Introduction
Hepatocellular carcinoma (HCC) is one of the most
common and lethal malignancies worldwide, especially in Asia and
Africa (1). The main cause of
cancer-related deaths in HCC patients is tumor metastasis, which
relies on angiogenesis (2). It has
been proposed that the metastatic activity of tumors is related to
whether they have acquired an angiogenic phenotype (the ability to
recruit vasculature), which enables them to obtain adequate
nutrition and oxygen by angiogenesis (3,4). As
the pivotal component of tumor blood vessels, tumor endothelial
cells (TECs) perform a very crucial role during tumor angiogenesis.
The migratory and invasive capacities of TECs enable them to
degrade the surrounding ECM and migrate towards pro-angiogenic cues
to form neovessels. Thus, understanding the mechanisms that
regulate the migration and invasion of TECs is very important for
the antiangiogenic therapy of cancer.
C-X-C chemokine receptor type 7 (CXCR7) is a
G-protein-coupled chemokine receptor, and its ligands are CXCL12
and CXCL11. Reportedly, CXCR7 was upregulated in cancer cells and
mediates a broad range of cellular activities, including
proliferation, survival, and metastasis, by binding to CXCL12
(5,6). A recent study showed that CXCR7 was
overexpressed in TECs derived from renal cell carcinoma (7). Further study indicated that the
expression of CXCR7 in TECs derived from renal cell carcinoma is
important for the migration and tube formation of TECs and for
tumor angiogenesis in vivo (8). Moreover, upregulating CXCR7 in human
HCC was found to increase the angiogenic capacity of human
umbilical vein endothelial cells (HUVECs) in in vitro tube
formation assays (9). This
indicated that CXCR7 might be associated with the functional
regulation of TECs in HCC. However, the role of CXCR7 in regulating
the invasion and migration of TECs in HCC as well as the underlying
mechanisms have not been adequately described. Therefore, in the
current study, we investigated the effect of CXCR7 silencing on the
migration and invasion of TECs derived from HCC in vitro and
further explored the mechanisms involved in CXCR7-regulated
migration and invasion of TECs derived from HCC. Our data
demonstrate that CXCR7 is an important molecule in regulating the
migration and invasion of TECs derived from HCC by triggering the
signal transducer and activator of transcription 3 (STAT3)
pathway.
Materials and methods
Cell lines
TECs from HCC were purchased from Sixin Biotech
(Shanghai, China; cat. no. TCHU82), routinely maintained in DMEM
supplemented with 10% FBS and penicillin-streptomycin solution, and
cultured at 37°C in a humid atmosphere with 5% CO2.
CXCR7 knockdown and STAT3 over
expression
The TEC cells were seeded in a six-well plate at
1.0×106 cells/well. When cells were 70–90% confluent,
they were transfected with a short hairpin (sh)RNA plasmid
targeting CXCR7 (CXCR7-shRNA) or empty shRNA plasmid (NC-shRNA)
(both from Santa Cruz Biotechnology, Inc., Dallas, TX, USA) using
Lipofectamine 3000 reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) according to the manufacturer's protocol.
At 24 h after transfection, the cells were cultured in medium
containing 2 µg/ml puromycin for 7 days to select stably
transfected TECs. The expression of CXCR7 in the surviving clones
was measured using reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) and immunoblotting.
For STAT3 overexpression, the pcDNA3.0 plasmid
expressing activated STAT3 (pcDNA3.0-STAT3) or empty pcDNA3.0
plasmid (pcDNA3.0) (a kind gift from Dr. Fengsheng Li) was
transiently transfected into TECs with a stable downregulation of
CXCR7 using Lipofectamine 3000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. At 48 h
after transfection, the cells were harvested for subsequent
experiments.
RT-qPCR
Total RNA was extracted from TECs transfected with
or without pcDNA3.0-STAT3, pcDNA3.0, CXCR7-shRNA or NC-shRNA using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
First-strand cDNA was then generated using ALL-in-One™
First-Stand cDNA Synthesis kit (GeneCopoeia, Rockville, MD, USA)
according to the manufacturer's protocol. RT-qPCR was carried out
using Talent qPCR PreMix (SYBR-Green) kit (Tiangen Biotech,
Beijing, China) according to the manufacturer's protocol. The
primer sequences used in the current study are shown in Table I. The relative level of target mRNA
was determined by the 2−ΔΔCq method.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Forward sequence
(5′-3′) | Reverse sequence
(5′-3′) |
|---|
| CXCR7 |
CACTGCTACATCTTGAACCT |
GTTGATGGAGAAGATGAGGTGT |
| MMP2 |
ATGCCGTCGTGGACCTGC |
TGCTTCCAAACTTCACGCTCTT |
| VEGF |
GCACCCATGGCAGAAGGAGGAG |
GTGCTGACGCTAACTGACC |
| STAT3 |
ATCACGCCTTCTACAGACTGC |
CATCCTGGAGATTCTCTACCACT |
| β-actin |
ATTGCCGACAGGATGCAGAAG |
AGAAGCATTTGCGGTGGACG |
Immunoblotting
Total protein from TECs was collected by using a
lysis buffer (1% Triton X-100, 150 mmol/l NaCl, 50 mmol/l Tris, pH
8.0, 1 mmol/l EDTA, 10 mg/l henylmethylsulfonyl fluoride). The
protein concentration was determined by the BCA Protein Assay kit
(Tiangen Biotech) according to the manufacturer's protocol. Then,
50 µg protein was separated by 10% SDS-PAGE and transferred onto a
PVDF membrane. After blocking with a 5% BSA in PBS, the membrane
was incubated with anti-CXCR7 antibody (1:1,000; Abgent, San Diego,
CA, USA), anti-STAT3 (phospho Y705) antibody (1:1,000; Cell
Signaling Technology, Inc., Danvers, MA, USA), anti-matrix
metalloproteinase-2 (MMP2) antibody (1:1,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), anti-vascular endothelial
growth factor (VEGF) antibody (1:1,000; Abgent) or anti-Tubulin
antibody (1:5,000, YTHX Biotechnology Co. Ltd, Beijing, China)
overnight at 4°C. After washing with Tris-buffered saline (pH 7.2)
containing 0.05% Tween-20, the membrane was incubated with the
secondary antibody and subsequently visualized using the ECL
system.
