Introduction
Familial hypercholesterolemia [FH; Online Mendelian
Inheritance in Man (OMIM) reference no. 143890] is one of the most
common and most serious autosomal dominant, hereditary monogenic
diseases. Clinically, it is divided into two types: Homozygous and
heterozygous. Homozygous FH patients manifest rising plasma
low-density lipoprotein cholesterol (LDL-C), tendon xanthoma in
infancy or early childhood, early onset atherosclerosis, severe
coronary heart disease, or even death during youth and its
incidence is ~one in a million (1). Heterozygous FH patients' phenotypic
features are not obvious and it has an incidence rate of ~1 in 500
(2). The most reliable diagnosis
of FH may be made using phenotypic criteria and genetic testing.
Genetic testing increases the accuracy of an FH diagnosis. The
pathology of FH is associated with serum LDL-C metabolic pathway
abnormalities. Generally, FH results from mutations in the genes
encoding the LDL receptor (LDLR), apolipoprotein B (apoB), or
proprotein convertase subtilisin/kexintype 9 (PCSK9). Previous
studies have identified several LDLR mutations in FH and the
association between the phenotype and underlying genotype (3,4),
including a systematic review analyzing the characteristics,
distribution, gene frequency and association between the genotype
and phenotype of LDLR mutations especially in the Chinese
population (5). However, FH is a
very heterogeneous disease and there are different variants of LDLR
gene mutations throughout the gene. Although the phenotype is
supposed to be associated with the genotype, ~30% of patients with
the typical clinical phenotype still cannot be identified with
significant pathogenic genetic mutations (4,6–8),
which may be due to the extensive genetic heterogeneity of FH
patients or its clinical variability. Therefore, it is challenging
and essential to diagnose an FH patient with homozygous or
heterozygous FH. In the present study, the genetic variations in a
family with compound heterozygous FH were analyzed to investigate
the association between the phenotype and the genotype.
Materials and methods
Subjects
All the investigated subjects gave written informed
consent for all tests and the publication of associated data and
accompanying images, prior to taking part in the study and
participating in these examinations. The study was conducted
according to the Helsinki declaration (2013) and approved by the
Institutional Ethics Committees of Shanghai Jiao Tong University,
affiliated to Rui Jin Hospital (Shanghai, China).
Determination of lipids
Blood samples (20 ml per patient) were obtained from
the proband and her parents following a 12 h fast at Rui Jin
Hospital in March, 2014. The proband was 16 years old and her
parents were both 47 years old. All samples were collected
simultaneously, immediately placed on ice and subsequently
centrifuged within 30 min at 1,000 × g at 4°C for 20 min. Total
cholesterol (TC), triglycerides (TG), LDL-C and high-density
lipoprotein cholesterol (HDL-C) were measured using routine
commercial kits (Beckman Coulter, Inc., Brea, CA, USA) and a
Beckman AU 4500 automated biochemical analyzer (Beckman Coulter,
Inc.), according to the manufacturer's protocols.
Genotyping assays
Venous peripheral blood was collected from the
proband and her parents. Genomic DNA was prepared from the
peripheral white blood cells by phenol/chloroform extraction
(9). The polymerase chain reaction
(PCR) and direct sequencing were used to detect the fragment of 18
exons of the low-density lipoprotein receptor (LDLR; OMIM 606945)
and apolipoprotein B100 (apoB100) Q3500R of the proband and her
parents. The primer pairs for these gene fragments are presented in
Table I. The gene sequences were
compared with normal sequences downloaded from GenBank (https://www.ncbi.nlm.nih.gov/nuccore/DQ379956.1) to
identify mutations.
 | Table I.Primers of the gene fragments and
amplification fragment length of each polymerase chain reaction
system. |
Table I.
Primers of the gene fragments and
amplification fragment length of each polymerase chain reaction
system.
| Amplified
segments | Upstream primer
(5′-3′) F | Downstream primer
(5′-3′) R | Fragment length
(bp) |
|---|
| ApoB-100E26 |
gtggagggtagtcataacag |
acatacaagcaaagggaccg | 510 |
| LDLR promoter |
gagtgggaatcagagcttcacgggt |
cattgaaatgctgtaaatgacgtgg | 155 |
| LDLR E1 |
acccaaatacaacaaatcaagtcg |
agggtgagggaccgcatct | 629 |
| LDLR E2 |
ttagttggcaggaaatagaca |
gccaccacctctatcttgt | 598 |
| LDLR E3 |
gcctcagtgggtctttcc |
aatgacccacgtaacatgaag | 563 |
| LDLR E4-1 |
atagaatgggctggtgttgg |
aggacaaatctgacgaggaa | 440 |
| LDLR E4-2 |
cgaagatggctcgatga |
gcttattgggaaatcactgttt | 466 |
| LDLR E5 |
aaggtggcacgattatggc |
cttctcagagcctcacttcctt | 566 |
| LDLR E6 |
gattacaggcacaaaccacc |
ggaaagggactgagacatga | 339 |
| LDLR E7 |
attagcctgtcatggtcgtg |
aacaccttgggacgccga | 703 |
| LDLR E8 |
gggtgcgtaatccctgtaa |
ttaggacttgggcttgcc | 834 |
| LDLR E9 |
tttctgggtgcctcctctg |
ggaatggagggagcagga | 655 |
| LDLR E10 |
tccatcgcctacctcttctt |
tggttagtgggctgggca | 564 |
| LDLR E11 |
aaaacccaaacaagccaca |
ctgctccctgaaggtttcc | 381 |
| LDLR E12 |
aggtatgttcgcaggacagc |
ccttctccttggccgtct | 784 |
| LDLR E13 |
ccagtgtttaacgggatttg |
tgtgagaggaccaccctgag | 353 |
| LDLR E14 |
tcttccacaacctcacccag |
ctgctcatctgtcaaatgggta | 399 |
| LDLR E15 |
tgagactttcgtcattaggcg |
gtctaataacagttcttgccctc | 448 |
| LDLR E16 |
tgggaagttctccaagtgtc |
gtcccgtgctgtggaatag | 435 |
| LDLR E17 |
caaggttatggtacgatgcc |
cagaatgtcagcggacaatg | 418 |
| LDLR E18 |
actcaccgtctccctctgg |
gggagccagaacaggctt | 333 |
Detection of target DNA copy number
variations
The QX200 Droplet Digital PCR (ddPCR™) (10,11)
system was used to detect target DNA copy number variations of the
proband and her parents. The workflow was carried out based on the
manufacturer's protocol (http://www.bio-rad.com/zh-cn/product/qx200-droplet-digital-pcr-system).
