Introduction
Non-alcoholic fatty liver disease (NAFLD) is
becoming the most common hepatic disorder worldwide, and will be
the most common reason for liver transplantation by 2020 (1). NAFLD begins with simple hepatic
steatosis that may progress to non-alcoholic steatohepatitis
(NASH), potentially leading to liver cirrhosis and hepatic cell
carcinoma (1). Excessive fat
accumulation is the initiating factor in the development of NAFLD,
which is associated with diabetes, obesity and hyperlipidemia.
Immune and inflammatory responses are secondary processes in the
development of NAFLD (2). However,
the specific pathogenesis of NAFLD remains unclear.
Fatty acid translocase (known as FAT or CD36), which
may be induced in obesity, is a fatty acid receptor that plays an
essential role in modulating lipid and glucose use; the
upregulation of CD36 is associated with NASH (3). The mRNA expression of nuclear factor
(NF)-κB, a key regulator involved in the induction of inflammatory
mediators, can be significantly influenced by the manipulation of
CD36 expression (4). Nevertheless,
the results of studies regarding the function of CD36 in NAFLD have
been conflicting, and the mechanisms are not well understood.
Sirtuin 1 (SIRT1) is an NAD(+)-dependent
deacetylase, and a critical regulator in various metabolic
processes. SIRT1 overexpression reduces the level of oxygen
consumption in NAFLD. It has been reported that activation of SIRT1
by SRT1720 can alleviate high-fat diet (HFD)-induced liver
steatosis (5). SIRT1 can interact
with RelA/P65, subunits of NF-κB, directly, and inhibit NF-κB by
deacetylating RelA/P65 at lysine 310, leading to the IκBa-dependent
nuclear export of NF-κB (6).
Hepatic macrophages, known as Kupffer cells (KCs),
account for 80–90% of the inherent macrophages. KCs release various
inflammatory cytokines and play a critical role in the pathogenesis
of liver inflammation disease (7).
It has been reported that SIRT1 can control hepatic CD36 expression
and triglyceride accumulation. However, other studies have
suggested that SIRT1 increased the expression of proteins in the
CD36 metabolic pathway. Therefore, whether SIRT1 could control the
expression of CD36 in the KCs of free fatty acid (FFA)-reduced NASH
liver remains unknown. In the present study, we investigated the
effects of SIRT1 on the expression of CD36 and NF-κB, and the
protective effect of SIRT1 against NAFLD.
Materials and methods
Animals and diets
Male C57BL/6 mice (weighing 18–22 g) were provided
by the laboratory animal research center of Chongqing Medical
University (Chongqing, China). All the mice were housed in a
ventilated and temperature-controlled specific pathogen-free room.
Water and food were accessed ad libitum. Mice were randomized into
three groups, with 10 mice in each group: Normal diet (ND) group,
mice fed with an ND for 8 weeks; HFD group, mice fed with a HFD
(cat. no. D12492; Research Diets, Inc., New Brunswick, NJ, USA) for
8 weeks; and HFD+SRT1720 group, mice fed with a HFD for 8 weeks,
and SRT1720 (30 mg/kg/d) in the last 4 weeks. Ether inhalation was
used for anesthetizing mice prior to the experiment. Sections of
the liver tissues were formalin-fixed and embedded in paraffin for
analysis by hematoxylin and eosin (H&E) staining and
immunohistochemistry. Mouse serum was stored at −80°C for ELISA.
This study was conducted in accordance with the ethical guidelines
of the 1975 Declaration of Helsinki and was approved by the
Committee for Animal Subjects of Chongqing Medical University.
Animal experiments were performed in compliance with the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals.
Cell culture
Primary KCs were isolated from mouse livers as
previously described (8) for use
in qPCR and western blot analyses. Cells were cultured in DMEM
medium (high glucose) supplemented with 10% fetal bovine serum
(both Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) and 100
nM penicillin/streptomycin.
H&E staining and
immunohistochemistry
The paraffin embedded tissues were sectioned and
stained with H&E, and immunohistochemically stained with
specific antibodies against SIRT1, CD36 and P65, as follows: The
fixed liver samples were cut into 5-µm sections, which were treated
with 3% H2O2 and blocked with 3% normal goat
serum. Then, the sections were incubated with SIRT1, CD36 and P65
(all Santa Cruz Biotechnology, Inc., Dallas, TX, USA) rabbit
primary antibodies overnight at 4°C. A biotinylated secondary
antibody was detected by liquid aminoethylcarbazole or
diaminobenzidine, and hematoxylin was used for counterstaining. The
presence of brown particles in cells when observed under an optical
microscope was considered positive staining.