Cell migration assay
The migration of TECs was measured by both transwell
migration assay and wound healing assay, as described in previous
studies (10,11). For the transwell migration assay,
briefly, 5×104 TECs in 200 µl of serum-free DMEM was
added to the top compartment of the transwell insert-setup (Corning
Incorporated, Corning, NY, USA). Then, 500 µl of DMEM medium
supplemented with 10% FBS was added to the bottom chamber.
Following incubation for 12 h at 37°C in a humid atmosphere with 5%
CO2, the cells on the lower surface of the membrane were
fixed with 100% methanol and stained with Giemsa. Migrated cells
were counted and expressed as an average number of
cells/microscopic field. For the wound healing assay, TECs with
normal or downregulated CXCR7 expression were seeded into 6-well
plates and grown to confluence. After washing with serum-free
medium, the confluent cells were wounded with a 200 µl pipette tip
followed by washing with serum-free medium to remove the
non-adherent cells. The wounded monolayers were subsequently
incubated with serum-free DMEM at 37°C in a humid atmosphere with
5% CO2 for 24 h. The cells were observed by
phase-contrast microscopy, and images were captured (magnification,
×100).
Invasion assay
The invasive capacity of TEC cells was measured
using a transwell invasion assay. Briefly, 0.1 ml Matrigel (BD
Biosciences, Franklin Lakes, NJ, USA) diluted by serum-free DMEM
was added into the upper chamber. After the Matrigel proteins
polymerized, 5×104 TECs that had been grown in
serum-free DMEM medium for 24 h and suspended in 200 µl of FBS-free
DMEM were added to the upper chamber. Then, 500 µl of DMEM with 10%
FBS was added to the lower chamber. The cell invasion chambers were
incubated for 20 h at 37°C in a humid atmosphere with 5%
CO2. The cells on the lower surface of the membrane were
fixed, stained and counted as described for the cell migration
assay.
Statistical analysis
The experimental data are presented as the mean ±
standard deviation. One-way ANOVA followed by Student-Newman-Keuls
test was performed to analyze the differences among groups using
the statistical software SPSS 19.0 (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
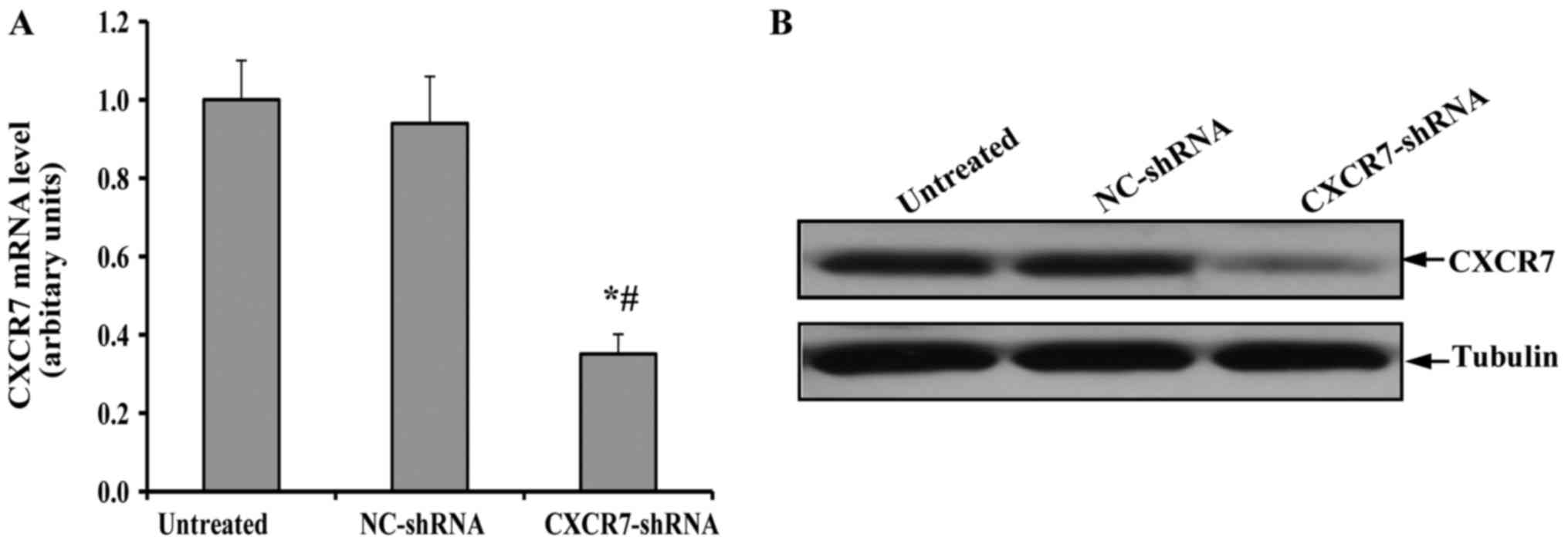
The generation of TECs with the stable
downregulation of CXCR7
After selection with puromycin, the expression of
CXCR7 in the surviving TEC clones was detected by RT-qPCR and
immunoblotting. As shown in Fig.
