Introduction
An abdominal aortic aneurysm (AAA) is an
irreversible widening of the blood vessels caused by weakening of
different layers in the vascular wall. The primary complication of
AAA is aortic rupture, which has an overall mortality rate of 90%
(1). Currently, there is no
accepted drug treatment to limit AAA rupture. The underlying
mechanism of the pathogenesis of AAA remains uncertain. The
pathological hallmarks of AAA are chronic inflammation, vascular
smooth muscle cell (VSMC) apoptosis, extracellular matrix (ECM)
degradation and thrombosis, which are additionally involved in
atherosclerotic plaque formation (2). A variety of cytokines promote the
destruction of the aortic wall in AAA and arteriosclerosis
(3). High concentrations of the
glycoprotein osteoprotegerin (OPG) have been demonstrated in human
AAA biopsies (4,5).
OPG, additionally termed tumor necrosis factor
receptor superfamily member 11B (TNFRSF11B), is a cytokine receptor
that may function as a soluble decoy receptor for the receptor
activator of nuclear factor-κB (RANK) and TNF-related
apoptosis-inducing ligand (6–8). The
RANK/RANK ligand (L)/OPG axis is involved in bone remodeling, and
regulates the differentiation and activation of osteoclasts and,
therefore, there is a crucial balance between bone formation and
bone resorption (9). RANK is
located on the surface of osteoclast precursors, including
monocytes, macrophages and dendritic cells (10,11).
RANKL is expressed on the surface of stromal cells, osteoblasts and
T cells (12). OPG functions as a
soluble decoy receptor by binding to RANKL, and competitively
inhibiting the interaction between RANKL and its receptor (13).
Emerging evidence has suggested that OPG is not
merely a protective factor for bone, but that it may additionally
act as a protective modulator in the cardiovascular system. Studies
have reinforced the idea of OPG as a novel biomarker for
cardiovascular disease (CVD) and arteriosclerosis in humans
(14–16). The concentration of OPG is in fact
associated with aortic diameter, and pro-inflammatory proteases and
lymphocyte markers of aneurysmal disease (17).
OPG is expressed in the vascular system, including
VSMCs and endothelial cells (18),
and its release may be modulated by pro-inflammatory cytokines,
including interleukin-1β (IL-1β) and tumor necrosis factor-α
(TNF-α) (19,20). OPG inactivation in apolipoprotein-E
(ApoE)-knockout mice accelerates advanced atherosclerotic lesion
progression and vascular calcification (21), which is in accordance with an
earlier study that reported profound calcification of the large
arteries, including intimal and medial proliferation, in mice with
targeted disruption of OPG (22).
In line with these findings, transgenic expression of OPG prevented
the formation of calcified lesions in the arteries of adolescent
mice (23). Furthermore, high
levels of OPG were demonstrated to promote the accumulation of
VSMCs and the formation of collagen, although it did not affect the
inflammatory properties of atherosclerotic lesions (24). In a study by Candido et al
(25), low levels of OPG (1 µg
every 3rd week) increased the number of VSMCs in aortic
plaques.
A total of two independent studies have investigated
the effects of OPG and AAA in mice. Moran et al (26) used ApoE/OPG double knockouts with
angiotensin II (angII)-induced AAA that resulted in a reduction in
pro-inflammatory markers and an inhibition of aortic dilation and
rupture. By contrast, results by Bumdelger et al (27) demonstrated that OPG-knockout in
mice using a CaCl2-induced AAA model promoted AAA
formation.
The impact of treatment with OPG in the development
of CVD and AAA remains unclear. Based on the previous findings, the
present study hypothesized that OPG may be involved in key events
of AAA development, including stabilization of the vessel wall
through compensatory production of collagen.
In the present study, two different functional mouse
models of AAA were used with the aim of assessing the effects of
low levels of OPG in AAA development in mice.
Material and methods
Mouse experiments
All mouse experiments were approved by Stockholm
North Ethical Committee on Animal Research (Stockholm, Sweden).
Mice were treated with the analgesic buprenorphine (Temgesic; 0.1
mg/kg; Indivior UK Limited, Slough, UK) two times per day for 72 h
following surgery. Operated animals were inspected every day for
any sign of pain or disability. No abnormal behaviour indicating
pain or distress, or surgical site infection was observed in any of
the mice after surgery during the study. If any animals would
suffer from the above they would be sacrificed. However, this was
never needed. Mice (n=64; male; weighing 20–30 g) were fed a normal
chow diet and water was provided ad libitum during the whole
observation period. Animals were housed in a controlled temperature
of 22°C, a 12/12 h light/dark cycle and a humidity of 40–60%. For
histology and gene expression analysis, the aortas of the mice were
removed and immediately fixed in either 4% zinc formaldehyde at
room temperature for 24 h or in RNAlater (Ambion; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 4°C for 24 h, and
subsequently frozen at −70°C.
AngII-induced AAA in mice
A total of 2 weeks prior to the induction of AAA
using angII, mice were randomized to be administered injections of
recombinant human (rh)-OPG (1 µg twice a week; R&D Systems,
Inc., Minneapolis, MN, USA) or 0.9% NaCl intraperitoneally during
the two weeks. In eight week-old male ApoE−/− mice
(Taconic Biosciences, Bomholt, Denmark), AAA was induced by chronic
infusion of 1,000 ng/kg/min angII (cat no. 9525; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) or 0.9% NaCl as a control via
mini-osmotic pumps (Model 1004; Alzet, Cupertino, CA, USA). Mice
were anesthetized with 2% isoflurane and 0.5 l/min oxygen through a
nose mask. The pumps were implanted subcutaneously into the right
flank of the mice to release angII over a period of 28 days. Rh-OPG
was loaded into the mini-osmotic pumps for additional continuous
administration of 7.1 µg OPG during the 28 days of the experiment.
Groups were divided as follows: NaCl and NaCl (n=5), angII and NaCl
(n=9), NaCl and rh-OPG (n=7), and angII and rh-OPG (n=10).
