Introduction
Schwartz-Jampel syndrome type 1 (SJS1; Online
Mendelian Inheritance in Man #255800) is a type of autosomal
recessive skeletal dysplasia characterized by permanent myotonic
myopathy and skeletal dysplasia, which result in short stature,
dystrophy of epiphyseal cartilages, joint contractures,
blepharophimosis, unusual pinnae, myopia and pigeon breast. In
addition, myotonic (leading to progressive stiffness of the face
and limbs), persistent bowing of the limbs, and severe
kyphoscoliosis may develop. Electromyograms exhibit continuous
discharges at rest (1–3). Furthermore, walking becomes
increasingly more difficult and secondary contractures of the large
joints may develop in patients with SJS1; by late adolescence,
severely affected patients may be confined to a bed and a
wheelchair (2). Adult height
varies from 140 to 170 cm, and ~25% of patients with SJS1 present
with mental retardation (4,5). In
addition, procainamide therapy has been shown to help muscle
function (6).
SJS1 is caused by mutations in heparan sulfate
proteoglycan 2 (HSPG2; also known as basement
membrane-specific heparin sulfate). The HSPG2 gene encodes
the perlecan protein, a ubiquitous HSP, which serves essential
roles in multiple biological activities, including forming the
vascular extracellular matrix, maintaining endothelial barrier
function, promoting growth factor and being a potent inhibitor of
smooth muscle cell proliferation (7,8).
Alternative splicing of this gene results in multiple transcript
variants, which may cause SJS1, Silverman-Handmaker type
dyssegmental dysplasia, and tardive dyskinesia (9).
In the present study, one SJS1 proband from a
Chinese family, who was diagnosed by X-ray and physical