1A, the transfection with NC-shRNA did not change the
expression of CXCR7 mRNA in TECS compared to the untreated TECs. In
contrast, the level of CXCR7 mRNA in TECs transfected with
CXCR7-shRNA was significantly decreased. More importantly, a
similar expression pattern of CXCR7 protein was observed in the
immunoblotting assay (Fig. 1B).
These results indicated that appropriate control and CXCR7
knock-down TECs were generated.
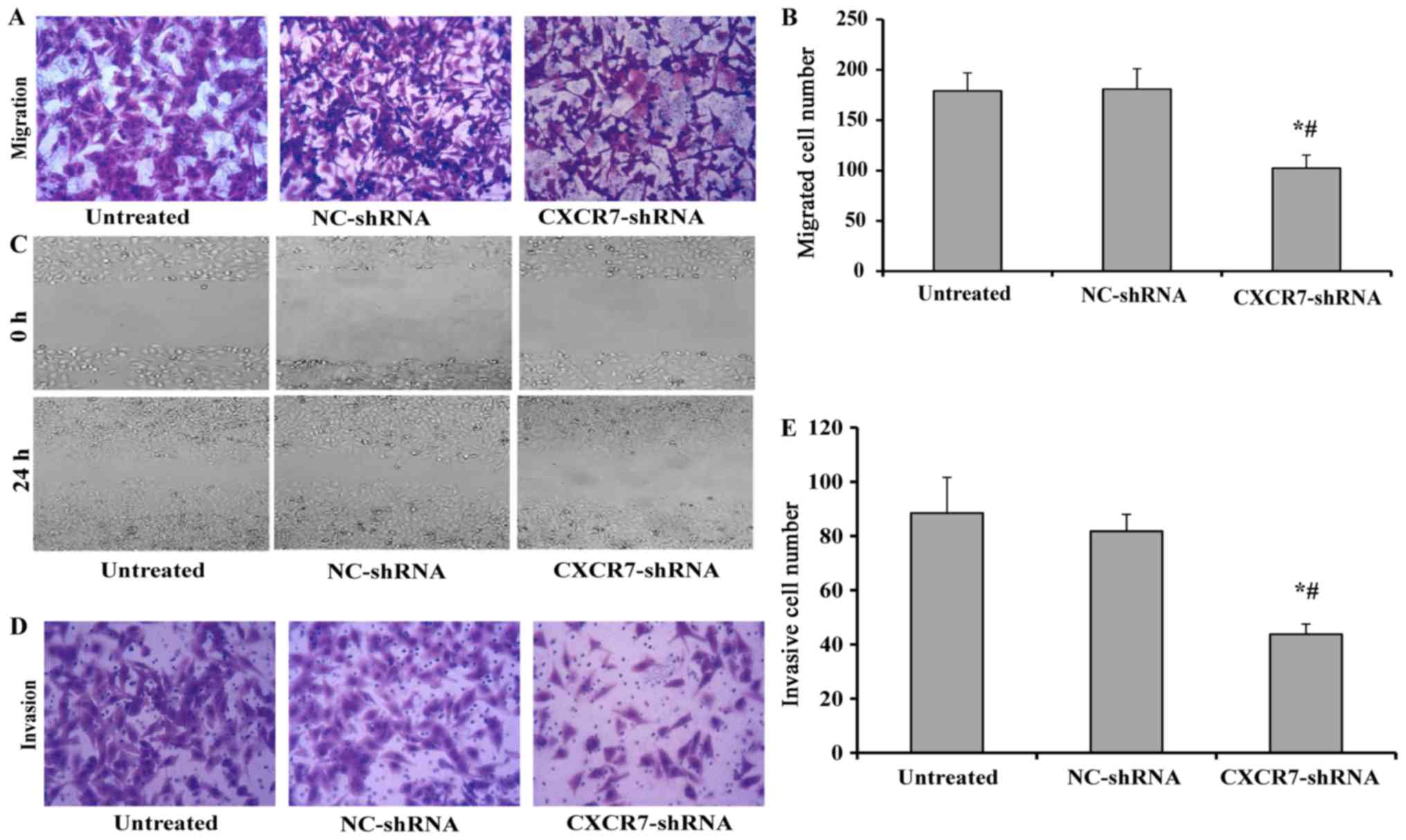
CXCR7 silencing inhibits the migration
and invasion of TECs
The migratory capacity of TECs with stable CXCR7
downregulation was investigated using a transwell migration assay.
As shown in Fig. 2A and B, TECs
with stable NC-shRNA expression showed a similar migratory ability
to that of the untreated TECs, but the migratory capacity of TECs
with stable down-regulation of CXCR7 was significantly impaired
compared to both untreated TECs and NC-shRNA-treated TECs, which
was further confirmed by the wound healing assay (Fig. 2C). The invasive ability of TECs
transfected with CXCR7 shRNA was further examined by a transwell
invasion assay. Similarly, the transfection of NC-shRNA did not
affect the invasive ability of TECs, but the TECs transfected with
CXCR7 shRNA showed significantly impaired invasion (Fig. 2D and E).