CaCl2-induced AAA in
mice
C57Bl/6J mice of 8 weeks of age, purchased from
Jackson Laboratory (Bar Harbor, ME, USA), were anesthetized with 2%
isoflurane and 0.5 l/min oxygen through a nose mask, and were
randomized to receive rh-OPG and periaortic application of
CaCl2. A small compress strip soaked with 0.5 M
CaCl2 or saline as a control was applied to the isolated
abdominal aorta, which was covered for 15 min. Thereafter, the
compress was removed, and the treated area was dried and washed
twice with saline. At 2 weeks, the mice were sacrificed. Three
groups were divided as follows: CaCl2 and saline (n=10),
CaCl2 and rh-OPG (n=10), saline and saline (n=10). The
intraperitoneal rh-OPG (1 µg twice a week; R&D Systems) or
saline treatment was administrated four times before and four times
following surgery to induce aneurysm using the
CaCl2-model (two injections/week). On the day of
surgery, the mice received an rh-OPG/vehicle injection. In total,
the mice had nine intraperitoneal injections of rh-OPG/vehicle
during the 4 weeks.
Measurement of mouse abdominal aortic
diameter
Following dehydration, paraffin embedding and
sectioning (5 µm), aortas were incubated in Bouin's solution (Sigma
Aldrich; Merck KGaA) overnight at room temperature, counterstained
with Weigert's hematoxylin (HistoRx, New Haven, CT, USA) for 10 min
at room temperature and stained with the Masson's Trichrome kit
(Sigma Aldrich; Merck KGaA), according to the manufacturer's
protocol (phosphomolybdic acid fuchsin solution for 10 min; aniline
blue for 5 min) at room temperature. Whole sections were observed
under a light microscope (magnification, ×10 and ×40). Aortic
circumference was measured for the outer adventitial area and for
the inner luminal area. The vessel wall thickness was determined
from the outer adventitial diameter and the inner luminal diameter.
Definition of an aneurysm was set as a 1.5-fold enlargement of the
aortic wall. To measure aortic outer diameter, the largest part of
the aorta was taken for this purpose.
Quantification of collagen and
elastin
Aortic sections were stained with Picro-sirius red
(Sigma Aldrich; Merck KGaA) for 1 h at room temperature to
visualize thin and thick collagen fibers and washed with 1% acetic
acid twice for 10 min at room temperature. Measurements were made
using the LeicaQWin image analysis software (Leica Microsystems
GmbH, Wetzlar, Germany) to measure the amount of red-orange and
yellow-green color relative to the aortic area. Thick collagen
fibers presented an intense birefringence resulting in a red-orange
color, whereas thin collagen fibers presented a weak birefringence
with a yellow-green color when observing through a light microscope
under polarized light (magnification, ×10 and ×40).
For elastin, paraffin embedded abdominal aortic
sections (5 µm) were stained with Verhoeff's hematoxylin for 1 h,
differentiated in 2% ferric chloride for 2 min and counterstained
with Van Gieson solution for 5 min (all at room temperature) to
identify elastic fibers in the aortic tissue. For analysis, a
scoring system between 1 and 4 was used, and a score of 1 was
defined as intact elastin, a score of 2 as low elastin degradation,
a score of 3 as intermediate elastin degradation and a score of 4
as high elastin degradation.
Cell culture of human aortic smooth
muscle cells
Human vascular aortic SMCs, obtained from the
American Type Culture Collection (Manassas, VA, USA), were
maintained in smooth muscle growth medium-2 containing 5% fetal
bovine serum (FBS; CC-3182; Clonetics; Cambrex Bio Science
Walkersville, Inc.; Lonza Group Ltd., Basel, Switzerland). The
cells were used at passages 3–5. All cells were incubated in 5%
CO2 at 37°C and stimulated with 20 ng/ml recombinant
TNF-α, 20 ng/ml interferon-γ (IFN-γ) or 10 ng/ml IL-1β (R&D
Systems) for 4, 24 and 48 h at 37°C.
Reverse transcription-quantitative
polymerase chain reaction (PCR)
Total RNA was extracted from aortas and VSMCs using
TRIzol (Thermo Fisher Scientific, Inc.) and an RNeasy Mini kit
(Qiagen GmbH, Hilden, Germany). RNA was reverse transcribed with
random primers and Superscript III, according to the manufacturer's
protocol (cat. nos. 48190-011, 18427-013 and 18080093; Invitrogen;
Thermo Fisher Scientific, Inc.). Briefly, total RNA, primers and
dNTPs were heated to 65°C for 5 min, incubated on ice for 1 min and
then mixed with first strand buffer, dithiothreitol and Superscript
III, and then further incubated at 50°C for 30 min. Following this,
the reaction was inactivated via incubation at 70°C for 15 min.
From each PCR product, using the standard curve method in a
fluorescent temperature cycler, cDNA (1.0 ng) was amplified for 40
cycles (denaturation at 95°C for 3 sec and extension at 60°C for 30
sec) in a 20 µl PCR reaction volume using the TaqMan Universal PCR
Mastermix (Applied Biosystems; Thermo Fisher Scientific, Inc.) in
96-well fast plates on a 7500 Fast Real-time PCR Sequence Detector
(Applied Biosystems; Thermo Fisher Scientific, Inc.) in duplicate.
The following gene expression assays, all purchased from Applied
Biosystems; Thermo Fisher Scientific, Inc., were used: Adiponectin
(cat no. Mm00456425_m1); integrin subunit αM (CD11b; cat no.
Mm00434455_m1); CD68 (cat no. Mm03047340_m1); cathepsin K (cat no.
Mm00484036_m1); cathepsin S (cat no. Mm00457902_m1); TNF-α (cat no.
Mm00443258_m1); α-actin (cat no. Mm1187533_m1); transgelin (Sm22α;
cat no. Mm00441660_m1); lysyl oxidase (Lox; cat no. Mm01265612) and
OPG (cat no. Hs00900358_m1). The results were normalized to the
values of murine TATA-box binding protein (cat no. Mm00446973_m1)
or human ribosomal protein lateral stalk subunit P0 (cat no.