examination, was recruited and genetic sequencing was performed.
Our study suggested that the compound heterozygous mutations in
HSPG2 were responsible for SJS1 and demonstrated the
genotype-phenotype relationship between mutations in the
HSPG2 gene and characteristics of SJS1.
Case report
The Ethics Committee of Nanjing Drum Tower Hospital
(Jiangsu, China) approved the present study. Written informed
consent was obtained from all subjects or their parents/guardians.
The 10-year-old female proband was recruited to the present study
for genetic testing between December 2016 and June 2017. The
proband was the only child born to healthy parents of Chinese Han
descent; there was no SJS1 or other skeletal diseases in the
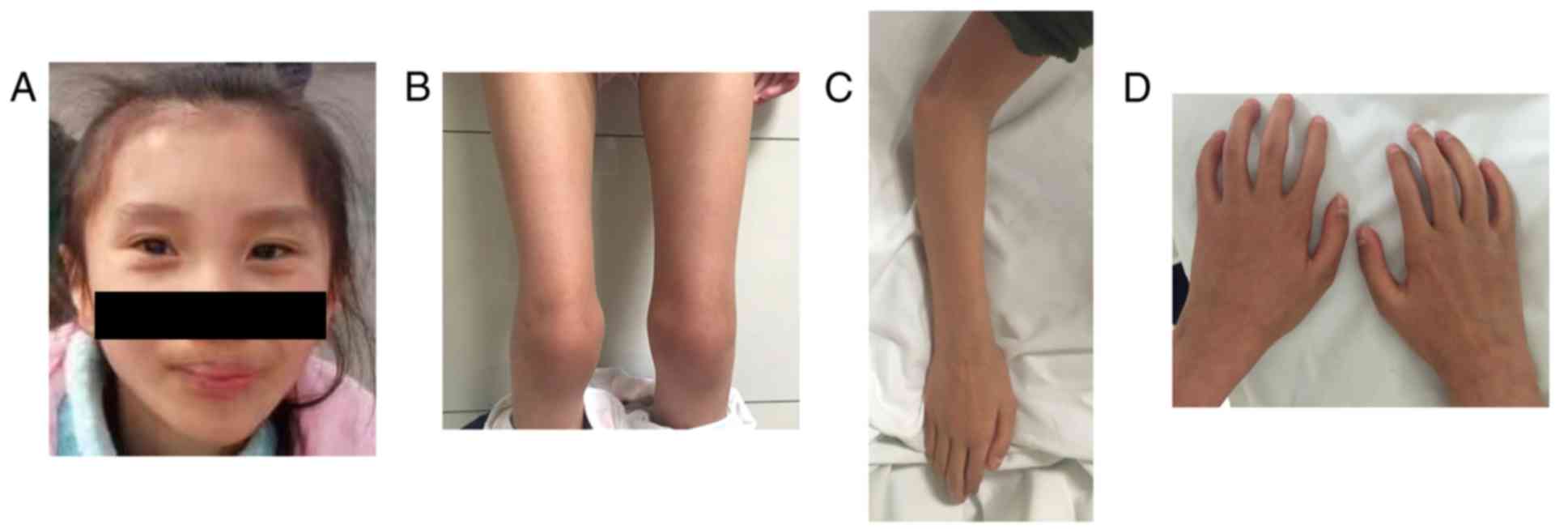
proband's family history. Upon physical examination of the proband,
a peculiar facial appearance with pursed lips was noted. A small
mouth, blepharophimosis, unusual pinnae and myopia were not
presented (Fig. 1A); however,
severe kyphoscoliosis, and progressive stiffness of the face, limbs
and hands were observed in the proband (Fig. 1B-D). In addition, the patient did
not exhibit anterior hypoplasia of cervical bodies with cervical
kyphosis. At the age of 2, the proband exhibited hypotonia with
poor muscle bulk and proximal leg weakness. Myotonic features,
which resulted in progressive stiffness, were noticed during the
proband's early childhood. At the age of 8, the proband began to
suffer from contractures of the large joints, including the elbow,
knee and hip, and walking became difficult; a lordotic gait was
also present at this time. The results of neurological and other