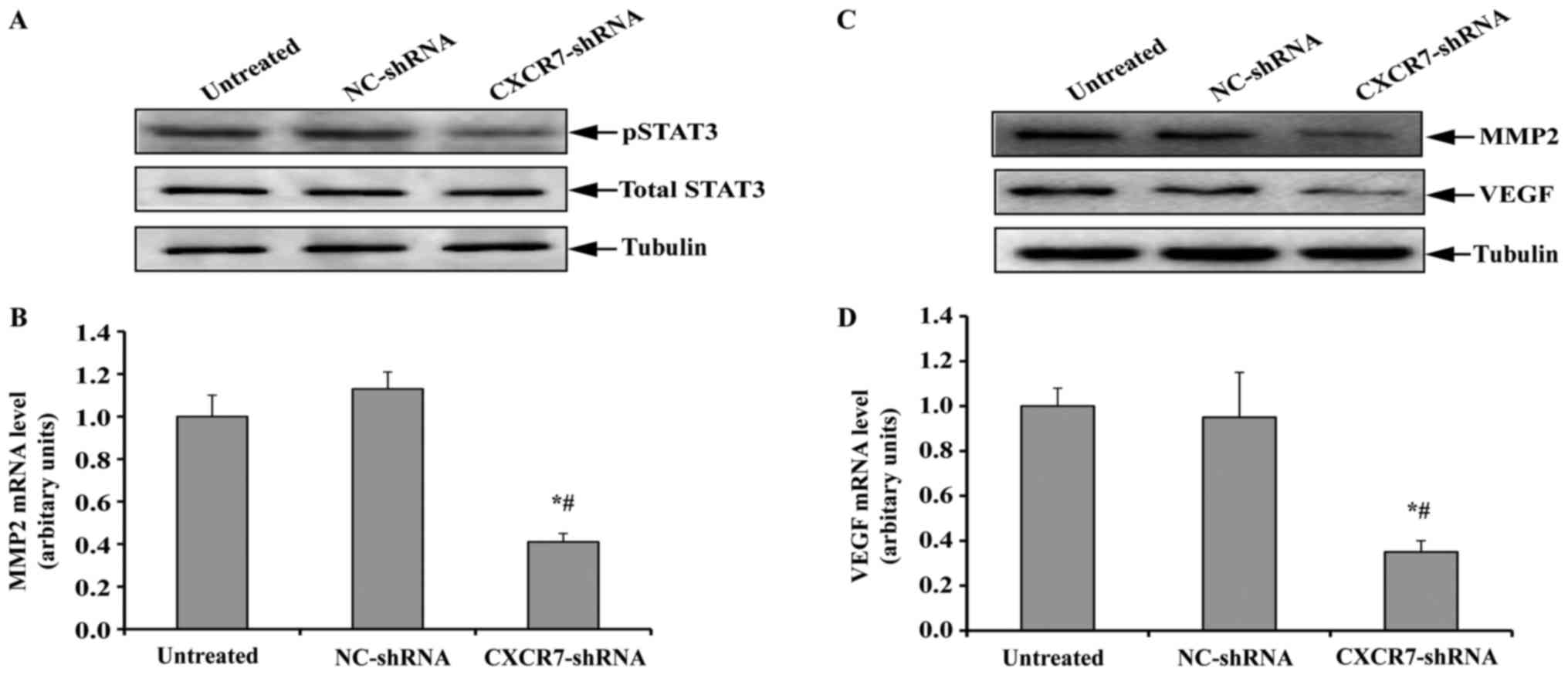
CXCR7 silencing suppresses the STAT3
pathway in TECs
Previous studies have suggested that CXCR7 can
trigger STAT3 expression, which has been found to contribute to
cell migration, invasion, and tumor metastasis by transcriptionally
regulating the expression of genes such as MMP2 and VEGF (12–14).
We therefore detected the expression of total and phosphorylated
STAT3 in the TECs with stable CXCR7 downregulation. As shown in
Fig. 3A, the inhibition of CXCR7
did not change the level of total STAT3 in TECs compared to
untreated or NC-shRNA-transfected TECs. However, phosphorylated
STAT3 was significantly decreased after CXCR7 silencing.
The expression of MMP2 and VEGF, which are 2
downstream genes of STAT3, in TECs was further investigated. As
anticipated, the expression of MMP2 was significantly inhibited in
TECs with stable CXCR7 downregulation at both the mRNA and protein
levels compared to untreated or NC-shRNA-transfected TECs (Fig. 3B and C). Similarly, the
downregulation of CXCR7 resulted in a significant decrease in the
expression of VEGF mRNA and protein (Fig. 3C and D). These results indicated
that the inhibition of CXCR7 suppressed the STAT3 pathway in
TECs.