Hs00420895_gH).
Measurement of OPG from VSMCs
Secreted levels of OPG were measured in the muscle
growth medium-2 containing 5% FBS from cultured VSMCs from three
separate experiments that had been cultured for 48 h prior to
analysis using an ELISA kit, according to the manufacturer's
protocol (cat no. DY805; human OPG; R&D Systems, Inc.).
Statistical analysis
Statistical analyses were performed using SPSS
software version 24 (IBM Corp., Armonk, NY, USA) and GraphPad Prism
version 7 software (GraphPad Software, Inc., La Jolla, CA, USA).
Data are presented as the mean ± standard deviation. Differences
between groups were analyzed by two-tailed unpaired t-test and one
way analysis of variance followed by the Bonferroni method post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Minor effects of treatment with OPG in
angII-induced AAA development
Infusion of angII induced AAA in eight out of nine
vehicle control and nine out of ten OPG-treated mice. Sections were
not available for aortic investigation in three mice of the vehicle
and one mouse of the OPG-treated mice. In total, one mouse
succumbed to rupture in the vehicle-treated angII group and one in
the OPG-treated angII group. Treatment with OPG in angII infused
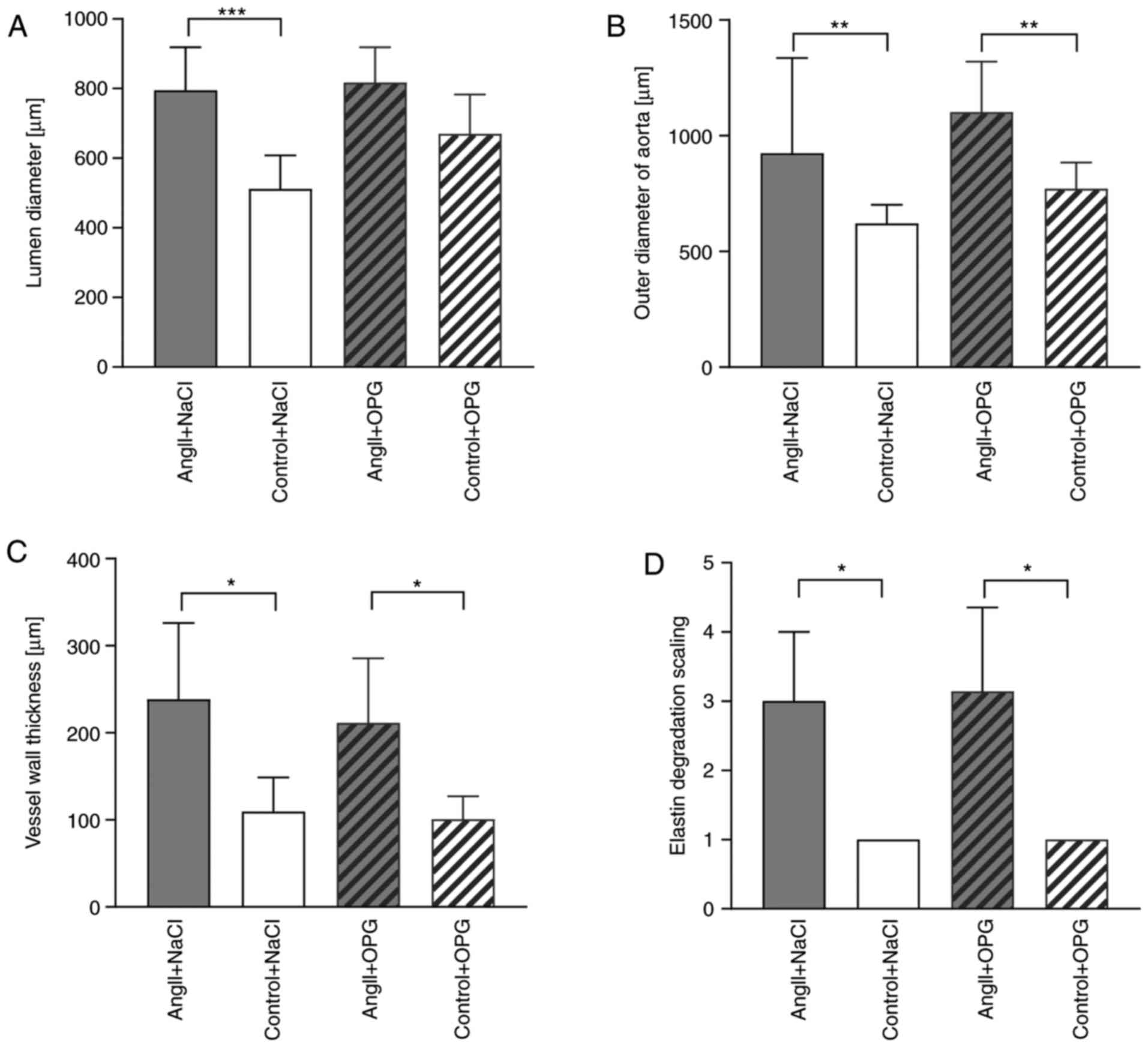
mice had no significant effect on luminal diameter (Fig. 1A), outer diameter of the aorta
(Fig. 1B) or vessel wall thickness
(Fig. 1C) compared with
angII-infused saline treated mice. Treatment with OPG led to
significantly increased aortic diameter in ApoE−/−