examinations were within the normal range, and the proband's height
was 85, 3.5 cm below than the average height of Han Chinese girls
of the same age.
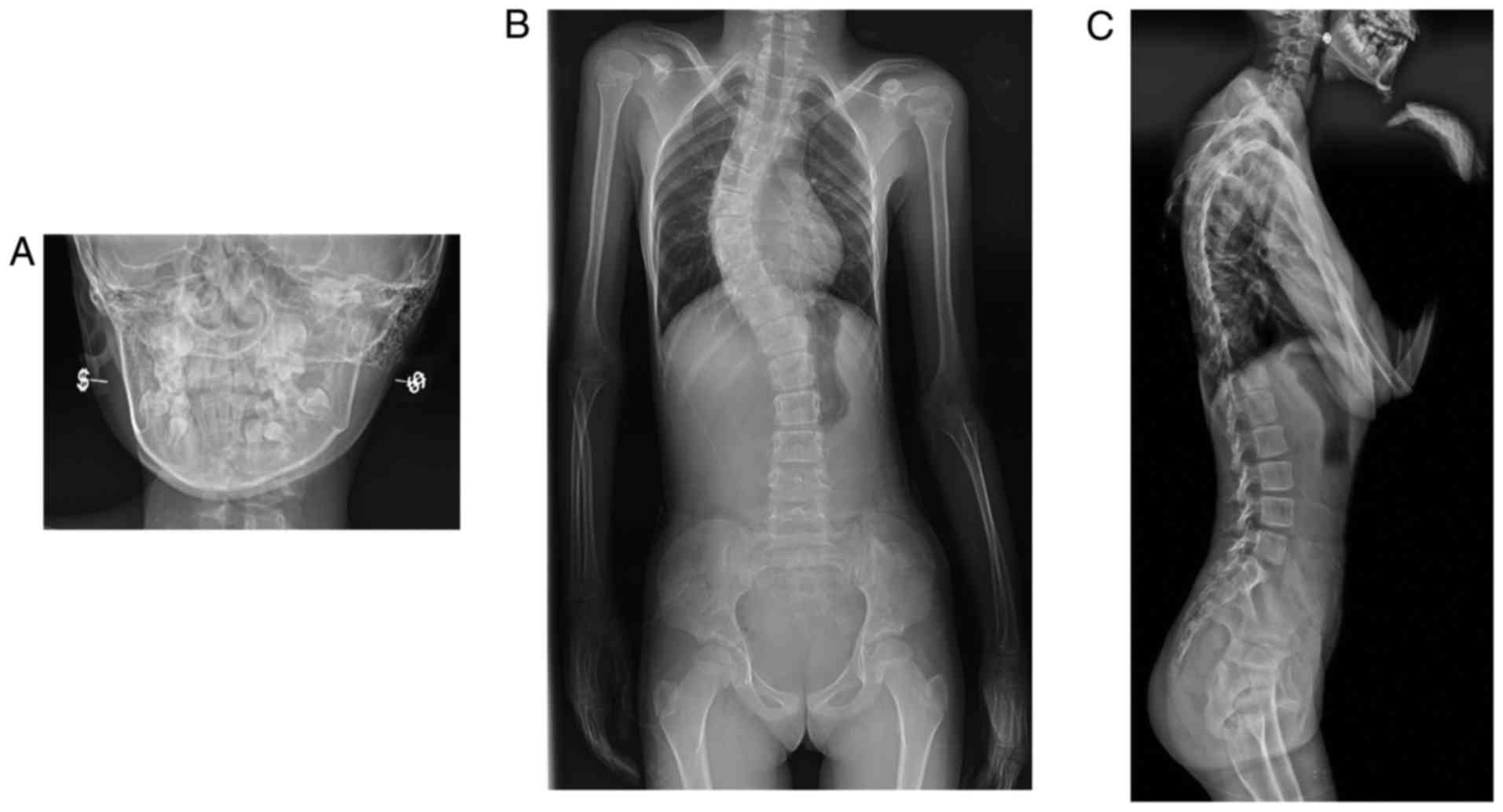
X-ray imaging performed when the patient was 10
years of age revealed micrognathia and a narrow upper thoracic
inlet (Fig. 2). No flattened
vertebral bodies with regular end-plates were noted (Fig. 2). There was significant
kyphoscoliosis, progression of epiphyseal changes and generalized
osteopenia. Bowed femora and large round capital femoral epiphyses
were also observed (Fig. 2).
These radiographic results of the patient were
similar to those described in previous reports of SJS1 (1,10,11);
however, due to the clinical and radiographic similarities of
skeletal abnormalities, patients with other skeletal diseases, such
as Stuve-Wiedemann syndrome or focal femoral hypoplasia-unusual
facies syndrome, may be misdiagnosed with SJS1. Therefore, the
present study performed molecular analysis of the patient and
parents.
Once written informed consent was obtained, genomic
DNA was extracted from the peripheral blood of the patient and
family members using a Qiagen DNA Mini kit (Qiagen GmbH, Hilden,
Germany). Whole exome sequencing (WES) of the proband was
conducted. Briefly, genomic DNA was divided into smaller fragments
of 200–250 bp using an ultrasonic instrument (Covaris LE220;
Covaris, Inc., Woburn, MA, USA). Subsequently, purification with
Ampure Beads (Beckman Coulter, Inc., Brea, CA, USA) was performed
to add poly A/joint reaction to the end of the purified DNA
fragments. The gene-trapping chip (Roche NimbleGen, Madison, WI,
USA) was hybridized with the purified DNA fragments of the proband.