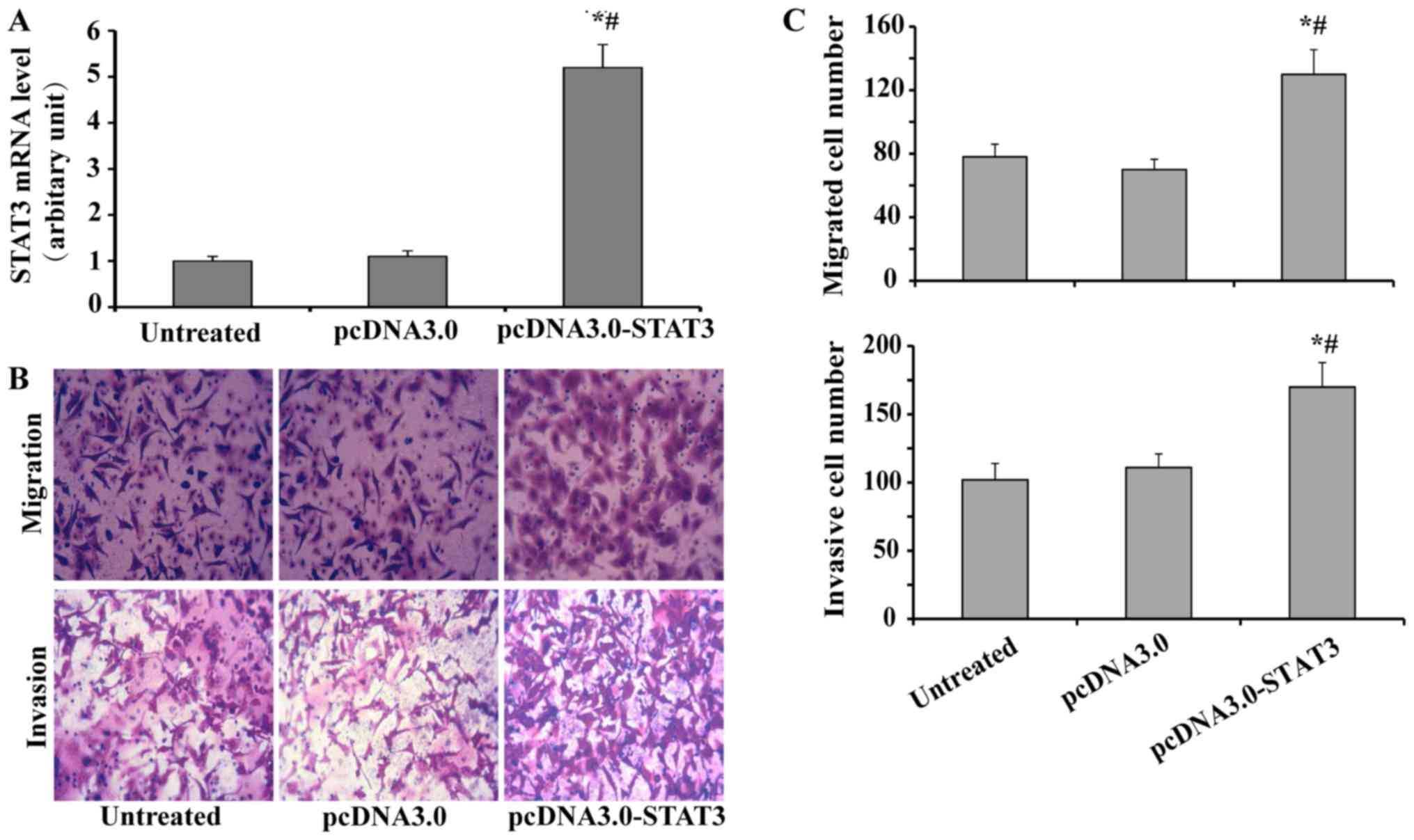
CXCR7 silencing suppresses the STAT3
pathway and decreases migration and invasion of TECs
We firstly examined the role of STAT3 in regulating
migration and invasion of TECs. The TECs were transfected with or
without pcDNA3.0-STAT3 of pcDNA3.0, and subsequently subjected to
RT-qPCR, migration assay or invasion assay. As shown in Fig. 4A, the expression of STAT3 in TECs
was significantly up regulated by transfection with pcDNA3.0-STAT3,
compared to both pcDNA3.0 and untreated control; while treatment
with pcDNA3.0 did not affect the expression of STAT3 in TECs,
compared to untreated control. Further study showed that
transfection with pcDNA3.0-STAT3 increased the migration and
invasion of TECs (Fig. 4B and C),
which indicated that STAT3 was involved in the regulation of TEC
migration and invasion.
To investigate whether the inhibition of the STAT3
pathway was necessary for the CXCR7 silencing-mediated decreases in
TEC migration and invasion, the TECs with stable CXCR7
downregulation were transfected with pcDNA3.0-STAT3 or empty
pcDNA3.0 plasmid. As shown in Fig.
5A, transfection with the empty pcDNA3.0 plasmid did not result
in a significant change in phosphorylated STAT3 compared to TECs
with stable CXCR7 downregulation. In contrast, treatment with
pcDNA3.0-STAT3 significantly abolished the CXCR7-shRNA-induced
downregulation of phosphorylated STAT3, accompanied by a
significant increase in total STAT3. The levels of phosphorylated
and total STAT3 in TECs treated with CXCR7-shRNA plus
pcDNA3.0-STAT3 were even higher than those observed in untreated
TECs. Further study showed that the decreased migratory and
invasive capacities of TECs with stable CXCR7 downregulation were
also abolished after the expression of phosphorylated STAT3 was
restored (Fig. 5B and C). Compared
to TECs transfected with NC-shRNA, TECs transfected with
CXCR7-shRNA plus pcDNA3.0-STAT3 seemed to possess increased
migratory and invasive capacities, but there was no significant
difference in migratory or invasive capacity between the
groups.