The TaqMan® Copy Number Reference Assay, with RNase P
(Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), is recommended as the standard reference assay for copy
number analysis, which is present at 1 copy per haploid genome and
was labeled with VIC fluorescent dye. The 1st target site was
located at exon 18 of the LDLR gene. The forward primer was
5′-TTGGCAGAGACAGATGGTCAGT-3′ and the reverse primer was
5′-CGGGACTCCAGGCAGATG-3′; the probe was
FAMTGGAGGATGACGTGGCGTMGBNFQ. The 2nd target site was located at a
distal region of the exon 18. The forward primer was
5′-GATTGACGAAATTGGTGCTCAA-3′ and the reverse primer was
5′-GCCATCTGAGCCCGCTTA-3′; the probe was
FAMCAAGTGGCAAACAGGMGBNFQ.
CopyCaller™ Software, v2.0 (Thermo Fisher
Scientific, Inc.), was used to analyze the ratios of the FAM and
VIC fluorescence number, including RNase P in the 1st target site
and that in the 2nd target site, respectively. The ratios obtained
represented the DNA copy number at the target site, compared with a
haploid genome.
Cell culture, plasmid construction and
transfection
The LDLR wild-type gene was cloned as well as genes
with specific mutations including C377G and L855fs, from the GV230
vector (Shanghai GeneChem, Co., Ltd., Shanghai, China) separately,
which has an N-terminal green fluorescent protein (GFP) tag. Prior
to being used, the integrity of all constructs were verified by
quantitative (q)PCR and western blot analysis.
The wild type (WT) and mutant plasmids (20 µg/ml)
were transfected into 293T cells (4×105/ml; Shanghai
GeneChem, Co., Ltd.), which do not have endogenous LDL-R
expression, using Lipofectamine 2000 (Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. One day prior to
transfection, cells (2×105 cells/well) were seeded in a
six-well plates. Transfection efficiency was estimated by counting
the GFP positive cells using a fluorescent microscope, which was
observed to be ~65–70% in all experiments, following the counting
of 7 randomly selected fields of view (magnification, ×400).
Transfected cells were cultured in RPMI 1640 (cat. no. 22400-089;
Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
lipoprotein deprived serum (Gibco; Thermo Fisher Scientific, Inc.)
for 48 h prior to analysis of LDLR expression and function by flow
cytometry. Cells were maintained at 37°C and a humidity of 95% in
an incubator with an atmosphere of 5% CO2.
Reverse transcription (RT)-qPCR
analysis of LDLR
Total RNA was isolated from the cultured 293T cells
using the acid-phenol extraction method in the presence of
chaotropic salts (TRIzol™; Thermo Fisher Scientific, Inc.) and
subsequent isopropanol-ethanol precipitation. Reverse transcription
of mRNA encoding LDLR as well as GAPDH was performed on 2 µg total
RNA using the SuperScript First-Strand Synthesis System (Thermo
Fisher Scientific, Inc.), following the manufacturer's protocol.
qPCR was conducted using the SYBR Green PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.) and ABI Prism 7500
Sequence Detection System (Applied Biosystems; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. The
thermocycling conditions included initial activation at 95°C for 10
min, followed by 40 cycles of denaturation for 15 sec at 95°C and
an annealing/extension step for 1 min at 60°C. The equation
2−ΔΔCq measurement was used to analyze the results
(12). Primer sets were as
follows: LDL-R: Forward, 5′-AACGAATGCTTGGACAACAAC-3′, reverse
5′-CTTCCTCACACTGGCACTTG-3′. GAPDH: Forward,
5′-TGACTTCAACAGCGACACCCA-3′, reverse
5′-CACCCTGTTGCTGTAGCCAAA-3′.
Western blot analysis
Cells were lysed in a radioimmunoprecipitation assay
lysis buffer containing 100 mM NaCl and 10 nM EDTA (cat. no. 9803;
Cell Signaling Technology, Inc., Danvers, MA, USA). Protein
concentration was determined using the BCA assay and following
this, protein samples (15 µg) were loaded and separated by 10%
SDS-PAGE and transferred onto nitrocellulose membranes.
Non-specific binding sites were blocked in Tris-buffered
saline/Tween buffer (TBST; 0.1%, v/v) supplemented with 5% non-fat
dry milk for 1 h at room temperature and the membrane was then
probed with primary antibodies to LDLR (1:5,000; cat. no. ab52818;
Abcam, Cambridge, UK) and GAPDH (1:2,000; cat. no. sc-32233; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) in TBST supplemented
with 1% bovine serum albumin (BSA; Beyotime Biotechnology,
Shanghai, China) at 4°C overnight. Following three subsequent
washes in TBST, membranes were incubated with the following
horseradish peroxidase (HRP)-conjugated secondary antibodies for 80
min at room temperature: Anti-rabbit HRP (cat. no. 7074; 1:3,000;
Cell Signaling Technology, Inc.) and anti-mouse HRP (cat. no. 7076;
1:3,000; Cell Signaling Technology, Inc.). Following another three
washes in TBST, specific bands were detected using an enhanced
chemiluminescent reagent (cat. no. B18005; Selleck Chemicals,
Houston, TX, USA), and the obtained images were analyzed using
ImageJ (version 1.48; National Institutes of Health, Bethesda, MD,
USA).
Flow cytometric analysis
Following transfection, cells (1×106)
were harvested by trypsinization and suspended by PBS for flow
cytometric analysis. The cell suspension was then divided into two
groups: The first group was used for LDL binding ability analysis;
the second was used for LDL internalization analysis. The cells
undergoing LDL binding analysis were placed in 20 mg/ml
1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
(Dil) labeled LDL (Molecular Probes; Thermo Fisher Scientific,
Inc.) for 1 h at 4°C. Following the incubation, the medium was
removed, then the cells were washed twice in ice-cold PBS with 0.5%
BSA (Beyotime Biotechnology, Inc.). The same cells were incubated
in 20 mg/ml Dil labeled LDL for 1 h at 37°C for LDLR
internalization analysis, followed by medium removal. Following
that, cells were washed twice in ice-cold PBS with 0.5% BSA prior
to flow cytometry analysis. LDLR function was analyzed by a BD FACS
Flow Cytometer with an argon laser operating at 514 nm (Dil). All
experiments and analyses were repeated three times. For analyses,
histogram plots presented Dil-LDL positive cells gated on GFP
tagged LDL-R and the x-axis demonstrated the activity of Dil-LDL
binding and of Dil-LDL internalization. All data were collected and
analyzed using FlowJo (version 9.3.2; FlowJo LLC, Ashland, OR,
USA).