Following hybridization, captured DNA was sequenced with an
Illumina HiSeq2500 Analyzer (Illumina, Inc., San Diego, CA, USA)
and read using Illumina Pipeline software (version 1.3.4; Illumina,
Inc.). The present study used BWA v0.59 (12) to align sequence reads to the human
genome reference (build 37) and removed duplicated reads from
subsequent analyses. Sequence variants were identified via
comparisons with the NCBI reference sequence NM 005529.5 and
annotated by the current version of ANNOVAR (2017 Jul 16;
annovar.openbioinformatics.org/en/latest/). The
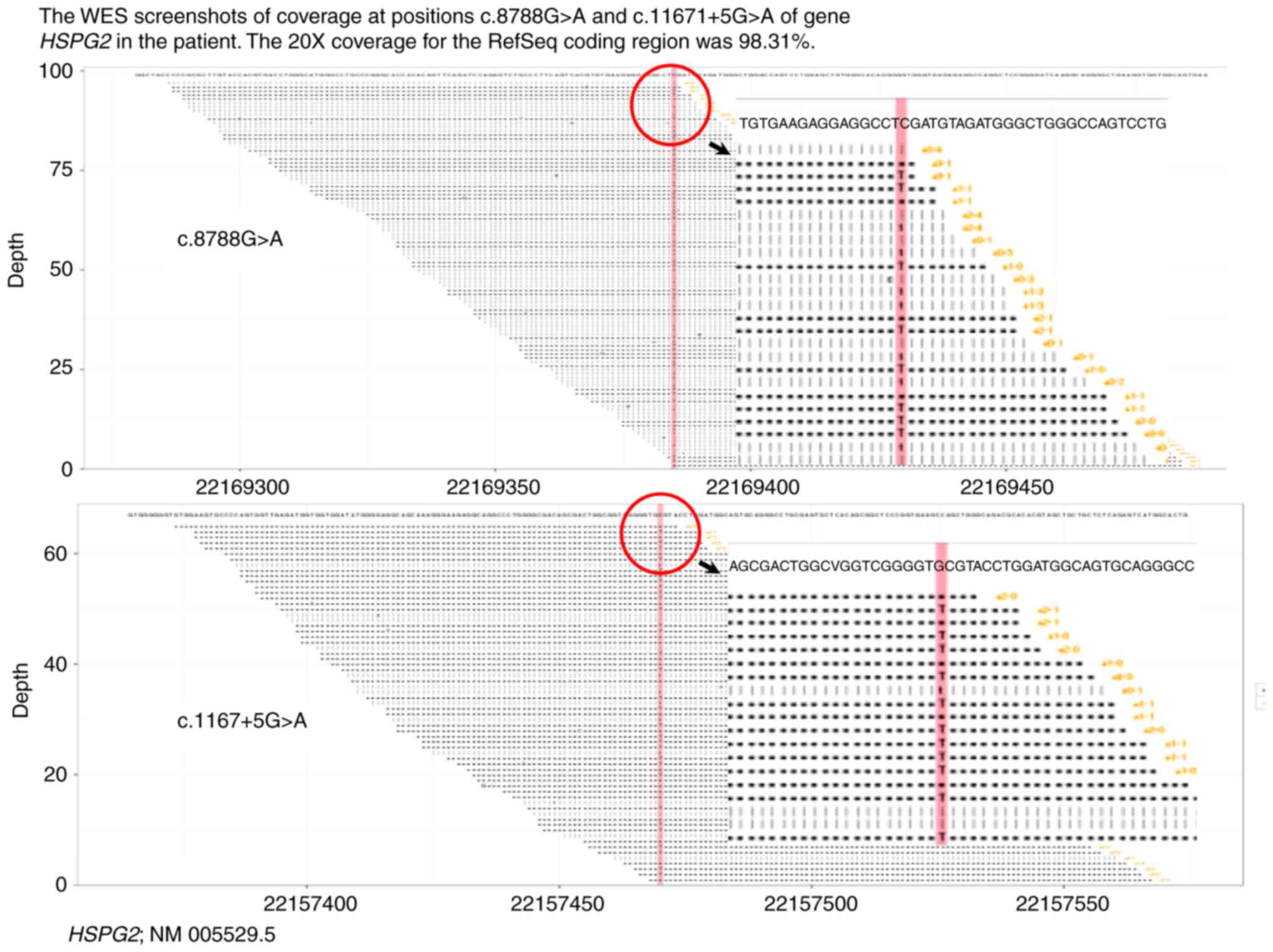
20X coverage for the RefSeq coding region was 98.31% (Fig. 3).