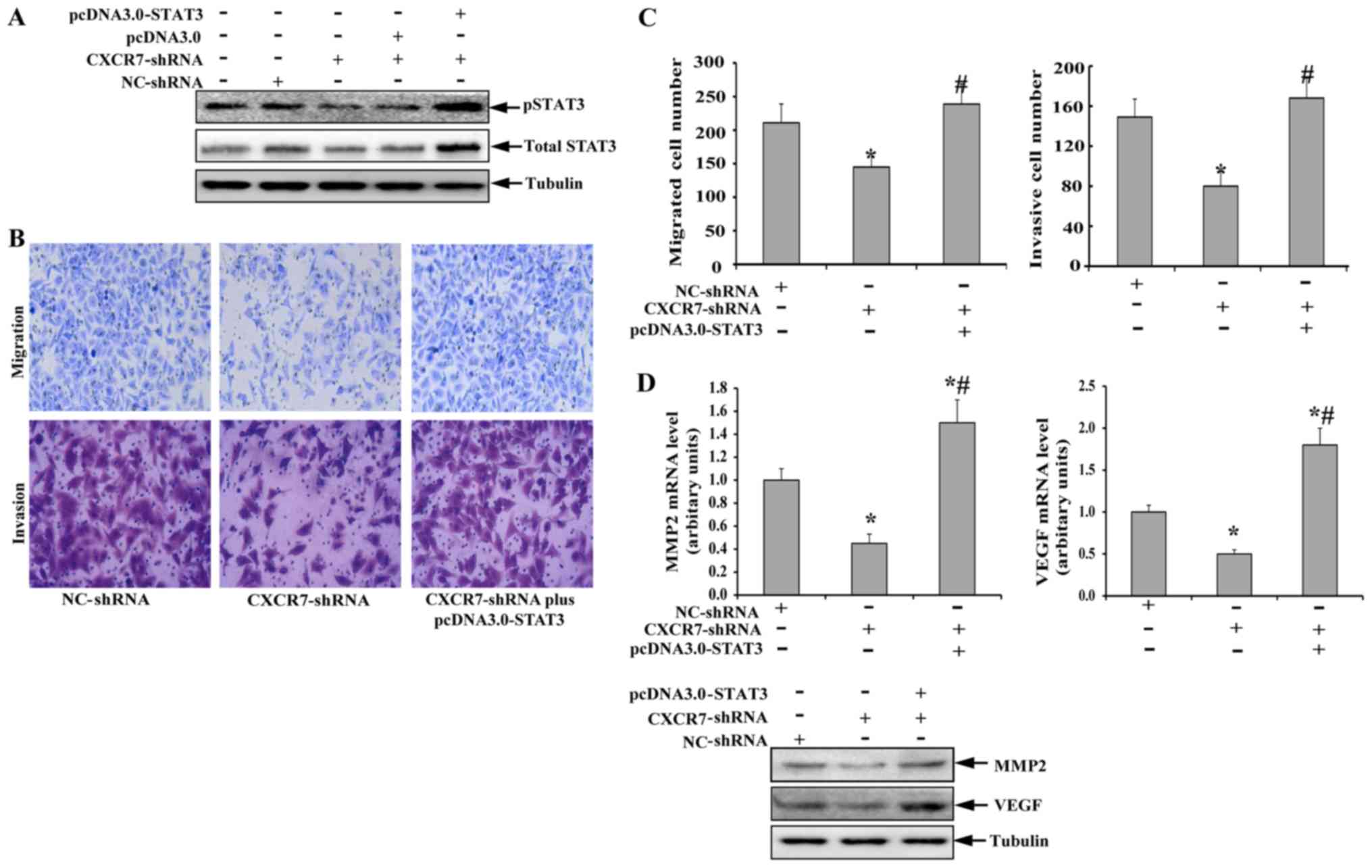 | Figure 5.(A-D) Restoration of STAT3 activity
abolishes the CXCR7 silencing-mediated decrease of TEC migration
and invasion. TECs with stable CXCR7 downregulation were
transfected with pcDNA3.0 or pcDNA3.0-STAT3. Following transfection
for 48 h, the cells were harvested and subjected to an
immunoblotting assay to detect the expression of phosphorylated and
total (A) STAT3, (D, lower panel) MMP2, and VEGF. (B) Transwell
migration and invasion assays were used to examine the migratory
and invasive capacity (magnification, ×200), respectively. RT-qPCR
was used to investigate the mRNA levels of MMP2 and VEGF (D, upper
panel). (C) Quantification of the migrated cells in the Transwell
migration assay and the invasive cells in Transwell invasion assay.
*P<0.05 vs. NC-shRNA; #P<0.05 vs. CXCR7-shRNA.
CXCR7, C-X-C chemokine receptor type 7; TEC, tumor endothelial
cell; shRNA, short hairpin RNA; MMP, matrix metalloproteinase;
VEGF, vascular endothelial growth factor. |
Finally, the expression of MMP2 and VEGF in TECs
with decreased CXCR7 expression was also investigated after the
restoration of phosphorylated STAT3 expression. As shown in
Fig. 5D, the downregulation of
MMP2 and VEGF in TECs induced by CXCR7 silencing was also abolished
at both the mRNA and protein levels by transfection with
pcDNA3.0-STAT3. Interestingly, the overexpression of constructively
active STAT3 in TECs with CXCR7 silencing resulted in a
significantly higher expression of MMP2 and VEGF than that of TECs
with normal CXCR7 expression. These data indicated that the CXCR7
silencing mediated the decreased TEC migration and invasion
abilities by suppressing the STAT3 pathway.