Statistical analysis
Data are presented as mean ± standard error of the
mean. Comparisons between two groups were made using a Student's
t-test or Mann-Whitney U test, while the data obtained from
multiple groups were compared using Kruskal-Wallis test with Dunn's
multiple comparison test. A minimum of three repeats per experiment
was performed. All data were analyzed using SPSS software, version
20.0 (IBM SPSS, Armonk, NY, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Clinical features of the proband and
her family members
The proband was a 16-year-old girl, who presented
with elevated serum lipid levels and a xanthoma for ~13 years. At
the age of 3, her parents identified a soy bean-sized xanthoma on
the joints of her limbs and her lab results demonstrated an
abnormally high level of serum lipids. The proband was prescribed
Lipitor therapy, 10 mg orally once daily for 4 years, with
accompanying dietary and lifestyle alterations. However, her serum
lipid levels remained high, with a TC of 20.15 mmol/l and LDL-C of
13.16 mmol/l. Her xanthoma also increased gradually and therefore,
she was prescribed more anti-hyperlipidemic drugs: Lipitor, 20 mg
twice a day, along with 4 g cholestyramine thrice a day. The lipid
levels were still poorly controlled, although the growth rate of
the xanthoma slowed slightly.
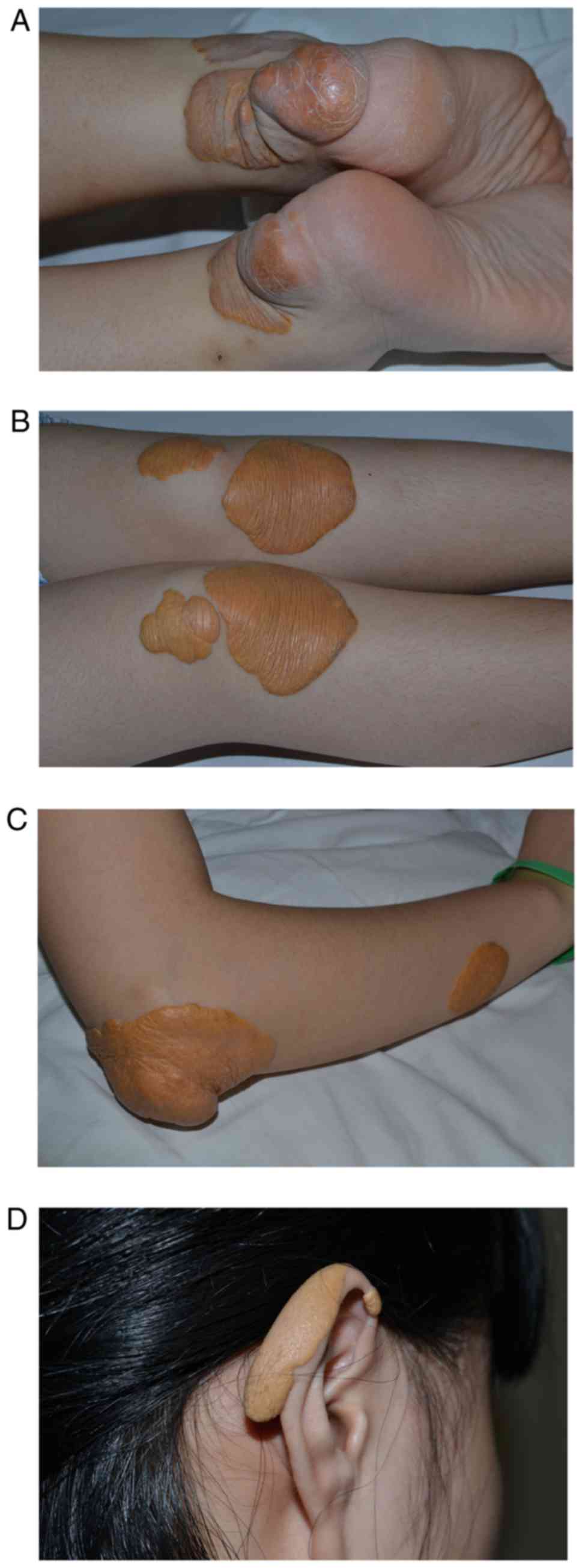
A physical examination revealed that the proband was
of good nutritional status and normal development, and her BMI was
20.7 kg/m2. Flat or nodular, yellow protruding rashes,
5–10 cm in diameter, were visible on her Achilles tendon, knees,
elbows and ears (Fig. 1). Systolic
murmurs of the left carotid artery were detected and a jet-like
murmur around the aortic valve area was heard.
Echocardiography demonstrated severe, supravalvular
aortic stenosis 7 mm from the aortic root. The narrowest diameter
was ~5 mm and there were multiple echogenic plaques on the
irregular aortic arch sidewall. Vascular ultrasonography
demonstrated bilateral carotid atherosclerotic plaque formation.
The percentage stenosis of the right internal carotid artery was
~50% and the percentage occlusion of the left internal carotid
artery was ~60%. The right common carotid artery had a soft plaque
of ~2.6×27.2 mm and left common carotid artery had a soft plaque of
~2.8×16.4 mm. Coronary computed tomography angiography demonstrated
that there were multiple plaques on the wall of the left main stem,
the middle of the left anterior descending artery and the right
coronary with mild luminal stenosis; a mixed plaque was also seen
on the proximal left circumflex artery with moderate luminal
stenosis. The aortic root had multiple plaques, leading to
supravalvularaortic stenosis.
Furthermore, the proband's parents had no apparent
clinical features upon physical examination. They had a
non-consanguineous marriage and the family had no similar diseases.
The proband had no secondary causes of hypercholesterolemia,
including proteinuria, hypothyroidism or medications.
The proband's lipid values remained high, with a TC
of 14.21 mmol/l, a LDL-C of 12.22 mmol/l and a TG of 1.63 mmol/l,
even following treatment with 40 mg atorvastatin per day. Although
her parents had no apparent clinical features upon physical
examination, they also suffered from hyperlipidemia and their serum
lipid levels were mildly elevated, accompanied by high TC and LDL-C
levels. The results of the serum lipid profile of the primary
family members are presented in Table
II.
| Table II.Serum lipid profile of primary family
members. |
Table II.