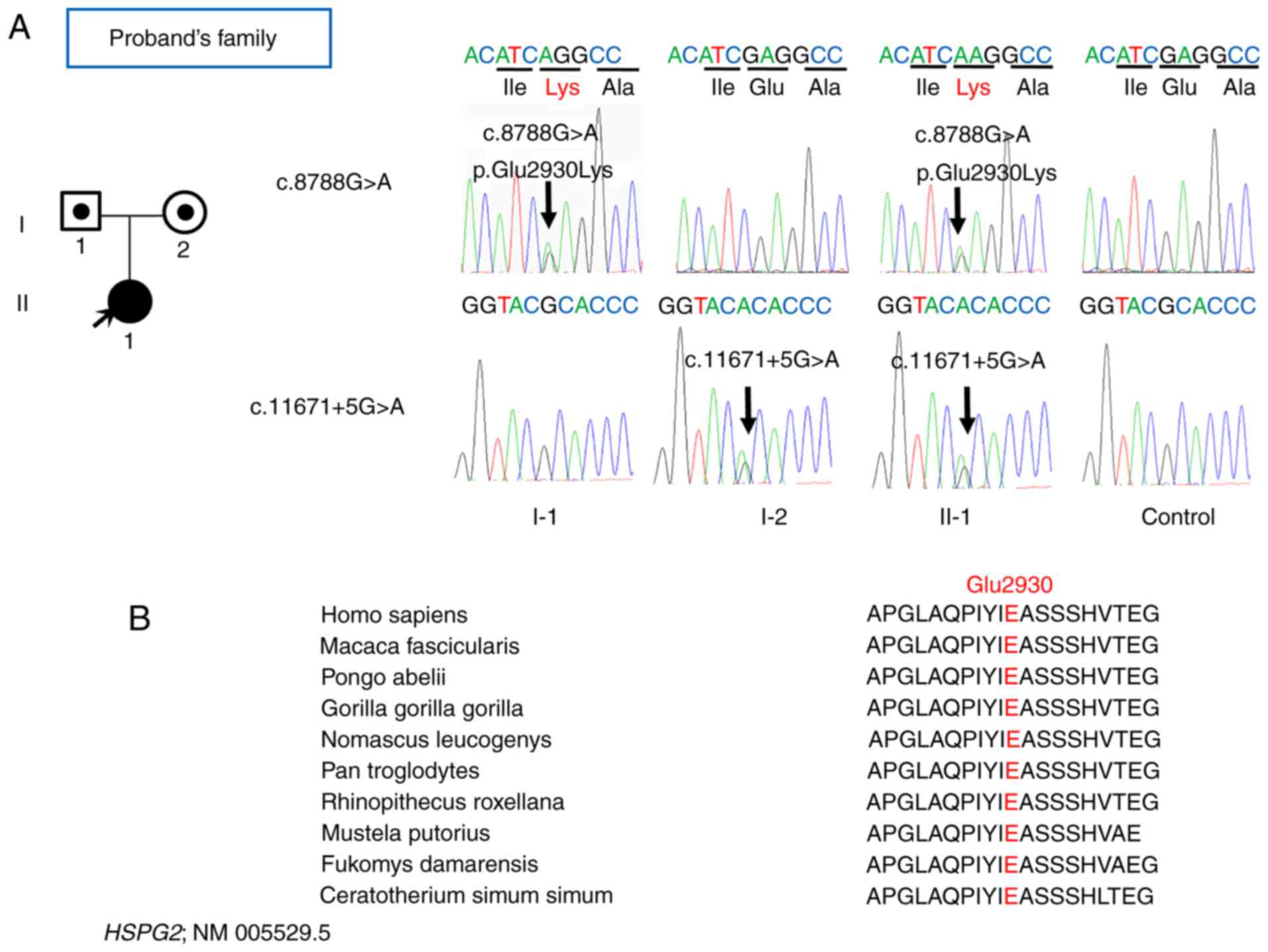
A novel missense mutation (c.8788 G>A;
p.Glu2930Lys) in exon 67 of HSPG2 and a mutation in intron
85 (c.11671+5G>A) the splicing region, were detected in the
proband (Fig. 4A). One guanine
ribonucleotide was altered to an adenine ribonucleotide in codon
8788, which caused a change in the reading frame from glutamine to
lysine. The Polyphen-2 tool (13)
predicted the mutation as likely to cause damage to the HSPG2
protein function (Table I). To the
best of our knowledge, the mutations c.8788G>A and
c.11671+5G>A have not been reported in SJS1 previously. The
mutation c.8788G>A; p.Glu2930Lys was located in the domain
Immunoglobulin_2 in HSPG2, while the mutation
c.11671+5G>A was present in the splicing region near the
Epidermal Growth Factor Calcium-binding domain. These 2 mutations
may result in the loss-of-function of HSPG2. In the 1,000
Genomes database (www.internationalgenome.org), dbSNP database
(www.ncbi.nlm.nih.gov/SNP/), Exome
Sequencing Project 6500 database (evs.gs.washington.edu/EVS/) and Exome Aggregation
Consortium EAS database (exac.broadinstitute.org), the allele frequencies of
the c.8788G>A mutation were 0, 0, 0 and 0.000155, respectively,
while the allele frequencies of c.11671+5 g> A mutation were 0,
0, 0.000077 and 0.01684, respectively (Table I).
 | Table I.Identification of single nucleotide
variations in the heparan sulfate proteoglycan 2 gene through whole
exome sequencing. |
Table I.
Identification of single nucleotide
variations in the heparan sulfate proteoglycan 2 gene through whole
exome sequencing.
| Variable | c.11671+5G>A
mutation | c.8788G>A
mutation |
|---|
| Chromosome | 1 | 1 |
| Position | 22,157,470 | 22,169,385 |
| Predicted protein
variants | – | p.Glu2930Lys |