Western blotting
Total protein was extracted with RIPA buffer, and
the concentration of protein was determined using a Bradford Assay
Kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Equivalent
amounts of protein were separated by electrophoresis on 10%
polyacrylamide gels and transferred onto polyvinylidene fluoride
membranes. The membranes were blocked with 5% bovine serum albumin
for 1 h, and were incubated with primary antibodies against
β-actin, SIRT1, CD36 and P65 (all Santa Cruz Biotechnology, Inc.)
overnight at 4°C, followed by a corresponding horseradish
peroxidase (HRP)-conjugated secondary antibody for 2 h. Using an
enhanced chemiluminescence detection kit (Pierce; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), protein bands on the membranes
were exposed on autoradiographic film (Kodak, Rochester, NY, USA)
and quantified by an image analysis system (Bio-Rad Gel Doc 2,000;
Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative PCR
analysis
Total RNA was extracted from KCs using the RNAiso
Plus kit (Takara Bio, Inc., Otsu, Japan), according to the
manufacturer's instructions. The concentration of RNA was
quantitated by measuring A260/A280 with a
spectrophotometer. Equivalent amounts of RNA were reverse
transcribed into cDNA using a PrimeScript RT reagent kit (Takara
Bio, Inc.). cDNA was amplified by quantitative real-time PCR using
the Bio-Rad iCycler (Bio-Rad Laboratories, Inc.) with SYBR-Green
DNA fluorescent dye (Takara Bio, Inc.) as previously described
(9). β-actin served as endogenous
normalization control. Bio-Rad CFX Manager software (version 2.0;
Bio-Rad Laboratories, Inc.) was utilized for data analysis. Primers
were designed by Sangon Biotech Co., Ltd. (Shanghai, China), with
sequences as follows: SIRT1 forward, 5′-CGGCTACCGAGGTCCATATAC-3′
and reverse, 5′-ACAATCTGCCACAGCGTCAT-3′; TNF forward,
5′-CCTCACACTCACAAACCACCA-3′ and reverse,