Serum lipid profile of primary family
members.
| Subjects | Age (year) | TC (mmol/l) | HDL-C (mmol/l) | LDL-C (mmol/l) | TG (mmol/l) | Treatment |
|---|
| Proband | 16 | 14.21 | 0.84 | 12.22 | 1.63 | Atorvastatin |
|
|
|
|
|
|
| 40 mg/day |
| Father | 47 | 7.34 | 1.35 | 4.40 | 1.4 | None |
| Mother | 47 | 8.33 | 1.79 | 4.98 | 0.97 | None |
Genetic mutations
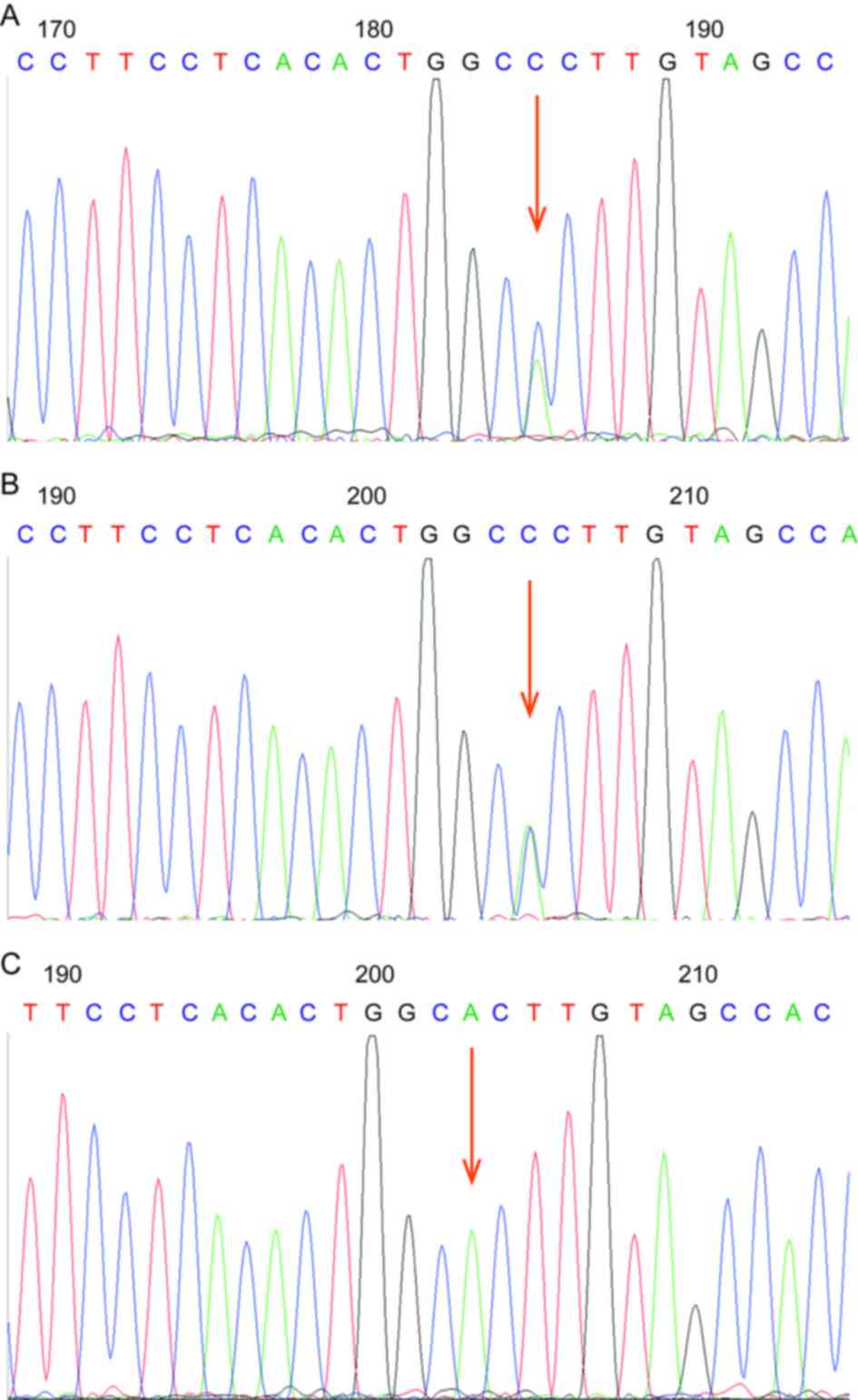
The genetic screening results demonstrated that the
proband and her mother had the same heterozygous missense mutation
C377G, 28893T>G in exon 8 (Fig. 2A
and B), whereas her father did not have this mutation (Fig. 2C). Therefore, the proband does not
have a homozygous mutation in her genotype. However, it was
demonstrated that in exons 16 and 18 of the proband and her
parents, gene sequences were mismatched. Multiple AA alleles were
present in the father, whereas GG alleles were present in the
mother and proband (rs13306501, rs2304182, rs2116899, rs2116897,
rs2116898 and rs14158; Table
III), which didn't obey the law of inheritance. According to
the laws of inheritance, if the DNA fragment was complete in the
associated exon region of the father, AG alleles would be expected
to be present in the proband (A from her father and G from her
mother). However, GG alleles were present in both the mother and
proband, which suggests that the proband did not inherit said
alleles in the same region from her father as she did from her
mother. As such, it was hypothesized that a large DNA fragment had
been deleted in the exon 16 and 18 regions of both the father and
the proband. There was no mutation observed in apoBQ3500R.
| Table III.Mismatched alleles in the low-density
lipoprotein-receptor gene exon of the family detected by DNA
sequencing. |
Table III.