| Variants type | Het | Het |
| Gene region | Splicing | Exon |
| Ref Seq | NM_005529.5 | NM_005529.5 |
| SNP ID | rs77527456 | rs368020528 |
| 1000g_chbs | – | – |
| dbSNP | 0 | – |
| ExAC_EAS | 0.01684 | 0 |
| ESP6500 | 0.000077 | 0.000155 |
| Polyphen | – | ‘Probably
damaging’ |
In the present study, Sanger sequencing was also
performed for the proband's family, which confirmed the compound
heterozygous mutation in the proband and the heterozygous status of
the father and mother (Fig. 4A);
there was no evidence of SJS1 or of other skeletal diseases in the
proband's parents. Furthermore, the mutations c.8788G>A;
p.Glu2930Lys and c.11671+5G>A were highly conserved among a
diverse range of species (Fig.
4B). Thus, the present study identified 2 novel HSPG2
mutations in a child case of SJS1.
Discussion
SJS1 is a rare, autosomal recessive progressive
disorder that is characterized by clinical features including short
stature, myotonic myopathy, dystrophy of epiphyseal cartilages,
joint contractures, blepharophimosis, unusual pinnae, myopia,
pigeon breast, and progressive stiffness of the face and limbs
(14–16). This disorder can be further divided
into the subtypes: SJS1A, a milder phenotype with an onset during
infancy to early childhood and relatively milder chondrodysplasia;
and SJS1B, a more severe phenotype with neonatal onset and
significant chondrodysplasia (17). Mask-like facies typically manifest
as blepharophimosis, with a pursed mouth and a fixed face with a
sad appearance. Chondrodysplasia consists of metaphyseal widening,
slender diaphysis and kyphoscoliosis (18).
Recessive inheritance is consistent with the
loss-of-function nature of the 2 mutations (c.8788 G>A;
p.Glu2930Lys, and c.11671+5G>A) and the previously reported
HSPG2 mutations in SJS (14–18).