5′-ACAAGGTACAACCCATCGGC-3′; IL-6 forward,
5′-CCAGTTGCCTTCTTGGGACT-3′ and reverse,
5′-GGTCTGTTGGGAGTGGTATCC-3′; CD36 forward,
5′-TTGTGGAGCTCAAAGACCTG-3′ and reverse, 5′-TGCAAGAAGCGGATGTAGTC-3′;
β-actin forward, 5′-CATTGTGATGGACTCCGGAG-3′ and reverse,
3′-ATATGATGACCTGGCCGTC-5′. The relative mRNA expression was
calculated by the Vandesompele method.
ELISA
The serum levels of TNF-α and IL-6 were measured by
specific ELISA kits (Abcam, Hong Kong, China) according to the
manufacturer's protocols. Briefly, samples and standards were
inserted into a pre-coated 96-well plate. The inflammatory
mediators were bound to the wells by immobilized antibodies. A
biotinylated antibody for TNF-α or IL-6 was added separately.
HRP-conjugated streptavidin and TMB substrate solution were added
successively after washing, and the addition of Stop Solution
changed the color to yellow. Finally, the absorbance was measured
at 450 nm, and the levels of TNF-α and IL-6 were calculated by the
optical density and standard curve.
Statistical analysis
Experimental data were presented as the mean ±
standard deviation of at least three independent experiments, and
the differences between groups were compared by Student's t test or
one way ANOVA with the Student-Newman-Keuls post-hoc test in SPSS
18.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
SIRT1 activation alleviates hepatic
steatosis and inflammation
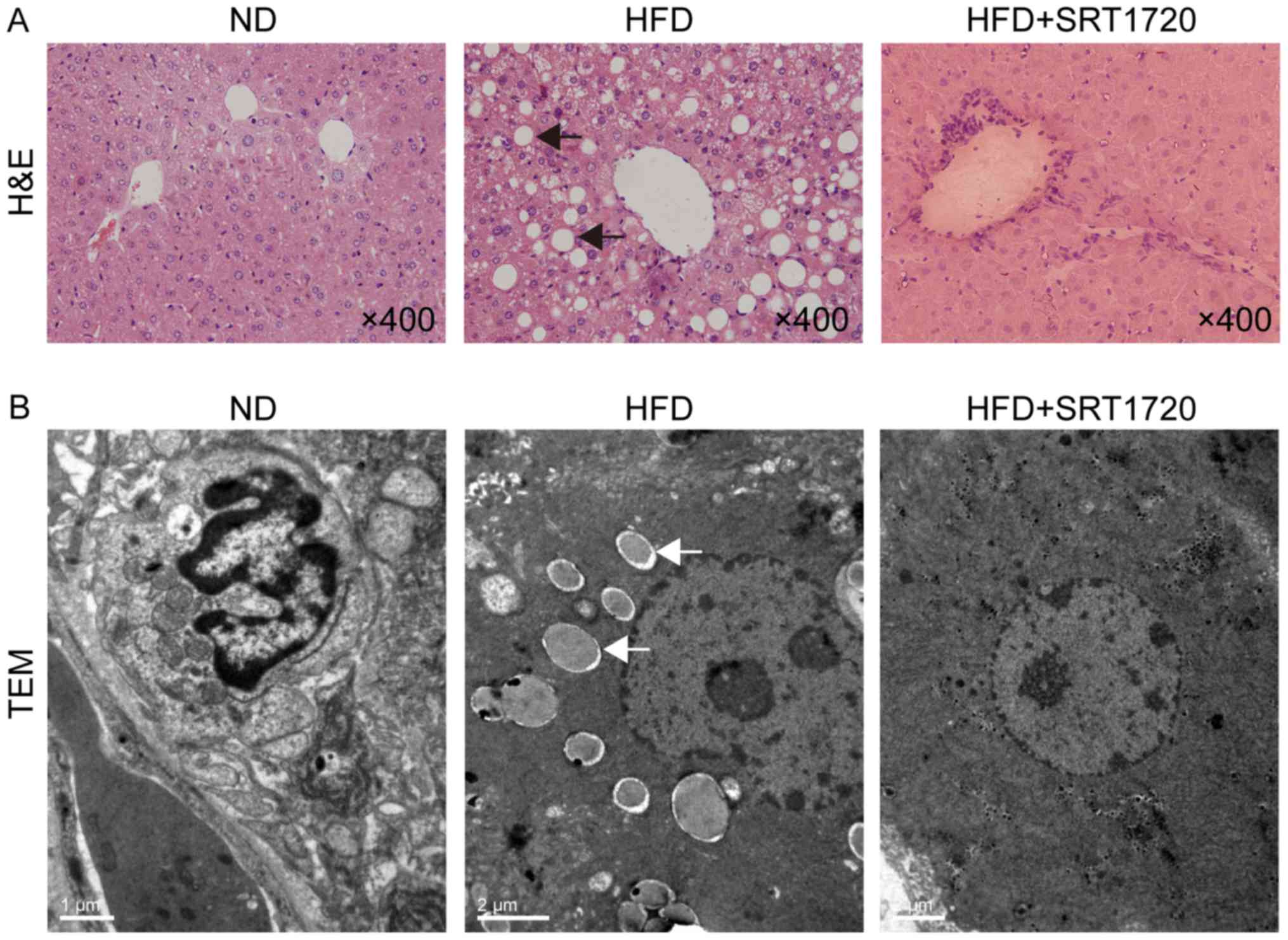
H&E-stained sections of liver were observed
under a light microscope to assess the effect of SRT1720 on the
hepatic histopathology. The normal-appearing hepatic lobule
structures and hepatocytes were evident in the ND group, while the
morphological changes were clearly observable in the HFD group
(Fig. 1A). Hepatic lobule
structures were swollen and severely damaged, with the ballooning
degeneration of hepatocytes and inflammatory cell infiltration.