control mice that were treated with saline instead of angII,
although this did not remain significant after adjustment for
multiple comparison (data not shown). Treatment with OPG in control
mice did not markedly alter the diameter of the lumen or vessel
wall thickness (Fig. 1A and C)
compared with control mice treated with saline. Both angII-infused
groups of mice (angII+NaCl and angII+OPG) exhibited significantly
increased degradation of elastin compared with their control
littermates that were not treated with angII, and treatment with
OPG did not contribute to any alterations in elastin degradation as
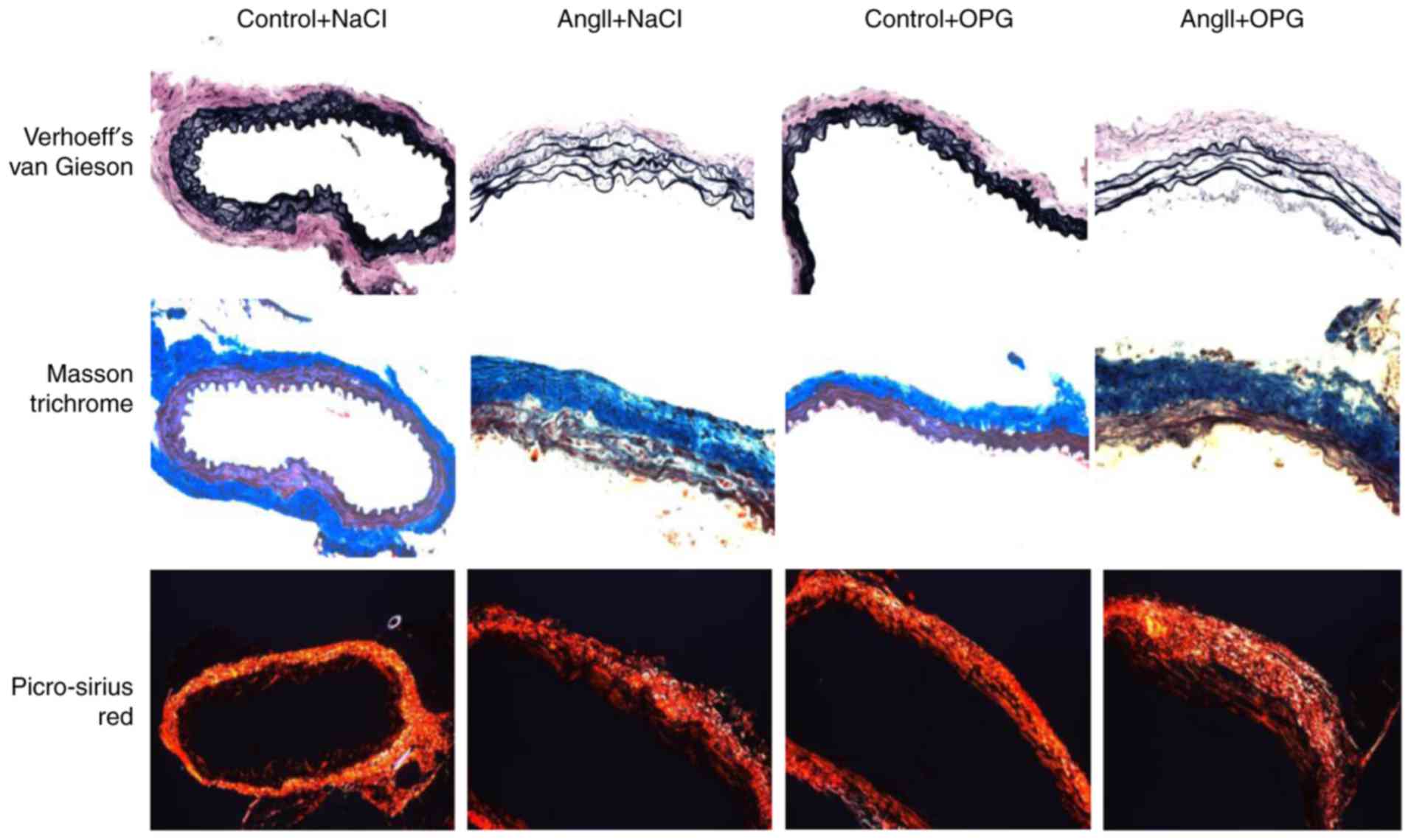
indicated by Verhoeff's van Gieson staining (Fig. 1D and upper panel of Fig. 2). No differences were observed in
the amount of collagen between OPG-treated and untreated mice as
indicated by Masson trichrome staining (Fig. 2).
Treatment with OPG increases vessel
wall thickness and collagen levels in the aorta in
CaCl2-induced dilated aortas
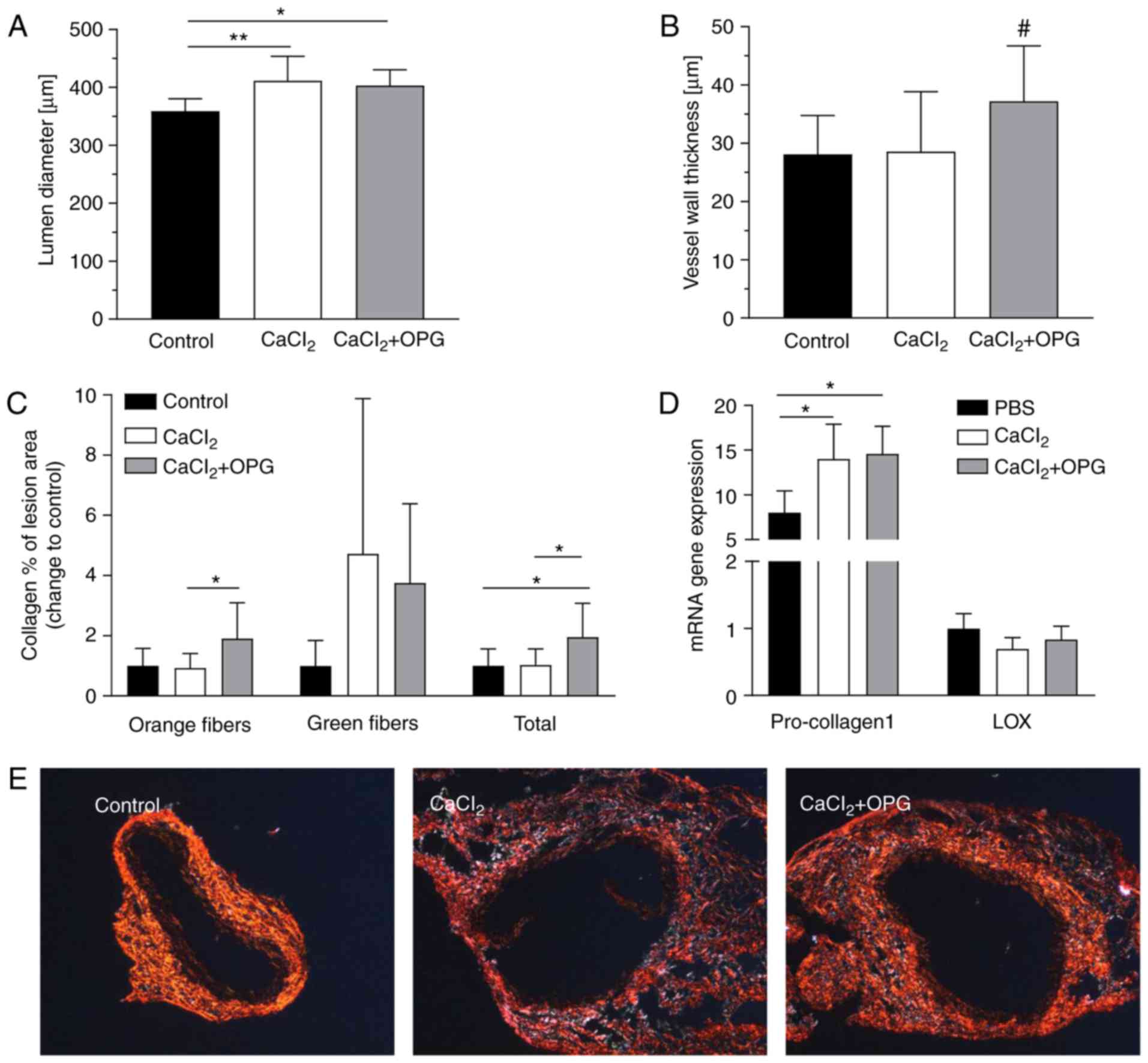
Periaortic application of CaCl2 resulted
in a small, although significant, dilatation of the aorta compared
with the control (Fig. 3A). No
animals succumbed to rupture (data not shown). Furthermore,
treatment with OPG led to significantly increased vessel wall
thickness of the aorta in the CaCl2-induced AAA mice
compared with control mice (Fig.
3B); however, this did not remain significant after adjustment
of multiple comparison. Total collagen composition, primarily