Discussion
One of key steps in the growth and metastasis of
tumor is angiogenesis, which enables tumor cells to obtain
sufficient nutrition and oxygen, and TECs are a critical component
of angiogenesis, which makes them a major target of anti-angiogenic
therapies (15). Understanding the
mechanisms underlying various biologic behaviors of TECs, such as
cell migration and invasion, is helpful for developing novel
anti-angiogenic agents. The current study reveals that CXCR7 is
involved in the migration and invasion of TECs derived from HCC.
Further study showed that silencing CXCR7 resulted in a decrease in
both phosphorylated STAT3 at Tyr705 and its downstream genes, MMP2
and VEGF, in TECs. Restoring STAT3 phosphorylation abolished the
CXCR7-shRNA-mediated decrease of TEC migration and invasion and
restored the expression of MMP2 and VEGF.
CXCR4, the ‘canonical’ receptor of CXCL12, has been
demonstrated to promote tumor growth and metastasis in various
tumor types by binding to CXCL12 (16,17).
CXCR7 has been recently identified as a second receptor for CXCL12
(18). It has been confirmed that
CXCR7 is elevated in both malignant cells and tumor-associated
blood vessels in human breast and lung cancer tissue (18). Although the overexpression of CXCR7
in tumor cells was shown to promote tumor growth, progression of
metastasis, and epithelial-mesenchymal transition (EMT) (18,19),
the role of CXCR7 in TECs was not fully understood. Yamada et
al first reported that CXCR7 was involved in migration,
angiogenesis, and resistance to serum starvation in TECs derived
from murine tumor A375SM xenografts (8). Consistently, our data showed that
inhibiting CXCR7 in TECs derived from HCC suppressed cell migration
and invasion in vitro. Because the migration and invasion of
TECs are essential for angiogenesis in both primary and metastatic
HCC lesions, this finding indicated that targeting TECs in HCC
might be a potential strategy for treating HCC.
Various molecules have been reported to be
downstream of CXCR7, including extracellular regulated protein
kinases 1/2 (ERK1/2) in epithelial ovarian carcinomas (20), mitogen-activated protein kinase
(MAPK) in HCC (21), AKT in
epithelial ovarian carcinomas and HCC (9,20),
and STAT3 in breast cancer (12).
STAT3 belongs to the STAT protein family, whose role has been
demonstrated to transmit various stimulatory signals from the cell
membrane to the nucleus, where it transcriptionally regulates the
expression of diverse target genes to response to the stimuli
(22). STAT3 has been proven to
promote the development and metastasis of various tumor types by
elevating the expression of its target genes, such as VEGF, MMP2
and survivin (23–25). Wani et al (12) reported that CXCR7 expression
enhanced the growth and metastasis of breast cancer by activating
the STAT3/MMP2 pathway. In the current study, we revealed that
inhibiting CXCR7 in TECs suppressed the activity of STAT3, followed
by the downregulation of MMP2 and VEGF. This result indicated that
STAT3 was one of downstream molecules of CXCR7 in TECs derived from
HHC. The restoration of activated STAT3 in TECs abolished the CXCR7
inhibition-mediated decrease of cell migration and invasion as well
as the downregulation of MMP2 and VEGF, indicating that CXCR7
regulates the migration and invasion of TECs via STAT3. Moreover,
the overexpression of constitutively activated STAT3 resulted in
significantly increased MMP2 and VEGF expression in TECs with CXCR7
silencing, which was accompanied by an enhanced migratory and
invasive capacity of these cells. This was in line with the finding
that TECs with CXCR7 silencing and STAT3 overexpression have a
higher level of phosphorylated STAT3.
In conclusion, the current study demonstrates that
CXCR7 plays an important role in regulating the migration and
invasion of TECs derived from HCC. STAT3 is responsible for
CXCR7-mediated migration and invasion in TECs derived from HCC.
Targeting CXCR7 may be an effective antiangiogenic strategy for the
treatment of HCC. Further in vivo studies are needed in
future research.
Acknowledgements
The authors would like to thank Dr. Chenyi Guo from
General Hospital of the PLA Rocket Force (Beijing, China) for
helpful suggestions during statistical analysis.
Funding
This study was supported by the Key Project of The
Affiliated Hospital of Inner Mongolia Medical University (grant no.
NYFY ZD 2014008).
Availability of data and materials
The data that support the findings of this study are
available from The Affiliated Hospital of Inner Mongolia Medical
University but restrictions apply to the availability of these
data, which were used under license for the current study, and so
are not publicly available. Data are available from the authors
upon reasonable request and with permission of The Affiliated
Hospital of Inner Mongolia Medical University.
Authors' contributions
YW, LT and SX performed research and analyzed the
data. YX, MZ and JZ contributed to the data collection and
analysis. ZW contributed to the initial design of the project and
the manuscript preparation. All the authors have approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McGlynn KA, Petrick JL and London WT:
Global epidemiology of hepatocellular carcinoma: An emphasis on
demographic and regional variability. Clin Liver Dis. 19:223–238.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zetter BR: Angiogenesis and tumor
metastasis. Annu Rev Med. 49:407–424. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Folkman J: Role of angiogenesis in tumor
growth and metastasis. Semin Oncol. 29 6 Suppl 16:S15–S18. 2002.