SRT1720 administration alleviated the histological presentation of
edema, ballooning degeneration and inflammation in the liver
(Fig. 1A).
The ultrastructures of the liver samples were
examined by TEM, demonstrating that the number of lipid droplets
were significantly increased in the hepatocyte cytoplasm,
accompanied by a compressed hepatic sinusoid, in the HFD group
(Fig. 1B). The ultrastructures
were less abnormal in the HFD+SRT1720 group than in the HFD group
(Fig. 1B). These results indicated
that SRT1720 can attenuate hepatic steatosis and inflammation
induced by HFD, and the upregulation of SIRT1 may be a protective
event in NAFLD.
SRT1720 improves liver function and
decreases inflammatory factor levels in serum
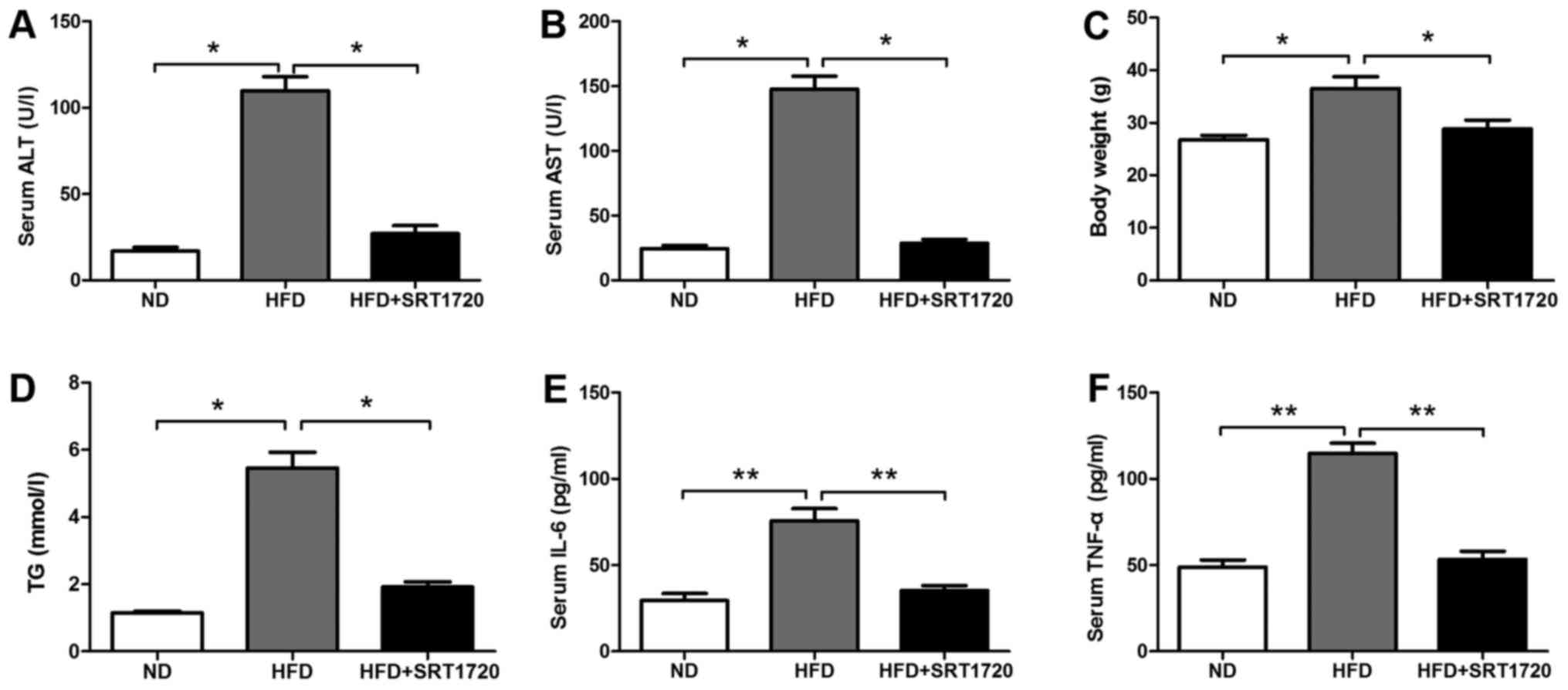
The levels of ALT and AST were significantly
increased in the HFD group compared with the ND group. However, the
levels of ALT and AST were significantly decreased in the
HFD+SRT1720 group, suggesting that SRT1720 played a protective role
in the progression of NAFLD (Fig. 2A
and B). Additionally, the mouse body weight and triglyceride
levels in the HFD+SRT1720 group were lower than in the HFD group
(Fig. 2C). The serum triglyceride
level in the HFD group was significantly higher than those in the
control and HFD+SRT1720 group (Fig.
2D). Afterwards, we examined the expression of the inflammatory
factors, IL-6 and TNF-α, in the mouse serum from each group. IL-6
and TNF-α remained at low levels in the ND group, and were
significantly increased in the HFD group compared with the ND group
(P<0.05). Meanwhile, the levels of IL-6 and TNF-α were
significantly decreased in the HFD+SRT1720 group compared with the
HFD group (P<0.05) (Fig. 2E and
F). These data suggested that SRT1720 effectively inhibited the
production of inflammatory factors induced by a HFD, further
inhibiting the progression of NAFLD.
SIRT1 upregulation suppresses the