accounted for by mature fibers, was twice as high throughout the
lesions in the aorta of OPG-treated mice compared with
vehicle-treated controls and CaCl2-induced mice
(Fig. 3C). At the mRNA level,
pro-collagen1 expression was significantly increased in
AAA-developing mice compared with controls, although there was no
significant difference in OPG-treated mice (Fig. 3D). Treatment with OPG exerted no
effect on Lox expression (Fig.
3D). Fig. 3E demonstrates that
collagen composition was higher in lesions of OPG-treated mice
compared with control mice. There were no detectable differences in
elastin degradation in mice following treatment with OPG (data not
shown).
Low levels of OPG do not affect
inflammatory and ECM gene expression in the abdominal aortic wall
of CaCl2-treated aortas
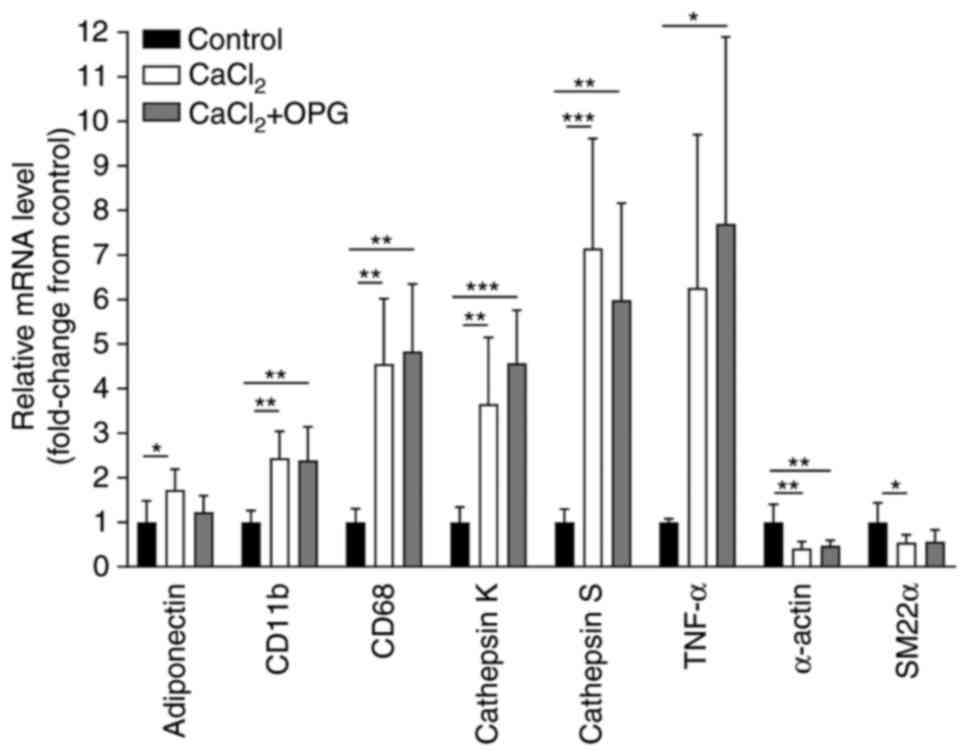
Significantly increased mRNA expression levels of
markers of inflammation, including (TNF-α), macrophages (Cd68),
leukocytes (Cd11b), adipokines (adiponectin) and proteases
(cathepsin K and cathepsin S) was observed, and a significant
reduction in the mRNA expression levels of VSMC markers (α-actin
and Sm-22α) in the mice with periaortic CaCl2
application compared with their littermate controls (Fig. 4). However, treatment with OPG did
not affect the mRNA expression of the genes compared with
CaCl2 mice without OPG injection (Fig. 4). An exception was observed in the
expression of adiponectin, which was not increased in the
CaCl2-treated mice administered OPG compared with the
control group.