View Article : Google Scholar
|
|
4
|
Volpert OV, Dameron KM and Bouck N:
Sequential development of an angiogenic phenotype by human
fibroblasts progressing to tumorigenicity. Oncogene. 14:1495–1502.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liberman J, Sartelet H, Flahaut M,
Mühlethaler-Mottet A, Coulon A, Nyalendo C, Vassal G, Joseph JM and
Gross N: Involvement of the CXCR7/CXCR4/CXCL12 axis in the
malignant progression of human neuroblastoma. PLoS One.
7:e436652012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang J, Shiozawa Y, Wang J, Wang Y, Jung
Y, Pienta KJ, Mehra R, Loberg R and Taichman RS: The role of
CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate
cancer. J Biol Chem. 283:4283–4294. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maishi N, Ohga N, Hida Y, Akiyama K,
Kitayama K, Osawa T, Onodera Y, Shinohara N, Nonomura K, Shindoh M
and Hida K: CXCR7: a novel tumor endothelial marker in renal cell
carcinoma. Pathol Int. 62:309–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yamada K, Maishi N, Akiyama K, Alam Towfik
M, Ohga N, Kawamoto T, Shindoh M, Takahashi N, Kamiyama T, Hida Y,
et al: CXCL12-CXCR7 axis is important for tumor endothelial cell
angiogenic property. Int J Cancer. 137:2825–2836. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen Y, Teng F, Wang G and Nie Z:
Overexpression of CXCR7 induces angiogenic capacity of human
hepatocellular carcinoma cells via the AKT signaling pathway. Oncol
Rep. 36:2275–2281. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu C, Shen SQ, Cui ZH, Chen ZB and Li W:
Effect of microRNA-1 on hepatocellular carcinoma tumor endothelial
cells. World J Gastroenterol. 21:5884–5892. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cao Z, Zheng L, Zhao J, Zhuang Q, Hong Z
and Lin W: Anti-angiogenic effect of Livistona chinensis seed
extract in vitro and in vivo. Oncol Lett. 14:7565–7570.
2017.PubMed/NCBI
|
|
12
|
Wani N, Nasser MW, Ahirwar DK, Zhao H,
Miao Z, Shilo K and Ganju RK: C-X-C motif chemokine 12/C-X-C
chemokine receptor type 7 signaling regulates breast cancer growth
and metastasis by modulating the tumor microenvironment. Breast
Cancer Res. 16:R542014. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hao M, Zheng J, Hou K and Wang J, Chen X,
Lu X, Bo J, Xu C, Shen K and Wang J: Role of chemokine receptor
CXCR7 in bladder cancer progression. Biochem Pharmacol. 84:204–214.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li F, Gao L, Jiang Q, Wang Z, Dong B, Yan
T and Chen X: Radiation enhances the invasion abilities of
pulmonary adenocarcinoma cells via STAT3. Mol Med Rep. 7:1883–1888.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hida K, Maishi N, Torii C and Hida Y:
Tumor angiogenesis-characteristics of tumor endothelial cells. Int
J Clin Oncol. 21:206–212. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Müller A, Homey B, Soto H, Ge N, Catron D,
Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al:
Involvement of chemokine receptors in breast cancer metastasis.
Nature. 410:50–56. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Teicher BA and Fricker SP: CXCL12
(SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 16:2927–2931.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miao Z, Luker KE, Summers BC, Berahovich
R, Bhojani MS, Rehemtulla A, Kleer CG, Essner JJ, Nasevicius A,
Luker GD, et al: CXCR7 (RDC1) promotes breast and lung tumor growth
in vivo and is expressed on tumor-associated vasculature. Proc Natl
Acad Sci USA. 104:15735–15740. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu YC, Tang SJ, Sun GH and Sun KH: CXCR7
mediates TGFβ1-promoted EMT and tumor-initiating features in lung
cancer. Oncogene. 35:2123–2132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu H, Zhang L and Liu P: CXCR7 signaling
induced epithelial-mesenchymal transition by AKT and ERK pathways
in epithelial ovarian carcinomas. Tumour Biol. 36:1679–1683. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin L, Han MM, Wang F, Xu LL, Yu HX and
Yang PY: CXCR7 stimulates MAPK signaling to regulate hepatocellular
carcinoma progression. Cell Death Dis. 5:e14882014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
O'Shea JJ, Holland SM and Staudt LM: JAKs
and STATs in immunity, immunodeficiency, and cancer. N Engl J Med.
368:161–170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xuan X, Li S, Lou X, Zheng X, Li Y, Wang
F, Gao Y, Zhang H, He H and Zeng Q: Stat3 promotes invasion of
esophageal squamous cell carcinoma through up-regulation of MMP2.
Mol Biol Rep. 42:907–915. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao G, Zhu G, Huang Y, Zheng W, Hua J,
Yang S, Zhuang J and Ye J: IL-6 mediates the signal pathway of
JAK-STAT3-VEGF-C promoting growth, invasion and lymphangiogenesis
in gastric cancer. Oncol Rep. 35:1787–1795. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang X, Qiu W, Zhang G, Xu S, Gao Q and
Yang Z: MicroRNA-204 targets JAK2 in breast cancer and induces cell
apoptosis through the STAT3/BCl-2/survivin pathway. Int J Clin Exp
Pathol. 8:5017–5025. 2015.PubMed/NCBI
|