protein expression of CD36 and P65
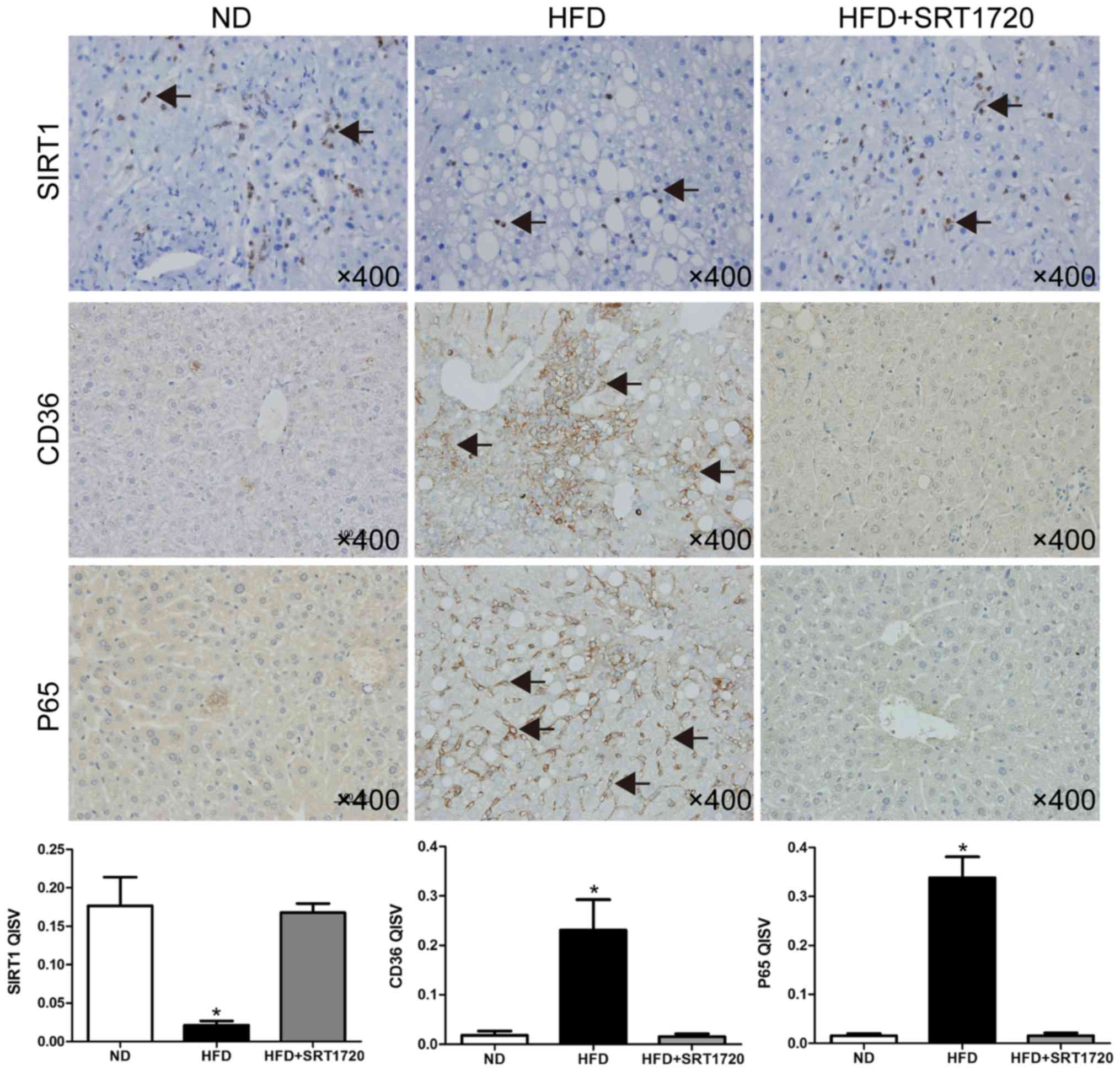
The expression levels of SIRT1, P65 and CD36 were
identified by immunohistochemistry. As shown in Fig. 3, a HFD reduced the SIRT1 protein
expression level, and induced the upregulation of CD36 and P65,
indicating that the protection of SIRT1 was lost, leading to the
expression of more lipid receptors, and inflammation. As expected,
the SIRT1 level was significantly upregulated following the
treatment with SRT1720, and P65 was simultaneously decreased.
Interestingly, the level of CD36 was also downregulated, compared
with the HFD group.
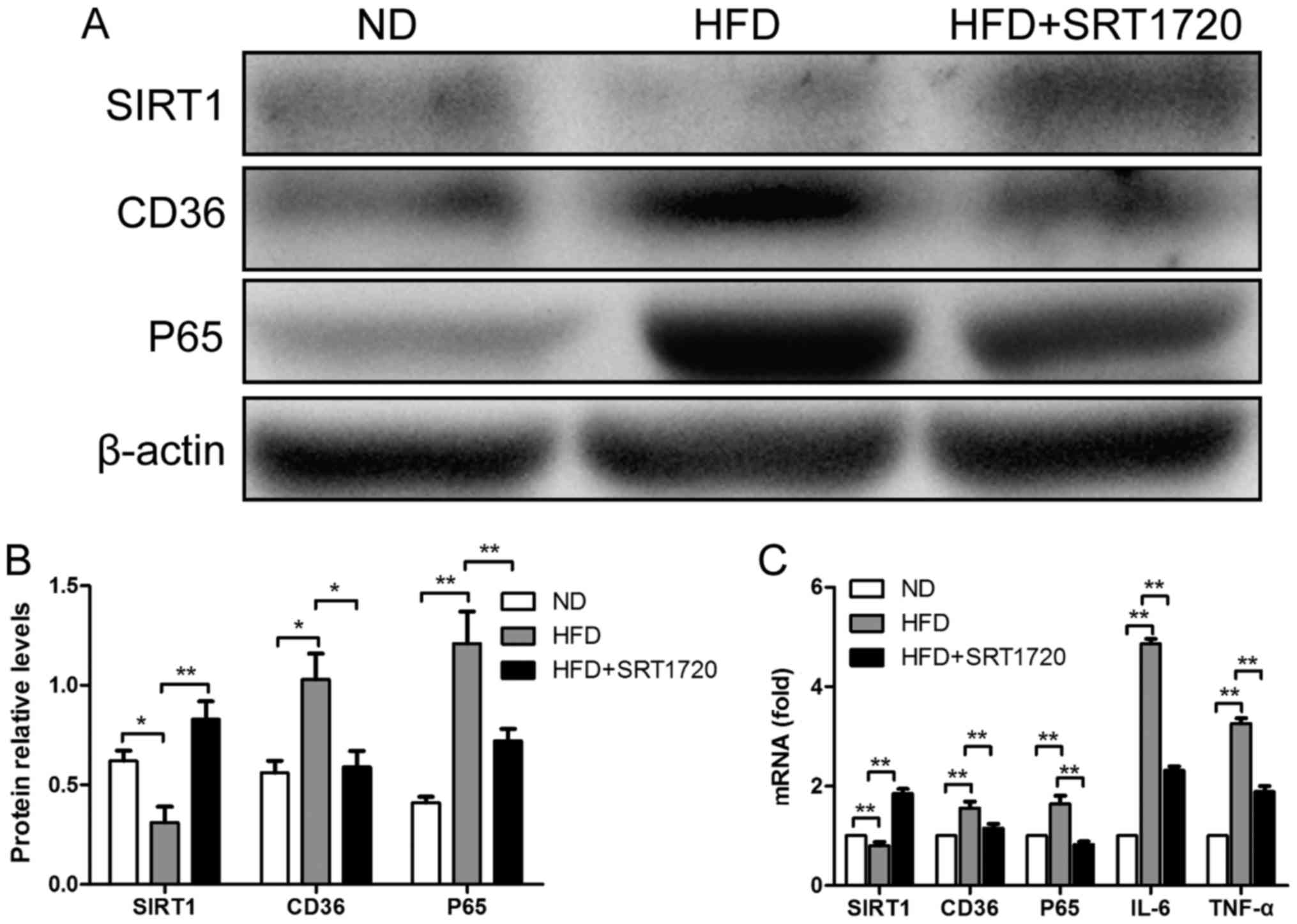
Furthermore, western blotting was performed to
quantify the protein expression levels of SIRT1, P65 and CD36; the
results were consistent with the immunohistochemistry results
(Fig. 4A and B). We found that the
KC levels of P65 and CD36 were significantly increased in the HFD
group, accompanied with a decrease in SIRT1, compared with the ND
group (P<0.01). Similarly, SRT1720 treatment remarkably
upregulated the SIRT1 level, leading to reductions in P65 and CD36
compared with the HFD group (P<0.05). Our results demonstrated
that the upregulation of SIRT1 by SRT1720 treatment can reduce the
P65 and CD36 protein expression levels induced by a HFD.
SIRT1 upregulation by SRT1720
suppresses mRNA expression levels of CD36 and NF-κB (P65)
To determine whether SRT1720 could attenuate hepatic
steatosis and inflammation by modulating SIRT1, P65 and CD36 at the
gene expression level, the mRNA expression of these genes in KCs
were detected by qPCR. As demonstrated in Fig. 4C, the mRNA levels of P65 and CD36
were elevated in the HFD group compared with the ND group, whereas
SIRT1 followed the opposite trend (P<0.01). However, the
elevation of P65 and CD36 levels was inhibited by SRT1720
treatment. P65 and CD36 were significantly decreased in the
