280delA
(p.Thr94LeufsTer32) in CHM was a pathogenic mutation in the
present choroideremia family as determined by captured exome
sequencing. Such targeted exome sequencing technology may provide a
promising tool for genetics testing and counseling among families
with choroideremia. Further functional and animal studies on the
mutant protein will be required to interpret its role in the
pathogenesis of the disease.
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science
Foundation of Hunan Province Project (grant no. 14JJ3041), the
National Natural Science Foundation of China (grant nos. 81700837,
81300798, and 61573380), and Department of Science and Technology,
Hunan (grant no. 2015TP2007).
Availability of data and materials
Our phenotype and variation data in this study are
available from ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/; accession no.
SCV000600017). Other datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
PO, FZ, CZ, and BZ performed the genetic studies,
participated in the sequence alignment and analyzed the data. YL
and JL participated in the sample collection and clinical
examination. LZ designed and supervised the study. LZ and PO wrote
the manuscript. All authors read and approved the final
manuscript.
Ethical approval and Informed consent
All procedures performed in studies involving human
participants were in accordance with the ethical standards of the
Second Xiangya Hospital Ethics Committee and with the 1964 Helsinki
declaration and its later amendments or comparable ethical
standards. Informed consent was obtained from all individuals
participants included in the study.
Consent for publication
The patient, the family members and the normal
control subjects provided their written informed consents for the
publication of any associated data and accompanying images.
Competing interests
All authors declare that they have no competing
interests.
References
|
1
|
Coussa RG and Traboulsi EI: Choroideremia:
A review of general findings and pathogenesis. Ophthalmic Genet.
33:57–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rudolph G, Preising M, Kalpadakis P,
Haritoglou C, Lang GE and Lorenz B: Phenotypic variability in three
carriers from a family with choroideremia and a frameshift mutation
1388delCCinsG in the REP-1 gene. Ophthalmic Genet. 24:203–214.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Renner AB, Kellner U, Cropp E, Preising
MN, MacDonald IM, van den Hurk JA, Cremers FP and Foerster MH:
Choroideremia: Variability of clinical and electrophysiological
characteristics and first report of a negative electroretinogram.
Ophthalmology. 113:e1–e10. 2006. View Article : Google Scholar
|
|
4
|
van den Hurk JA, van de Pol DJ, Wissinger
B, van Driel MA, Hoefsloot LH, de Wijs IJ, van den Born LI,
Heckenlively JR, Brunner HG, Zrenner E, et al: Novel types of
mutation in the choroideremia (CHM) gene: A full-length L1
insertion and an intronic mutation activating a cryptic exon. Hum
Genet. 113:268–275. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Radziwon A, Arno G, Wheaton K D, McDonagh
EM, Baple EL, Webb-Jones K, Birch G D, Webster AR and MacDonald IM:
Single-base substitutions in the CHM promoter as a cause of
choroideremia. Hum Mutat. 38:704–715. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Edwards TL, Williams J, Patrício MI,
Simunovic MP, Shanks M, Clouston P and MacLaren RE: Novel
non-contiguous exon duplication in choroideremia. Clin Genet.
93:144–148. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leung KF, Baron R and Seabra MC: Thematic
review series: Lipid posttranslational modifications.
geranylgeranylation of Rab GTPases. J Lipid Res. 47:467–475. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ali BR and Seabra MC: Targeting of Rab
GTPases to cellular membranes. Biochem Soc Trans. 33:652–656. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Larijani B, Hume AN, Tarafder AK and
Seabra MC: Multiple factors contribute to inefficient prenylation
of Rab27a in Rab prenylation diseases. J Biol Chem.
278:46798–46804. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Futter CE, Ramalho JS, Jaissle GB,
Seeliger MW and Seabra MC: The role of Rab27a in the regulation of
melanosome distribution within retinal pigment epithelial cells.
Mol Biol Cell. 15:2264–2275. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seabra MC, Ho YK and Anant JS: Deficient
geranylgeranylation of Ram/Rab27 in choroideremia. J Biol Chem.
270:24420–24427. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tolmachova T, Anders R, Abrink M, Bugeon
L, Dallman MJ, Futter CE, Ramalho JS, Tonagel F, Tanimoto N,
Seeliger MW, et al: Independent degeneration of photoreceptors and
retinal pigment epithelium in conditional knockout mouse models of
choroideremia. J Clin Invest. 116:386–394. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tolmachova T, Wavre-Shapton ST, Barnard
AR, MacLaren RE, Futter CE and Seabra MC: Retinal pigment
epithelium defects accelerate photoreceptor degeneration in cell
type-specific knockout mouse models of choroideremia. Invest
Ophthalmol Vis Sci. 51:4913–4920. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Allen RC, Zoghbi HY, Moseley AB,
Rosenblatt HM and Belmont JW: Methylation of HpaII and HhaI sites
near the polymorphic CAG repeat in the human androgen-receptor gene
correlates with X chromosome inactivation. Am J Hum Genet.
51:1229–1239. 1992.PubMed/NCBI
|
|
15
|
Beever C, Lai BP, Baldry SE, Peñaherrera
MS, Jiang R, Robinson WP and Brown CJ: Methylation of ZNF261 as an
assay for determining X chromosome inactivation patterns. Am J Med
Genet A. 120A:439–441. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li S, Guan L, Fang S, Jiang H, Xiao X,
Yang J, Wang P, Yin Y, Guo X, Wang J, et al: Exome sequencing
reveals CHM mutations in six families with atypical choroideremia
initially diagnosed as retinitis pigmentosa. Int J Mol Med.
34:573–577. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Renner AB, Fiebig BS, Cropp E, Weber BH
and Kellner U: Progression of retinal pigment epithelial
alterations during long-term follow-up in female carriers of
choroideremia and report of a novel CHM mutation. Arch Ophthalmol.
127:907–912. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou Q, Liu L, Xu F, Li H, Sergeev Y, Dong
F, Jiang R, MacDonald I and Sui R: Genetic and phenotypic
characteristics of three Mainland Chinese families with
choroideremia. Mol Vis. 18:309–316. 2012.PubMed/NCBI
|
|
19
|
Perez-Cano HJ, Garnica-Hayashi RE and
Zenteno JC: CHM gene molecular analysis and X-chromosome
inactivation pattern determination in two families with
choroideremia. Am J Med Genet A. 149A:2134–2140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Esposito G, De Falco F, Tinto N, Testa F,
Vitagliano L, Tandurella IC, Iannone L, Rossi S, Rinaldi E,
Simonelli F, et al: Comprehensive mutation analysis (20 families)
of the choroideremia gene reveals a missense variant that prevents
the binding of REP1 with Rab geranylgeranyl transferase. Hum Mutat.
32:1460–1469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Baron RA and Seabra MC: Rab
geranylgeranylation occurs preferentially via the pre-formed
REP-RGGT complex and is regulated by geranylgeranyl pyrophosphate.
Biochem J. 415:67–75. 2008. View Article : Google Scholar : PubMed/NCBI
|