Mismatched alleles in the low-density
lipoprotein-receptor gene exon of the family detected by DNA
sequencing.
| Exon | Nucleotide
(location) | Proband | Father | Mother |
|---|
| Exon 16 | rs13306501 | GG | AA | GG |
| Exon 16 | rs2304182 | GG | AA | GG |
| Exon 18 | rs2116899 | GG | AA | GG |
| Exon 18 | rs2116897 | GG | AA | GG |
| Exon 18 | rs2116898 | GG | AA | GG |
| Exon 18 | rs14158 | GG | AA | GG |
Abnormal target DNA copy number
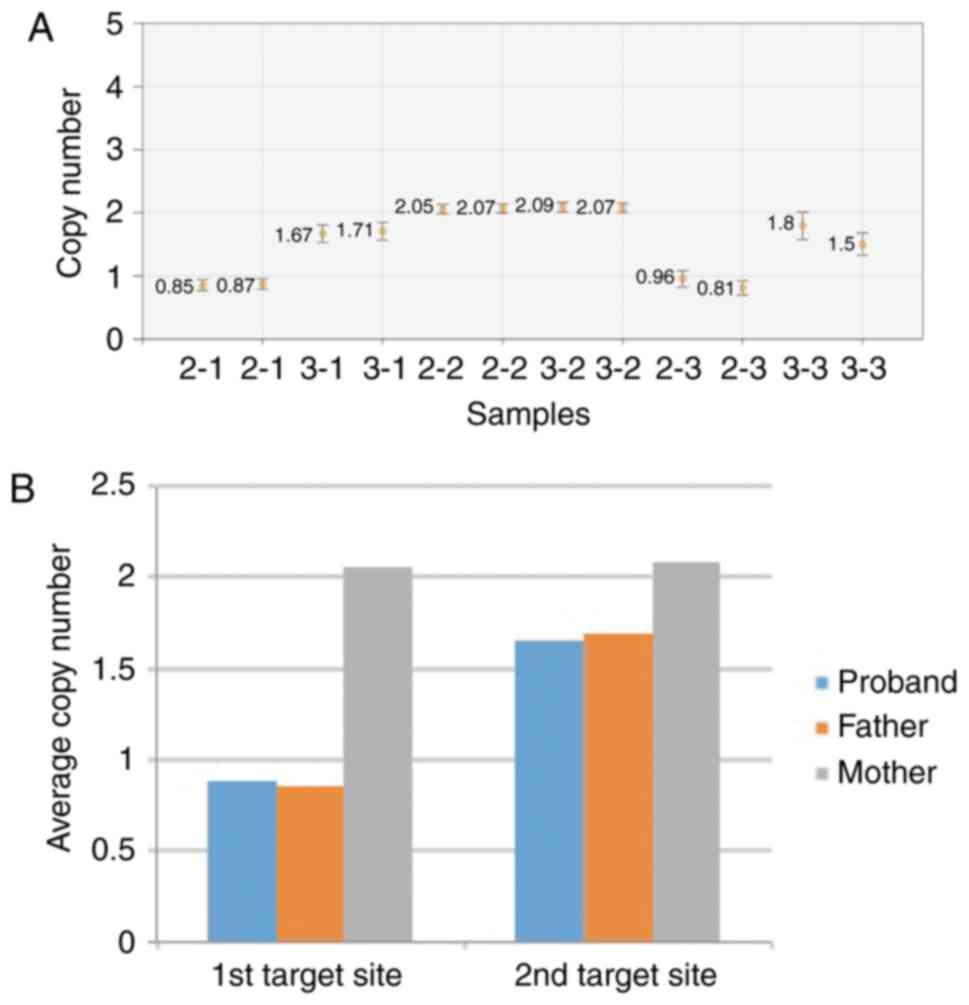
The results of the ddPCR™ system demonstrated that
the ratios of the father and the proband were close to 1 at the 1st
site, whereas the mother was close to 2 at the 1st site. The
results for the 2nd site demonstrated that the ratios for the
parents and the proband were all close to 2 (Fig. 3). These results indicated that the
father and proband were haploid at the 1st site, however diploid at
the 2nd site, whereas the mother was diploid at both the 1st and
2nd sites. This suggests that there were DNA fragment deletions at
exon 18 in both the proband and her father. The genotype of this
proband belongs to the compound heterozygous FH category.
Novel genetic mutations and fragment
deletions impair LDLR function
qPCR and western blot analysis of total cell lysates
from 293T cells transfected with WT and mutant LDLR expression
vectors were performed separately, including both C377G mutations
as well as the L855 deletion mutation. Compared with the WT, both
C377G missense mutations and L855 fragment deletion mutations led
to impaired expression of LDLR at the mRNA (Fig. 4A) and protein levels (Fig. 4B and C). To further demonstrate the
functional activity of these two lesions, 293T cells were used
without endogenous LDLR for LDL binding and internalization
analyses. Flow cytometry analysis demonstrated that compared with
WT control, these two mutations had significantly decreased LDL
binding (P<0.001; Fig. 4D and
E) and internalization ability (P<0.001; Fig. 4F and G).
 | Figure 4.Novel mutations impair LDLR expression
and function. (A) Quantitative polymerase chain reaction results of
293T cells transfected with WT, C377G, L855fs mutants and the
negative control vector. (B) Western blot analysis of total cell
lysates from cells transfected with WT and both C377G and L855fs
mutants, as well as the negative control vector. (C) Histogram of
different samples in western blotting analysis. (D)
Characterization of LDLR binding functions in transfected 293T
cells using flow cytometric measurements. Cells were transfected
with control vector, WT LDLR, C377G and L855fs mutant respectively.
The activity of Dil-LDL binding is given on the x-axis. (E) The
histogram exhibits the data as the percentage of mean fluorescence
compared with the WT. Bars and vertical lines indicate the mean of
five independent measurements and standard deviation, respectively.
(F) Characterization of LDLR internalization in transfected 293T
cells using flow cytometric measurements. Cells were transfected
with control vector, WT LDLR, C377G and L855fs mutant respectively.
The activity of Dil-LDL internalizing is given on the x-axis. (G)
Histogram of different samples as described previously.
**P<0.01; ***P<0.001. WT, wild type; NC, normal control;
LDLR, low-density lipoprotein receptor. |
Association between the phenotype and
genotype in this FH family
The genotype of this proband was the compound
heterozygous FH type, whereas her parents exhibited the simple
heterozygous genotype. Correspondingly, the proband demonstrated
severe clinical manifestations with an LDL-Cof <14 mmol/l,
tendon xanthoma, premature chronic heart diseases and peripheral
vascular disease, all which conformed to the homozygous FH
phenotype. Her parents only demonstrated mildly elevated serum
lipid levels with LDL-C between 4.0–4.9 mmol/l, without any other
clinical features, which met the heterozygous FH phenotype, in
accordance with The Dutch Lipid Network Criteria (13). Coincidentally, the phenotype was
associated with the genotype in this FH family (Table IV).
| Table IV.Association between the phenotype and
genotype in the familial hypercholestraemia family. |
Table IV.