HFD+SRT1720 group compared with the HFD group (P<0.05 and
P<0.01, respectively). These data revealed that the upregulation
of SIRT1 by SRT1720 inhibited the P65 and CD36 mRNA levels in
KCs.
Discussion
In the present study, we confirmed that the
activation of SIRT1 by SRT1720 treatment can protect against liver
injury induced by a HFD. Using a mouse NAFLD model fed with a HFD,
we observed that the liver morphology was damaged by swollen
hepatocytes and infiltrating inflammatory cells, and TNF-α and IL-6
in serum were significantly increased. SRT1720 treatment performed
favorably in relieving liver injury, and suppressing the levels of
TNF-α and IL-6 induced by a HFD through increasing the expression
of SIRT1, which resulted in the inhibition of NF-κB and CD36
expression in KCs.
SIRT1 has been shown to have a protective effect
against the pathophysiological mechanisms of NAFLD, and is a
candidate therapeutic target to prevent the development and
progression of NAFLD (10). It was
reported that treatment with SRT1720, an SIRT1 agonist, was
associated with a decreased prevalence of NAFLD (11). Mounting evidence has indicated that
SIRT1 is a protective factor, which maintained insulin sensitivity,
adjusted lipid homeostasis through antihyperlipidemic activity,
inhibited inflammation, positively influenced autophagy and
apoptosis, and promoted resistance to aging (12). Our study also demonstrated the
protective effects of SIRT1 against inflammation and steatosis,
which may be mediated by regulating NF-κB and CD36. However,
previous studies have reported that SIRT1 mediated cell survival by
deacetylating K382 on p53, and mediated cell death by deacetylating
K310 on P65, resulting in diminished NF-κB transcription and a
decrease in pro-survival gene products (13). Thus, the conclusions regarding the
function of SIRT1 are still controversial, and the mechanisms are
not fully understood.