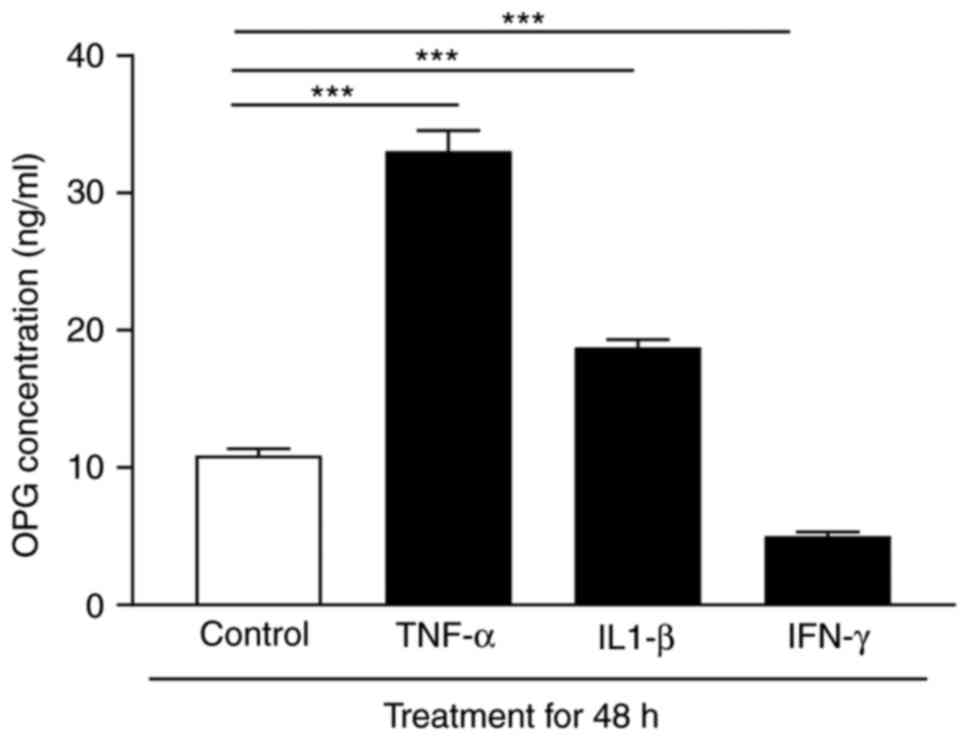
Cytokines cause altered OPG expression
in human aortic VSMCs
As VSMCs are primary components of the aortic wall
and are important producers of OPG, the regulatory effects of
different cytokines on OPG expression were analyzed in the media
from human aortic VSMCs in vitro. Stimulation of cultured
human aortic VSMCs with TNF-α and IL-1β led to a 2–3 fold higher
release of OPG, respectively, whereas IFN-γ downregulated the
levels of OPG following 48 h of stimulation compared with the
control group (Fig. 5). Similar
results were observed at 24 h of stimulation, whereas 4 h of
stimulation did not lead to any significant release (data not
shown).
Discussion
The present study investigated the role of low-dose
treatment with OPG in the development of CaCl2- and
angII-induced AAA. There has been increased interest in the role of
OPG as a predictor of CVD. Growing evidence has suggested that
circulating levels of OPG may serve as potential markers of CVD and
AAA risk; however, the role of this protein in the arterial wall in
relation to AAA remains unclear.
The present study focused on OPG, a glycoprotein
that serves as a decoy receptor with suggested protective
properties in the cardiovascular system. First, the present study
used the angII-induced AAA model on ApoE−/− background
mice to investigate the role of human rh-OPG in AAA development.
However, treatment with low levels of human rh-OPG did not alter
the progression of AAA in these mice. Bennett et al
(21) reported a protective role
of OPG via its inactivation in ApoE−/− mice that
accelerated advanced atherosclerotic lesion progression and
vascular calcification. These findings suggested that endogenous
OPG acts as an inhibitor of atherogenesis, whereas exogenous OPG
did not contribute to an improvement of advanced AAA development in
the ApoE−/− mouse model in the present study. This
result was confirmed by Morony et al (28), who concluded that high levels of
exogenously injected OPG did not alter the amount of
atherosclerosis in ApoE−/− mice. Candido et al
(25) demonstrated that low-dose
treatment with rh-OPG (1 µg injection every 3 weeks) resulted in a
slight increase in total aortic plaque area in diabetic
ApoE−/− mice with a significant increase in VSMC
content. Despite very low levels of rh-OPG, these researchers were
able to detect rh-OPG in the serum of treated animals at 1 and 7
days post-administration. In the experiments of the present study,
a similar approach was used by administering low levels of OPG (1
µg twice a week) and, consistent with Candido et al
(25), the present study detected
levels of rh-OPG in the serum of mice, despite the levels being
very low (data not shown). Differences in the results of previous
studies may be reflected in the dosage and administration time of
OPG, fusion or native OPG, in addition to the age and strain of
animals used in the experiments. However, a study by Moran et
al (26) using ApoE/OPG double
knockouts with the angII-induced AAA model resulted in an
inhibition of aortic dilation and rupture. A total of 30% of
angII-induced animals used in the Moran et al (26) study developed rupture, which was
greater than the 9% of angII-induced animals that developed
ruptures in the present study. One may speculate upon whether this
result was atherosclerosis-dependent as 6 month-old mice were used,
however, similar results were obtained in C57Bl/6 background mice,
thus suggesting that older animals are more prone to developing
ruptures.
To investigate method-specific effects of exogenous
administered rh-OPG, the present study used CaCl2 to
induce AAA in C57Bl/6J mice. The differences in the pathogenesis of
AAA between the CaCl2 and angII models were marked.