Association between the phenotype and
genotype in the familial hypercholestraemia family.
|
|
| Phenotype | Genotype |
|
|---|
|
|
|
|
|
|
|---|
| Subject | Age (year) | TC (mmol/l) | LDL-C (mmol/l) | Xanthoma |
C377G28893T>G | DNA fragments
deletions at exon 18 | Treatment |
|---|
| Proband | 16 | 14.21 | 12.22 | Yes | Yes | Yes | Atorvastatin |
|
|
|
|
|
|
|
| 40 mg/day |
| Father | 47 |
7.34 |
4.40 | None | None | Yes | None |
| Mother | 47 |
8.33 |
4.98 | None | Yes | None | None |
Discussion
The present study covered both phenotypic and
genotypic analyses of an FH patient and her family members. The
proband in the present study had the typical clinical
manifestations of homozygous FH, presenting with xanthoma at the
age of three; an LDL-C >14 mmol/l at the age of six despite
optimal Lipitor treatment; and early onset of severe supravalvular
aortic stenosis, carotid artery atherosclerotic plaques and
coronary atherosclerosis. Her diagnosis clearly conformed to the
homozygous FH clinical diagnostic criteria (14–17).
Based on clinical features, genetic testing is
helpful to confirm the diagnosis of homozygous FH. Currently,
>90% of the diagnosed molecular FH cases result from LDLR
genetic mutations (18), whereas
the others are primarily due to a pathogenic mutation in apoB or
PCSK9 (19,20). All the aforementioned defects will
ultimately affect lipid metabolism and result in higher circulating
LDL-C concentrations. Van der Graaf et al (21), analyzed 269 pediatric cases of
children diagnosed with FH due to gene defects. The results
demonstrated that LDLR mutations accounted for 95% of genetic
mutations, whereas apoB mutations accounted for 5% and no PCSK9
mutations were detected.
Sequencing results, obtained using PCR to amplify
the entire sequence of the amino acid coding region of the LDLR
gene, including 18 exons in the proband, exhibited a heterozygous
mutation in exon 8 (c. 28893 T>G, p. Cys 377 Gly), which
resulted in a substitution of Cys at codon 377 with Gly. LDLR gene
sequencing of exon 8 of the proband's mother revealed the same
genotype, whereas her father had no mutation at that site. Both the
proband and her mother's genotypes meet the criteria for
heterozygous FH diagnosis. However, the clinical manifestations of
the proband are more severe and conform to the homozygous FH
diagnosis, whereas those of her mother are consistent with
heterozygous FH. Therefore, 18 exons of the LDLR gene of the
parents were sequenced for comparison with normal sequences. It was
demonstrated that the proband and her parents' partial genes
mismatched in exon 16 and exon 18. For the father, multiple alleles
presented with AA, whereas GG was present in the mother and proband
(rs13306501, rs2304182, rs2116899 and rs2116898). The results from
the QX200 ddPCR™ system demonstrated that the patient and her
father were haploid in part of exon 18, indicating DNA fragment
deletions at these loci. The results revealed that the proband is
compound heterozygous with gene variation. The proband inherited a
heterozygous missense mutation in exon 8 of the LDLR gene from her
mother, and from her father, she inherited LDLR gene DNA fragment
deletions and variation in exon 18. As both of her chromosomes are
defective, she cannot make normal LDLR. In fact, the proband's gene
variation conforms to the diagnosis of compound heterozygous FH,
leading to clinical manifestations in line with the homozygous FH
phenotype.
The LDLR gene is located on the short arm of
chromosome 19 (Ch19pl3.1–13.3) and has a total length of 45 kb,
including 18 exons and 17 introns, which encodes an 839 amino acid
precursor protein with seven domains (22,23).
According to the Britain FH LDLR gene mutations database
(http://www.ucl.ac.uk/ldlr/LOVDv.1.1.0/), >1700 LDLR
gene mutations have been reported worldwide. The heterozygous gene
mutation of the proband and her mother was previously unknown (c.
28893 T>G, p. Cys 377 Gly); namely, this mutation in the LDLR
gene sequence coding region (CDS) at the 8th exon, no. 28,893,
leads to a alteration in the encoded protein, as Cys is replaced
with Gly. This type of mutation is located in the epidermal growth
factor precursor domain and it may lead to transport disorders of
LDLR from the endoplasmic reticulum to the Golgi, loss of function
for binding to LDL, or LDLR recycling disorders. A previous study
revealed that a pathogenic c.1129 T>G, C356G mutation in exon 8
results in 57% LDLR binding and 52% internalization activity in
transfected 293T cells (24).
The proband and her father's LDLR gene sequence CDS
of exon 18 exhibited deletion mutations, which occurred in the CDS
of the transmembrane domain and the cytoplasmic domains. These
deletions may easily lead to an inward deficient shift, where LDLR
may bind to LDL, however LDL cannot be transported to the cell
surface and accumulate in the coating lacuna. This kind of deletion
is identical to FH-Helsinki, which is a deletion of the LDLR gene
that deletes 9.6 kb from intron 15 to exon 18 and is very common
among Finnish FH subjects in Oslo, Norway (25,26).
Furthermore, in the present study, analyses using 293T cells
further demonstrated that both the C377G mutation and the fragment
deletions in exon 18 could affect expression and function of LDL-R
to some extent, leading to impaired LDL binding and
internalization. However, as the in vitro cell model
represents a simplified molecular environment compared with in
vivo conditions, such mutation and fragment deletions may cause
more significant dysfunction of LDL-R in in vivo
experiments.
The proband inherited her mother's exon 8
heterozygous mutation (c. 28893 T>G, p. Cys 377 Gly) and
inherited her father's exon 18 deletion mutation, which resulted in
two DNA strands that were unable to synthesize normal LDLR.
Therefore, she suffered from tendon xanthoma at an early age, a
large accumulation of LDL in plasma and severe atherosclerosis
during youth.
Regarding pathogenesis, due to a congenital
deficiency of LDLR, high amounts of cholesterol accumulated in the
body organs, resulting in severe supravalvular aortic stenosis,
carotid artery atherosclerotic plaques, coronary atherosclerosis
and tendon xanthoma. Given that patients with homozygous FH have a
severe or total deficiency in LDLR function, their response to LDL
lowering therapies is usually markedly attenuated (27). Although the proband did not reach
the target level of LDL during her 13 years of treatment, her LDL
level still decreased, through the application of statins. Statins
and ezetimibe are able to lower LDL-C in homozygous FH patients by
potentially decreasing hepatic secretion of apoB (1,28)
and upregulation of hepatic LDLRs (27). However, most of the homozygous FH
patients usually require apheresis (1,29).
Therefore, it was suggested that the proband take 100 mg aspirin
daily, 40 mg Lipitor every night, 10 mg ezetimibe daily and LDL
apheresis, based on her LDL level. As the proband currently has no
clinical symptoms of dizziness or chest tightness and no coronary
ostial lesions, she will be followed-up and surgery only suggested
when obvious clinical manifestations appear.