Previous findings have demonstrated that the
excessive fat accumulation in the liver induced by a HFD results
from an increased rate of hepatic de novo lipogenesis and
impaired fatty acid oxidation due to the inhibition of
5′-AMP-activated protein kinase (AMPK) activation through SIRT1
(14). Researchers have speculated
that in addition to the deacetylation of P65 K310, SIRT1 may bind
histone H3 by the CD36 promoter, leading to the downregulation of
CD36 in hepatocytes, which controls triglyceride accumulation
(15). In our experiments, a HFD
induced the upregulation of CD36 while inhibiting SIRT1 expression
in an NAFLD mice model, which was reversed by SRT1720 treatment.
These data were consistent with previous studies. In addition,
another study reported that SIRT1 expression was decreased in aged
mice, whereas CD36 was upregulated, which could explain the
increasing prevalence and progression of NAFLD associated with age
in the general human population (16). The AMPK-mediated effects of SIRT1
have been reported to promote the deacetylation of peroxisome
proliferator-activated receptor gamma coactivator 1α, which then
stimulates peroxisome proliferator-activated receptor-α, leading to
the initiation of CD36 transcription (17).
Studies have shown that CD36 contributes to the
development of NAFLD by modulating the rate of FFA uptake (18). Serum CD36, which is significantly
correlated with hepatic CD36 expression, is an independent factor
for the diagnosis of advanced steatosis in NAFLD (19). A decrease in hepatic CD36 level in
HFD-fed animals has been shown to be protective against
inflammation and insulin resistance (20). However, other experiments revealed
that CD36 depletion released LKB1, resulting in the constitutive
activation of AMPK, further impairing the AMPK lipid-sensing
ability (17). In addition, a
recent study reported that CD36 over-expression unexpectedly
attenuated hepatic steatosis, increased very low-density
lipoprotein secretion, and improved glucose tolerance and insulin
sensitivity (21). Some
researchers regard CD36 as a protective metabolic sensor in the
liver during lipid overload or metabolic stress.
Due to the complexity of their functions, the
relationship between CD36 and SIRT1 is quite intricate. In a
previous study, H9c2 rat cardiomyoblasts were treated with palmitic
acid (PA); during the experiment, CD36 and SIRT1 protein expression
levels were altered by PA treatment in a dose- and time-dependent
manner. A SIRT1 activator was then administered, significantly
increasing the expression of the CD36 metabolic pathway proteins to
counter the PA-induced switching from the SIRT1-CD36-fatty acid
pathway to the PKC zeta-GLUT4-glucose pathway (22).
At present, there is still no effective treatment
for NAFLD. Our results indicate that SRT1720 may be a promising
candidate for the treatment of NAFLD. However, before it can be
used in humans, its mechanisms of action need to be further
studied. In conclusion, we identified that the anti-steatosis and
anti-inflammatory properties of SRT1720-induced SIRT1 during NAFLD
may be associated with the inhibition of the NF-κB signaling
pathway and regulation of CD36.
Acknowledgements
The authors acknowledge the financial support for
this project by the National Natural Science Foundation of
China.
Funding
This project is funded by the National Natural
Science Foundation of China (grant nos. 81401622, 81301656 and
81601715).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
BN, KH, XZ and PL designed and performed the
experiments. BN, GR, SY and ZO analyzed the data. KH, PL, XZ, SY
and JG prepared all the figures. BN, KH and SY wrote the paper.
Ethics approval and consent to
participate
The present study study was conducted in accordance
with the ethical guidelines of the 1975 Declaration of Helsinki and
was approved by the Committee for Animal Subjects of Chongqing
Medical University. Animal experiments were performed in compliance
with the National Institutes of Health Guide for the Care and Use
of Laboratory Animals.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Charlton MR, Burns JM, Pedersen RA, Watt
KD, Heimbach JK and Dierkhising RA: Frequency and outcomes of liver
transplantation for nonalcoholic steatohepatitis in the United
States. Gastroenterology. 141:1249–1253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Michelotti GA, Machado MV and Diehl AM:
NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol.
10:656–665. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miquilena-Colina ME, Lima-Cabello E,
Sánchez-Campos S, García-Mediavilla MV, Fernández-Bermejo M,
Lozano-Rodríguez T, Vargas-Castrillón J, Buqué X, Ochoa B,
Aspichueta P, et al: Hepatic fatty acid translocase CD36
upregulation is associated with insulin resistance,
hyperinsulinaemia and increased steatosis in non-alcoholic
steatohepatitis and chronic hepatitis C. Gut. 60:1394–1402. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cao D, Luo J, Chen D, Xu H, Shi H, Jing X
and Zang W: CD36 regulates lipopolysaccharide-induced signaling
pathways and mediates the internalization of Escherichia coli in
cooperation with TLR4 in goat mammary gland epithelial cells. Sci
Rep. 6:231322016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Escande C, Chini CC, Nin V, Dykhouse KM,
Novak CM, Levine J, van Deursen J, Gores GJ, Chen J, Lou Z and
Chini EN: Deleted in breast cancer-1 regulates SIRT1 activity and
contributes to high-fat diet-induced liver steatosis in mice. J
Clin Invest. 120:545–558. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cao L, Liu C, Wang F and Wang H: SIRT1
negatively regulates amyloid-beta-induced inflammation via the
NF-κB pathway. Braz J Med Biol Res. 46:659–669. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wenfeng Z, Yakun W, Di M, Jianping G,
Chuanxin W and Chun H: Kupffer cells: Increasingly significant role
in nonalcoholic fatty liver disease. Ann Hepatol. 13:489–495.