AngII-induced AAA are located in the suprarenal areas and may
develop a thrombus, while CaCl2-induced AAA appear in
the infrarenal aorta with no thrombus formation, among other
differences. Composition of collagen and elastin vary in the
suprarenal and infrarenal parts of the aorta. In the present study,
the effects of CaCl2 on aneurysmal development were
smaller compared with the angII model, usually causing only
dilation of the aorta. However, this may be beneficial, as dilation
is developed prior to an aneurysm and is difficult to study in
models causing large aneurysms. In addition, with respect to the
studies performed by Moran and Bumdelger (26,27),
OPG knockout led to AAA inhibition in angII/ApoE and augmentation
of AAA in CaCl2/C57bl/6 model mice.
In the present study, a significant increase in the
vessel wall thickness through treatment with OPG was observed when
using the CaCl2 model. This may be contradictory to the
finding of Bucay et al (22) that OPG−/− mice develop
profound calcification of large arteries, including intimal and
medial proliferation. The results may be explainable by a
compensatory increase in the production of collagen in OPG-treated
mice, although treatment with OPG exerted no influence on
pro-collagen1 or Lox expression, which may contribute to collagen
fiber formation (29). Enhanced
plaque stability through collagen accumulation was promoted by
chronic treatment with OPG (24),
and this supports the hypothesis of the present study that a more
stable aneurysmal phenotype leads to increased collagen content.
Furthermore, one may speculate as to whether increased vessel wall
thickness depends on increased VSMC content due to increased
proliferation and/or decreased apoptosis, or a in phenotypic
switch. There were no observed effects on inflammation, the ECM or
VSMC remodeling in aortas from the mice treated with OPG in the
present study. A reason for this may potentially be explained by
the low dosage of OPG used in the present study. However, these
observations were partially in agreement with the results from
Ovchinnikova et al (24),
who reported a promotion of SMC accumulation, although no influence
on inflammation with chronic treatment with OPG when using a high
dosage of OPG. The lack of effect of OPG on inflammation in the
present study is in accordance with a report that subcutaneous
injections of human rh-OPG for 5 months led to decreased
atherosclerotic calcified lesions in LDLR−/− mice,
although they did not affect the total burden of atherosclerotic
lesions (28).
OPG is produced and secreted by VSMCs (18), in which it inhibits vascular
calcification (30). With regard
to the finding that high circulating OPG levels are associated with
the extent of CVD, the present study examined the effects of
pro-inflammatory cytokines on OPG secretion from human VSMCs to
investigate the possible regulatory effects of OPG. It is known
that rh-OPG promotes the proliferation of SMCs in both human and
rodent cells (9,25) and that OPG may be modulated by
pro-inflammatory cytokines (19,20).
The results of the present study demonstrated that stimulation of
cultured human aortic SMCs with TNF-α and IL-1β led to a 3-fold or
doubled increase in the release of OPG, whereas IFN-γ addition
significantly decreased OPG expression in human aortic SMCs by
one-half. Earlier in vitro studies demonstrated that the
expression and release of OPG was markedly upregulated in VSMCs in
response to inflammatory cytokines, including TNF-α and IL1-β
(31,32).
In conclusion, the results of the present study
indicated that C57Bl/6 mice treated with human rh-OPG have an
increased vessel wall thickness and collagen content. This
suggested a potential protective effect against dangerous rupture
in advanced AAA development. However, the present study did not
observe any ruptures using the CaCl2 model and,
therefore, this remains speculation. Although rupture occurred in
the angII model, many more animals than those included in the
present study would be required in each group to obtain any
differences with enough power. Further studies investigating
rupture models of AAA and using higher levels of OPG are required
to verify whether OPG exhibits any protective effects against
aneurysm rupture.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors' contributions
EV and DW contributed to concept and design of the
study. EV and DW performed experiments. EV, AK and DW analyzed the
data and contributed to the writing and revision of the manuscript,
as well as approving the final manuscript.
Ethics approval and consent to
participate
All mouse experiments were approved by Stockholm
North Ethical Committee on Animal Research (Stockholm, Sweden).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Assar AN and Zarins CK: Ruptured abdominal
aortic aneurysm: A surgical emergency with many clinical
presentations. Postgrad Med J. 85:268–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Toghill BJ, Saratzis A and Bown MJ:
Abdominal aortic aneurysm-an independent disease to
atherosclerosis? Cardiovasc Pathol. 27:71–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shimizu K, Libby P and Mitchell RN: Local
cytokine environments drive aneurysm formation in allografted
aortas. Trends Cardiovasc Med. 15:142–148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moran CS, McCann M, Karan M, Norman P,
Ketheesan N and Golledge J: Association of osteoprotegerin with
human abdominal aortic aneurysm progression. Circulation.
111:3119–3125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moran CS, Clancy P, Biros E, Blanco-Martin
B, McCaskie P, Palmer LJ, Coomans D, Norman PE and Golledge J:
Association of PPARgamma allelic variation, osteoprotegerin and
abdominal aortic aneurysm. Clin Endocrinol (Oxf).
72:128–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Emery JG, McDonnell P, Burke MB, Deen KC,
Lyn S, Silverman C, Dul E, Appelbaum ER, Eichman C, DiPrinzio R, et
al: Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J
Biol Chem. 273:14363–14367. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Simonet WS, Lacey DL, Dunstan CR, Kelley
M, Chang MS, Lüthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et
al: Osteoprotegerin: A novel secreted protein involved in the
regulation of bone density. Cell. 89:309–319. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pan G: An antagonist decoy receptor and a
death domain-containing receptor for TRAIL. Science. 277:815–818.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Ciriza Pérez C, Lawrie A and Varo N:
Osteoprotegerin in cardiometabolic disorders. Int J Endocrinol.
2015:5649342015.PubMed/NCBI
|
|
10
|
Lacey DL, Timms E, Tan HL, Kelley MJ,
Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S,
et al: Osteoprotegerin ligand is a cytokine that regulates
osteoclast differentiation and activation. Cell. 93:165–176. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hofbauer LC and Schoppet M: Clinical
implications of the osteoprotegerin/RANKL/RANK system for bone and
vascular diseases. JAMA. 292:490–495. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kong YY, Yoshida H, Sarosi I, Tan HL,
Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G,
Itie A, et al: OPGL is a key regulator of osteoclastogenesis,
lymphocyte development and lymph-node organogenesis. Nature.