In total, <40 patients with SJS have been reported and no
genotype-phenotype correlation is apparent. Although the splicing
variation c.11671+5G>A is a non-coding variant, it is highly
conserved among diverse species and several splicing-site variants
in HSPG2 have been reported to cause the aberrant splicing
in the exon region (14,15,18),
which form a considerable proportion of the reported variants. In
addition, the splice variation c.11671+5G>A in intron 85 of the
HSPG2 gene could highly affect the 5′ splice site, the base
5 nucleotides downstream of intron 85, increasing the likelihood of
reducing the content of effective mRNA that codes for the correct
protein. Finally, by combining the specific clinical information
and genetic evidence, the present study confirmed the diagnosis of
SJS1 in the proband.
The present study utilized WES, which allows
molecular results to be obtained faster, particularly when
traditional specific diagnosis takes a longer time to sequence a
large gene. HSPG2 is a very large gene, consisting of 97
exons, and encodes for the protein Perlecan, which is known to
serve an important role in maintaining cartilaginous tissue
integrity and regulating muscle excitability (2). Exome sequencing provides the ability
to identify rare variants, analyze the candidate gene and to check
for the presence of mutations in the genes at the same time
(19,20). In the present study, the
HSPG2 mutation spectrum was further expanded, thus
contributing to the earlier detection of the disease, which may
provide significant benefit to the patient and their family. In
addition, it may increase awareness of the growing number of SJS1
patients for future clinical diagnosis.
Acknowledgements
We thank the patient and her families who donated
their blood samples for this study.
Funding
The present study was supported by The Projects of
International Cooperation and Exchanges NSFC (grant no.
81420108021), Major Projects of NSFC (grant no. 8173000209),
Excellent Young Scholars NSFC (grant no. 81622033), Jiangsu
Provincial Key Medical Center Foundation, Jiangsu Provincial
Medical Talent Foundation and Jiangsu Provincial Medical
Outstanding Talent Foundation.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JD, XX, DS, DC, ZX and QJ performed the physical
examination and diagnosis of the patients. WY, XH and HT analyzed
and interpreted the WES data, and were major contributors to
writing and revising the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The Ethics Committee of Nanjing Drum Tower Hospital
(Jiangsu, China) approved the present study. Written informed
consent was obtained from all subjects or their
parents/guardians.
Consent for publication
Written informed consent was obtained from the
proband and their parents for the publication of the data and
images presented in the present study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mathur N and Ghosh PS: Schwartz-jampel
syndrome. Pediatr Neurol. 68:77–78. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nicole S, Davoine CS, Topaloglu H,
Cattolico L, Barral D, Beighton P, Hamida CB, Hammouda H, Cruaud C,
White PS, et al: Perlecan, the major proteoglycan of basement
membranes, is altered in patients with Schwartz-Jampel syndrome
(chondrodystrophic myotonia). Nat Genet. 26:480–483. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jablecki C and Schultz P: Single muscle
fiber recordings in the Schwartz-Jampel syndrome. Muscle Nerve.
5:S64–S69. 1982.PubMed/NCBI
|
|
4
|
Arikawa-Hirasawa E, Le AH, Nishino I,
Nonaka I, Ho NC, Francomano CA, Govindraj P, Hassell JR, Devaney
JM, Spranger J, et al: Structural and functional mutations of the
perlecan gene cause Schwartz-Jampel syndrome, with myotonic
myopathy and chondrodysplasia. Am J Hum Genet. 70:1368–1375. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rodgers KD, Sasaki T, Aszodi A and Jacenko
O: Reduced perlecan in mice results in chondrodysplasia resembling
Schwartz-Jampel syndrome. Hum Mol Genet. 16:515–528. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kashkouli MB, Shahrzad S, Jazayeri AA and
Abtahi MB: Treatment of blepharospasm in schwartz-jampel syndrome:
Botulinum toxin A injection or surgery. Ophthal Plast Reconstr
Surg. 33 3S Suppl 1:S47–S49. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stum M, Girard E, Bangratz M, Bernard V,
Herbin M, Vignaud A, Ferry A, Davoine CS, Echaniz-Laguna A, René F,
et al: Evidence of a dosage effect and a physiological endplate
acetylcholinesterase deficiency in the first mouse models mimicking
Schwartz-Jampel syndrome neuromyotonia. Hum Mol Genet.