2014.PubMed/NCBI
|
|
8
|
Li PZ, Li JZ, Li M, Gong JP and He K: An
efficient method to isolate and culture mouse Kupffer cells.
Immunol Lett. 158:52–56. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bustin SA, Benes V, Garson JA, Hellemans
J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL,
et al: The MIQE guidelines: Minimum information for publication of
quantitative real-time PCR experiments. Clin Chem. 55:611–622.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Colak Y, Yesil A, Mutlu HH, Caklili OT,
Ulasoglu C, Senates E, Takir M, Kostek O, Yilmaz Y, Enc Yilmaz F,
et al: A potential treatment of non-alcoholic fatty liver disease
with SIRT1 activators. J Gastrointestin Liver Dis. 23:311–319.
2014.PubMed/NCBI
|
|
11
|
Yamazaki Y, Usui I, Kanatani Y, Matsuya Y,
Tsuneyama K, Fujisaka S, Bukhari A, Suzuki H, Senda S, Imanishi S,
et al: Treatment with SRT1720, a SIRT1 activator, ameliorates fatty
liver with reduced expression of lipogenic enzymes in MSG mice. Am
J Physiol Endocrinol Metab. 297:E1179–E1186. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yeung F, Hoberg JE, Ramsey CS, Keller MD,
Jones DR, Frye RA and Mayo MW: Modulation of NF-kappaB-dependent
transcription and cell survival by the SIRT1 deacetylase. EMBO J.
23:2369–2380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim E, Choi Y, Jang J and Park T:
Carvacrol protects against hepatic steatosis in mice fed a high-fat
diet by enhancing SIRT1-AMPK signaling. Evid Based Complement
Alternat Med. 2013:2901042013.PubMed/NCBI
|
|
14
|
Cao Y, Xue Y, Xue L, Jiang X, Wang X,
Zhang Z, Yang J, Lu J, Zhang C, Wang W and Ning G: Hepatic menin
recruits SIRT1 to control liver steatosis through histone
deacetylation. J Hepatol. 59:1299–1306. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bertolotti M, Lonardo A, Mussi C, Baldelli
E, Pellegrini E, Ballestri S, Romagnoli D and Loria P: Nonalcoholic
fatty liver disease and aging: Epidemiology to management. World J
Gastroenterol. 20:14185–14204. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chau MD, Gao J, Yang Q, Wu Z and Gromada
J: Fibroblast growth factor 21 regulates energy metabolism by
activating the AMPK-SIRT1-PGC-1alpha pathway. Proc Natl Acad Sci
USA. 107:12553–12558. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jeppesen J, Albers PH, Rose AJ, Birk JB,
Schjerling P, Dzamko N, Steinberg GR and Kiens B:
Contraction-induced skeletal muscle FAT/CD36 trafficking and FA
uptake is AMPK independent. J Lipid Res. 52:699–711. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Samovski D, Sun J, Pietka T, Gross RW,
Eckel RH, Su X, Stahl PD and Abumrad NA: Regulation of AMPK
activation by CD36 links fatty acid uptake to β-oxidation.
Diabetes. 64:353–359. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Silverstein RL and Febbraio M: CD36, a
scavenger receptor involved in immunity, metabolism, angiogenesis,
and behavior. Sci Signal. 2:re32009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
García-Monzón C, Lo Iacono O, Crespo J,
Romero-Gómez M, García-Samaniego J, Fernández-Bermejo M,
Domínguez-Díez A, de Cía Rodríguez J, Sáez A, Porrero JL, et al:
Increased soluble CD36 is linked to advanced steatosis in
nonalcoholic fatty liver disease. Eur J Clin Invest. 44:65–73.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wilson CG, Tran JL, Erion DM, Vera NB,
Febbraio M and Weiss EJ: Hepatocyte-specific disruption of CD36
attenuates fatty liver and improves insulin sensitivity in HFD-fed
mice. Endocrinology. 157:570–585. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen YP, Tsai CW, Shen CY, Day CH, Yeh YL,
Chen RJ, Ho TJ, Padma VV, Kuo WW and Huang CY: Palmitic acid
interferes with energy metabolism balance by adversely switching
the SIRT1-CD36-fatty acid pathway to the PKC zeta-GLUT4-glucose
pathway in cardiomyoblasts. J Nutr Biochem. 31:137–149. 2016.
View Article : Google Scholar : PubMed/NCBI
|