397:315–323. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hofbauer LC, Shui C, Riggs BL, Dunstan CR,
Spelsberg TC, O'Brien T and Khosla S: Effects of immunosuppressants
on receptor activator of NF-kappaB ligand and osteoprotegerin
production by human osteoblastic and coronary artery smooth muscle
cells. Biochem Biophys Res Commun. 280:334–339. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lieb W, Gona P, Larson MG, Massaro JM,
Lipinska I, Keaney JF Jr, Rong J, Corey D, Hoffmann U, Fox CS, et
al: Biomarkers of the osteoprotegerin pathway: Clinical correlates,
subclinical disease, incident cardiovascular disease, and
mortality. Arterioscler Thromb Vasc Biol. 30:1849–1854. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Abedin M, Omland T, Ueland T, Khera A,
Aukrust P, Murphy SA, Jain T, Gruntmanis U, McGuire DK and de Lemos
JA: Relation of osteoprotegerin to coronary calcium and aortic
plaque (from the Dallas Heart Study). Am J Cardiol. 99:513–518.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kiechl S, Schett G, Wenning G, Redlich K,
Oberhollenzer M, Mayr A, Santer P, Smolen J, Poewe W and Willeit J:
Osteoprotegerin is a risk factor for progressive atherosclerosis
and cardiovascular disease. Circulation. 109:2175–2180. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koole D, Hurks R, Schoneveld A, Vink A,
Golledge J, Moran CS, de Kleijn DP, van Herwaarden JA, de Vries JP,
Laman JD, et al: Osteoprotegerin is associated with aneurysm
diameter and proteolysis in abdominal aortic aneurysm disease.
Arterioscler Thromb Vasc Biol. 32:1497–1504. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schoppet M, Preissner KT and Hofbauer LC:
RANK ligand and osteoprotegerin: Paracrine regulators of bone
metabolism and vascular function. Arterioscler Thromb Vasc Biol.
22:549–553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Collin-Osdoby P: Regulation of vascular
calcification by osteoclast regulatory factors RANKL and
osteoprotegerin. Circ Res. 95:1046–1057. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Venuraju SM, Yerramasu A, Corder R and
Lahiri A: Osteoprotegerin as a predictor of coronary artery disease
and cardiovascular mortality and morbidity. J Am Coll Cardiol.
55:2049–2061. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bennett BJ, Scatena M, Kirk EA, Rattazzi
M, Varon RM, Averill M, Schwartz SM, Giachelli CM and Rosenfeld ME:
Osteoprotegerin inactivation accelerates advanced atherosclerotic
lesion progression and calcification in older ApoE-/- mice.
Arterioscler Thromb Vasc Biol. 26:2117–2124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bucay N, Sarosi I, Dunstan CR, Morony S,
Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, et al:
Osteoprotegerin-deficient mice develop early onset osteoporosis and
arterial calcification. Genes Dev. 12:1260–1268. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Min H, Morony S, Sarosi I, Dunstan CR,
Capparelli C, Scully S, Van G, Kaufman S, Kostenuik PJ, Lacey DL,
et al: Osteoprotegerin reverses osteoporosis by inhibiting
endosteal osteoclasts and prevents vascular calcification by
blocking a process resembling osteoclastogenesis. J Exp Med.
192:463–474. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ovchinnikova O, Gylfe A, Bailey L,
Nordström A, Rudling M, Jung C, Bergström S, Waldenström A, Hansson
GK and Nordström P: Osteoprotegerin promotes fibrous cap formation
in atherosclerotic lesions of ApoE-deficient mice-brief report.
Arterioscler Thromb Vasc Biol. 29:1478–1480. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Candido R, Toffoli B, Corallini F,
Bernardi S, Zella D, Voltan R, Grill V, Celeghini C and Fabris B:
Human full-length osteoprotegerin induces the proliferation of
rodent vascular smooth muscle cells both in vitro and in vivo. J
Vasc Res. 47:252–261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moran CS, Jose RJ, Biros E and Golledge J:
Osteoprotegerin deficiency limits angiotensin II-induced aortic
dilatation and rupture in the apolipoprotein E-knockout mouse.
Arterioscler Thromb Vasc Biol. 34:2609–2616. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bumdelger B, Kokubo H, Kamata R, Fujii M,
Yoshimura K, Aoki H, Orita Y, Ishida T, Ohtaki M, Nagao M, et al:
Osteoprotegerin prevents development of abdominal aortic aneurysms.
PLoS One. 11:e01470882016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morony S, Tintut Y, Zhang Z, Cattley RC,
Van G, Dwyer D, Stolina M, Kostenuik PJ and Demer LL:
Osteoprotegerin inhibits vascular calcification without affecting
atherosclerosis in ldlr(−/-) mice. Circulation. 117:411–420. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mäki JM, Räsänen J, Tikkanen H, Sormunen
R, Mäkikallio K, Kivirikko KI and Soininen R: Inactivation of the
lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular
dysfunction, and perinatal death in mice. Circulation.
106:2503–2509. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Callegari A, Coons ML, Ricks JL, Rosenfeld
ME and Scatena M: Increased calcification in
osteoprotegerin-deficient smooth muscle cells: Dependence on
receptor activator of NF-κB ligand and interleukin 6. J Vasc Res.
51:118–131. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Olesen P, Ledet T and Rasmussen LM:
Arterial osteoprotegerin: Increased amounts in diabetes and
modifiable synthesis from vascular smooth muscle cells by insulin
and TNF-alpha. Diabetologia. 48:561–568. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang J, Fu M, Myles D, Zhu X, Du J, Cao X
and Chen YE: PDGF induces osteoprotegerin expression in vascular
smooth muscle cells by multiple signal pathways. FEBS Lett.
521:180–184. 2002. View Article : Google Scholar : PubMed/NCBI
|