In the present study, the proband's mother had a
heterozygous mutation in exon 8 (c. 28893 T>G, p. Cys 377 Gly);
this genotype belongs to the heterozygous FH diagnosis and
clinically manifests with mildly elevated blood lipids. The
patient's father had a single-stranded deletion in exon 18, causing
him to belong to the heterozygous FH diagnosis, either with mildly
elevated blood lipids or without clinical features upon physical
examination.
The proband inherited the variant genes of both her
father and her mother, so her genotype and clinical phenotype are
consistent with homozygous FH. She exhibited all of the typical
symptoms, including early manifestations of xanthoma,
hyperlipidemia, cardiovascular disease, ineffective administration
of lipid-lowering drugs and a need to reduce the level of
cholesterol with apheresis. To some extent, this indicated that
clinical manifestations and genotypes are associated. However, HF
is a heterogeneous disease with different mutations and the same
site mutation has occurred in different patients, however with
different clinical manifestations and symptoms of different
severity, particularly in those of the heterozygous genotype.
Epidemiological studies demonstrated that the clinical
manifestations of FH heterozygotes in China are much less severe
compared with patients in the West, which may be associated with
the traditional low-fat diet and some of the ‘lipid-regulating’
genes in China (1,28).
Regardless, once identified, FH patients should be
prescribed a high-potency statin therapy at the highest tolerated
dose. For the severe homozygous FH patients, lipoprotein apheresis
is further administered to remove lipoproteins. Several studies
have demonstrated that statins and lipoprotein apheresis may reduce
the progression of atherosclerosis and improve the outcome of FH
(30–33). Therefore, early diagnosis and early
treatment may delay the cumulative burden of elevated LDL-C,
reducing the risk of complications associated with atherosclerosis
and coronary heart disease (34).
Therefore, clinicians and society should raise awareness of FH and
the comprehensive screening strategy, including the clinical and
genetic aspects, in order to significantly improve the outcomes for
these patients (17,35).
In conclusion, in the present study, a compound
heterozygous family with FH were investigated. The family members
carried genetic defects in exon 8, with a heterozygous mutation (c.
28893 T>G, p. Cys 377 Gly) in one chromosome strand, and a
deletion mutation in exon 18 in another chromosome strand. To the
best of the author's knowledge, the heterozygous allelic mutation
has not yet been reported. The proband's genotype is different from
any of the previously reported FH genetic variations. The genotype
is associated with the phenotype, which may help to reasonably
explain the clinical manifestations.
Acknowledgements
The authors wish to acknowledge the contribution of
all the doctors of the Department of Cardiology at Rui Jin
Hospital, Shanghai Jiao Tong University School of Medicine,
(Shanghai, China) to the present study.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81670352 to RT),
Shanghai Municipal Education Commission-Gaofeng Clinical Medicine
Grant Support (grant no. 20152205 to RT) and Shanghai Municipal
Commission of Health and Family Planning (grant no. 201740248 to
FW). No relationships with industry are to be declared.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
FW, QF and RX contributed to all stages of the
present study, including conception and design of the study, as
well as the acquisition, analysis and interpretation of data. RT,
GG and RZ performed the experiments and assisted with data
acquisition, analysis and interpretation of the data. FW and QF
contributed to the writing of the article, together with RT, RX, RZ
and GG who additionally revised the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
All investigated subjects gave written informed
consent for all these tests and the publication of associated data
and accompanying images prior to taking part in the study and
participating in these examinations. The study was conducted
according to the Helsinki declaration (2013) and approved by the
Institutional Ethics Committees of Shanghai Jiao Tong University,
affiliated with Rui Jin Hospital (Shanghai, China).
Consent for publication
All investigated subjects gave written informed
consent for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Raal FJ and Santos RD: Homozygous familial
hypercholesterolemia: Current perspectives on diagnosis and
treatment. Atherosclerosis. 223:262–268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goldstein JL, Hobbs HH and Brown MS:
Familial hypercholesterolemiaThe metabolic basis of inherited
disease. McGraw-Hill; New York: pp. 2863–2913. 2001
|
|
3
|
Wang L, Lin J, Liu S, Cao S, Liu J, Yong
Q, Yang Y, Wu B, Pan X, Du L, et al: Mutations in the LDL receptor
gene in four Chinese homozygous familial hypercholesterolemia
phenotype patients. Nutr Metab Cardiovasc Dis. 19:391–400. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hooper AJ, Nguyen LT, Burnett JR, Bates
TR, Bell DA, Redgrave TG, Watts GF and van Bockxmeer FM: Genetic
analysis of familial hypercholesterolaemia in Western Australia.
Atherosclerosis. 224:430–434. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang L, Sun LY, Dai YF, Yang SW, Zhang F
and Wang LY: The distribution and characteristics of LDL receptor
mutations in China: A systematic review. Sci Rep. 5:172722015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Humphries SE, Norbury G, Leigh S, Hadfield
SG and Nair D: What is the clinical utility of DNA testing in
patients with familial hypercholesterolaemia? Curr Opin Lipidol.
19:362–368. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Taylor A, Wang D, Patel K, Whittall R,
Wood G, Farrer M, Neely RD, Fairgrieve S, Nair D, Barbir M, et al:
Mutation detection rate and spectrum in familial
hypercholesterolaemia patients in the UK pilot cascade project.
Clin Genet. 77:572–580. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lombardi MP, Redeker EJ, van Gent DH,
Smeele KL, Weerdesteijn R and Mannens MM: Molecular genetic testing
for familial hypercholesterolemia in the Netherlands: A stepwise
screening strategy enhances the mutation detection rate. Genet
Test. 10:77–84. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Poncz M, Solowiejczyk D, Harpel B, Mory Y,
Schwartz E and Surrey S: Construction of human gene libraries from
small amounts of peripheral blood: Analysis of beta-like globin
genes. Hemoglobin. 6:27–36. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Belgrader P, Tanner SC, Regan JF, Koehler
R, Hindson BJ and Brown AS: Droplet digital PCR measurement of HER2
copy number alteration in formalin-fixed paraffin-embedded breast
carcinoma tissue. Clin Chem. 59:991–994. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hindson BJ, Ness KD, Masquelier DA,
Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY,
Hiddessen AL, Legler TC, et al: High-throughput droplet digital PCR
system for absolute quantitation of DNA copy number. Anal Chem.