17:3166–3179. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kallunki P, Eddy RL, Byers MG, Kestilä M,
Shows TB and Tryggvason K: Cloning of human heparan sulfate
proteoglycan core protein, assignment of the gene (HSPG2) to
1p36.1–p35 and identification of a BamHI restriction fragment
length polymorphism. Genomics. 11:389–396. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dodge GR, Kovalszky I, Chu ML, Hassell JR,
McBride OW, Yi HF and Iozzo RV: Heparan sulfate proteoglycan of
human colon: Partial molecular cloning, cellular expression, and
mapping of the gene (HSPG2) to the short arm of human chromosome 1.
Genomics. 10:673–680. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stum M, Davoine CS, Vicart S, Guillot-Noël
L, Topaloglu H, Carod-Artal FJ, Kayserili H, Hentati F, Merlini L,
Urtizberea JA, et al: Spectrum of HSPG2 (Perlecan) mutations in
patients with Schwartz-Jampel syndrome. Hum Mutat. 27:1082–1091.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Spranger J, Hall BD, Häne B, Srivastava A
and Stevenson RE: Spectrum of Schwartz-Jampel syndrome includes
micromelic chondrodysplasia, kyphomelic dysplasia, and Burton
disease. Am J Med Genet. 94:287–295. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kelley LA and Sternberg MJ: Protein
structure prediction on the Web: A case study using the Phyre
server. Nat Protoc. 4:363–371. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Iwata S, Ito M, Nakata T, Noguchi Y, Okuno
T, Ohkawara B, Masuda A, Goto T, Adachi M, Osaka H, et al: A
missense mutation in domain III in HSPG2 in Schwartz-Jampel
syndrome compromises secretion of perlecan into the extracellular
space. Neuromuscul Disord. 25:667–671. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Das Bhowmik A, Dalal A, Matta D, Kandadai
RM, Kanikannan MA and Aggarwal S: Identification of a novel splice
site HSPG2 mutation and prenatal diagnosis in Schwartz Jampel
Syndrome type 1 using whole exome sequencing. Neuromuscul Disord.
26:809–814. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shah M and Scavina M: Novel mutations in
Schwartz-Jampel syndrome with successes in medical management.
Neurology. 84 Suppl 14:(P2): 2372015.
|
|
17
|
Giedion A, Boltshauser E, Briner J, Eich
G, Exner G, Fendel H, Kaufmann L, Steinmann B, Spranger J and
Superti-Furga A: Heterogeneity in Schwartz-Jampel chondrodystrophic
myotonia. Eur J Pediatr. 156:214–223. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arikawa-Hirasawa E, Le AH, Nishino I,
Nonaka I, Ho NC, Francomano CA, Govindraj P, Hassell JR, Devaney
JM, Spranger J, et al: Structural and functional mutations of the
perlecan gene cause Schwartz-Jampel syndrome, with myotonic
myopathy and chondrodysplasia. Am J Hum Genet. 70:1368–1375. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lim BC, Yoo SK, Lee S, Shin JY, Hwang H,
Chae JH, Hwang YS, Seo JS, Kim JI and Kim KJ: Hoyeraal-Hreidarsson
syndrome with a DKC1 mutation identified by whole-exome sequencing.
Gene. 546:425–429. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mansouri M, Kayserili H, Elalaoui SC,
Nishimura G, Iida A, Lyahyai J, Miyake N, Matsumoto N, Sefiani A
and Ikegawa S: Novel DDR2 mutation identified by whole exome
sequencing in a Moroccan patient with spondylo-meta-epiphyseal
dysplasia, short limb-abnormal calcification type. Am J Med Genet
A. 170A:460–465. 2016. View Article : Google Scholar : PubMed/NCBI
|