83:8604–8610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hartgers ML, Ray KK and Hovingh GK: New
approaches in detection and treatment of familial
hypercholesterolemia. Curr Cardiol Rep. 17:1092015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Watts GF, Sullivan DR, Poplawski N, van
Bockxmeer F, Hamilton-Craig I, Clifton PM, O'Brien R, Bishop W,
George P, Barter PJ, et al: Familial hypercholesterolaemia: A model
of care for Australasia. Atheroscler Suppl. 12:221–263. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Harada-Shiba M, Arai H, Oikawa S, Ohta T,
Okada T, Okamura T, Nohara A, Bujo H, Yokote K, Wakatsuki A, et al:
Guidelines for the management of familial hypercholesterolemia. J
Atheroscler Thromb. 19:1043–1060. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Williams RR, Hunt SC, Schumacher MC,
Hegele RA, Leppert MF, Ludwig EH and Hopkins PN: Diagnosing
heterozygous familial hypercholesterolemia using new practical
criteria validated by molecular genetics. Am J Cardiol. 72:171–176.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Watts GF, Gidding S, Wierzbicki AS, Toth
PP, Alonso R, Brown WV, Bruckert E, Defesche J, Lin KK, Livingston
M, et al: Integrated guidance on the care of familial
hypercholesterolaemia from the International FH Foundation:
Executive summary. J Atheroscler Thromb. 21:368–374.
2014.PubMed/NCBI
|
|
18
|
Nordestgaard BG, Chapman MJ, Humphries SE,
Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ,
Defesche JC, et al: Familial hypercholesterolaemia is
underdiagnosed and undertreated in the general population: Guidance
for clinicians to prevent coronary heart disease: Consensus
statement of the European Atherosclerosis Society. Eur Heart J.
34:a3478–a3490. 2013. View Article : Google Scholar
|
|
19
|
Tybjaerg-Hansen A and Humphries SE:
Familial defective apolipoprotein B-100: A single mutation that
causes hypercholesterolemia and premature coronary artery disease.
Atherosclerosis. 96:91–107. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abifadel M, Varret M, Rabès JP, Allard D,
Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich
D, et al: Mutations in PCSK9 cause autosomal dominant
hypercholesterolemia. Nat Genet. 34:154–156. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
van der Graaf A, Avis HJ, Kusters DM,
Vissers MN, Hutten BA, Defesche JC, Huijgen R, Fouchier SW, Wijburg
FA, Kastelein JJ and Wiegman A: Molecular basis of autosomal
dominant hypercholesterolemia: Assessment in a large cohort of
hypercholesterolemic children. Circulation. 123:1167–1173. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lindgren V, Luskey KL, Russell DW and
Francke U: Human genes involved in cholesterol metabolism:
Chromosomal mapping of the loci for the low density lipoprotein
receptor and 3-hydroxy-3-methylglutaryl-coenzyme A reductase with
cDNA probes. Proc Natl Acad Sci USA. 82:8567–8571. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamamoto T, Davis CG, Brown MS, Schneider
WJ, Casey ML, Goldstein JL and Russell DW: The human LDL receptor:
A cysteine-rich protein with multiple Alu sequences in its mRNA.
Cell. 39:27–38. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu WF, Sun LY, Pan XD, Yang SW and Wang
LY: Use of targeted exome sequencing in genetic diagnosis of
Chinese familial hypercholesterolemia. PLoS One. 9:e946972014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rødningen OK, Tonstad S, Ose L, Berg K and
Leren TP: Effects of a 9.6-kb deletion of the LDL receptor gene (FH
Helsinki) on structure and levels of mRNA. Hum Mutat. 12:95–102.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rødningen OK, Røsby O, Tonstad S, Ose L,
Berg K and Leren TP: A 9.6 kilobase deletion in the low density
lipoprotein receptor gene in Norwegian familial
hypercholesterolemia subjects. Clin Genet. 42:288–295. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ooi EM, Barrett PH and Watts GF: The
extended abnormalities in lipoprotein metabolism in familial
hypercholesterolemia: Developing a new framework for future
therapies. Int J Cardiol. 168:1811–1818. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gagné C, Gaudet D and Bruckert E:
Ezetimibe Study G: Efficacy and safety of ezetimibe coadministered
with atorvastatin or simvastatin in patients with homozygous
familial hypercholesterolemia. Circulation. 105:2469–2475. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thompson GR, Catapano A, Saheb S,
Atassi-Dumont M, Barbir M, Eriksson M, Paulweber B, Sijbrands E,
Stalenhoef AF and Parhofer KG: Severe hypercholesterolaemia:
Therapeutic goals and eligibility criteria for LDL apheresis in
Europe. Curr Opin Lipidol. 21:492–498. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smilde TJ, van Wissen S, Wollersheim H,
Trip MD, Kastelein JJ and Stalenhoef AF: Effect of aggressive
versus conventional lipid lowering on atherosclerosis progression
in familial hypercholesterolaemia (ASAP): A prospective,
randomised, double-blind trial. Lancet. 357:577–581. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Versmissen J, Oosterveer DM, Yazdanpanah
M, Defesche JC, Basart DC, Liem AH, Heeringa J, Witteman JC,
Lansberg PJ, Kastelein JJ and Sijbrands EJ: Efficacy of statins in
familial hypercholesterolaemia: A long term cohort study. BMJ.
337:a24232008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Raal FJ, Pilcher GJ, Panz VR, van Deventer
HE, Brice BC, Blom DJ and Marais AD: Reduction in mortality in
subjects with homozygous familial hypercholesterolemia associated
with advances in lipid-lowering therapy. Circulation.
124:2202–2207. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Thompson GR: The evidence-base for the
efficacy of lipoprotein apheresis in combating cardiovascular
disease. Atheroscler Suppl. 14:67–70. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wiegman A, Gidding SS, Watts GF, Chapman
MJ, Ginsberg HN, Cuchel M, Ose L, Averna M, Boileau C, Borén J, et
al: Familial hypercholesterolaemia in children and adolescents:
Gaining decades of life by optimizing detection and treatment. Eur
Heart J. 36:2425–2437. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cuchel M, Bruckert E, Ginsberg HN, Raal
FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps
OS, Steinhagen-Thiessen E, et al: Homozygous familial
hypercholesterolaemia: New insights and guidance for clinicians to
improve detection and clinical management. A position paper from
the Consensus Panel on Familial Hypercholesterolaemia of the
European Atherosclerosis Society. Eur Heart J. 35:2146–2157. 2014.
View Article : Google Scholar : PubMed/NCBI
|