Introduction
Spontaneous bacterial peritonitis (SBP) is one of
the serious complications that can occur in cirrhotic patients and
has high mortality and morbidity. Although the identification of
pathogen(s) is essential for the management of infectious diseases,
ascites fluid cultures often fail to provide positive results, even
when using ascites samples from patients who develop clinical
manifestations of SBP. Due to the necessity for early recognition
and treatment, SBP is usually diagnosed on the basis of an absolute
polymorphonuclear neutrophil (PMN) count in ascites with an optimal
cut-off value of ≥250/mm3, even if the pathogen is not
detected (1–4).
To overcome the difficulty experienced in obtaining
direct evidence of bacterial infection in SBP ascites, an
in-situ hybridization (ISH) method was recently developed
and its potential clinical utility reported (5). Bacterial DNA detection and sequencing
have been applied to the diagnosis of several infectious diseases.
Molecular techniques can detect small amounts of bacterial DNA
within a few h, and polymerase chain reaction (PCR)-based tests
that target the bacterium-specific 16S-ribosomal (r)RNA gene would
therefore provide a number of advantages. The highly conserved
sequences of the gene allow broad-range detection of almost all
bacterial species, while the hypervariable sequences can be used
for pathogen-specific identification (6–9).
In the studies that have investigated ascites
samples, PCR-based methods have demonstrated various positive rates
and highly conserved sequences of 16S rRNA have been detected in
~30–60% of non-infectious ascites samples (10–14).
However, with the detection of bacterial DNA in non-infectious
clinical samples, serious criticisms of the contamination of PCR
systems with bacterial DNA have been made (15–18).
For instance, commercially available DNA polymerases can be
contaminated with bacterial DNA, possibly as the products are
generated as recombinant proteins in bacterial cells (15,16).
Other commercial products including the reaction tubes for PCR
analysis have also been reported to contain contaminating DNA
fragments (17,18). However, if the amplification of 16S
rRNA gene is caused by contamination in these commercially
available products, bacterial DNA should be amplified in all the
samples tested. Previous studies have demonstrated that
conventional PCR detects bacterial genomic DNA in ~30–60% of the
non-SBP ascites samples tested (10–14).
Therefore, it is also suggested that amplification of the 16S rRNA
gene may reflect early detection of bacterial translocation in
cirrhotic ascites.
Since previous studies have reported varying
positive rates for PCR amplification, interpretation of PCR-based
detection of the 16S rRNA gene in non-infectious ascites remains
problematic. The present study developed a novel, highly sensitive
PCR protocol and analyzed the amplification obtained using
conventional PCR for the highly conserved sequences of the 16S rRNA
gene.
Patients and methods
Study population
Cirrhotic patients with ascites who were admitted to
Hyogo College of Medicine (Nishinomiya, Japan) between January 2010
and April 2013 were included in the present study. The study
protocol conformed to the ethical guidelines of the 1975 Helsinki
declaration and patients who agreed to the research use of ascites
were enrolled following their informed consent. Cirrhosis was
diagnosed on the basis of the histological results, clinical
(laboratory or imaging) data, or both. Patients with any
intra-abdominal, surgically treatable source of infection were
excluded. Patients who received antibiotic treatment and patients
with peritonitis carcinomatosa were also excluded from the
analysis. The present study was approved by the Ethics
Committee/Institutional Review Board of the Hyogo College of
Medicine.
Paracentesis
Cirrhotic patients underwent diagnostic paracentesis
under aseptic conditions using standard procedures for evaluation
of the presence or absence of SBP. The routine biochemical
variables and PMN count of the ascitic fluid were investigated.
Ascites samples with a high PMN count (≥250/µl) and low PMN count
(<250/µl) were considered as SBP ascites and non-SBP ascites,
respectively. Blood samples were also collected to perform routine
clinical studies.
DNA extraction
Genomic DNA was isolated from the bacterial strains
according to previously reported methods (5). The following bacterial strains were
obtained from Microbe Division/Japan Collection of Microorganisms
RIKEN BioResource Research Center (Tsukuba, Ibaraki, Japan):
Escherichia coli (cat. no. JCM1649), Klebsiella
pneumoniae subsp. pneumoniae (cat. no. JCM1662),
Enterobacter cloacae subsp. cloacae (cat. no.
JCM1232), Pseudomonas aeruginosa (cat. no. JCM5962),
Bacteroides fragilis (cat. no. JCM11019), Enterococcus
faecalis (cat. no. JCM5803), Enterococcus faecium (cat.
no. JCM5804), Streptococcus pyogenes (cat. no. JCM5674) and
Streptococcus agalactiae (cat. no. JCM5671). The following
bacterial strains were obtained from American Type Culture
Collection, (Manassas, VA, USA): Staphylococcus aureus (cat.
no. ATCC12600), Staphylococcus epidermidis (cat. no.
ATCC14990) and Streptococcus pneumoniae (cat. no.
ATCC39938). DNA was isolated from ascitic fluids according to the
methods described by Such et al (10). In brief, 200 µl of an ascites
sample was treated with an enzyme (lysozyme/proteinase K)
containing buffer for 2 h, and DNA was extracted using QIAamp Spin
Columns (QIAamp DNA Mini kit; Qiagen GmbH, Hilden, Germany)
according to the manufacturer's protocols (10).
Detection of bacterial DNA and DNA
sequencing
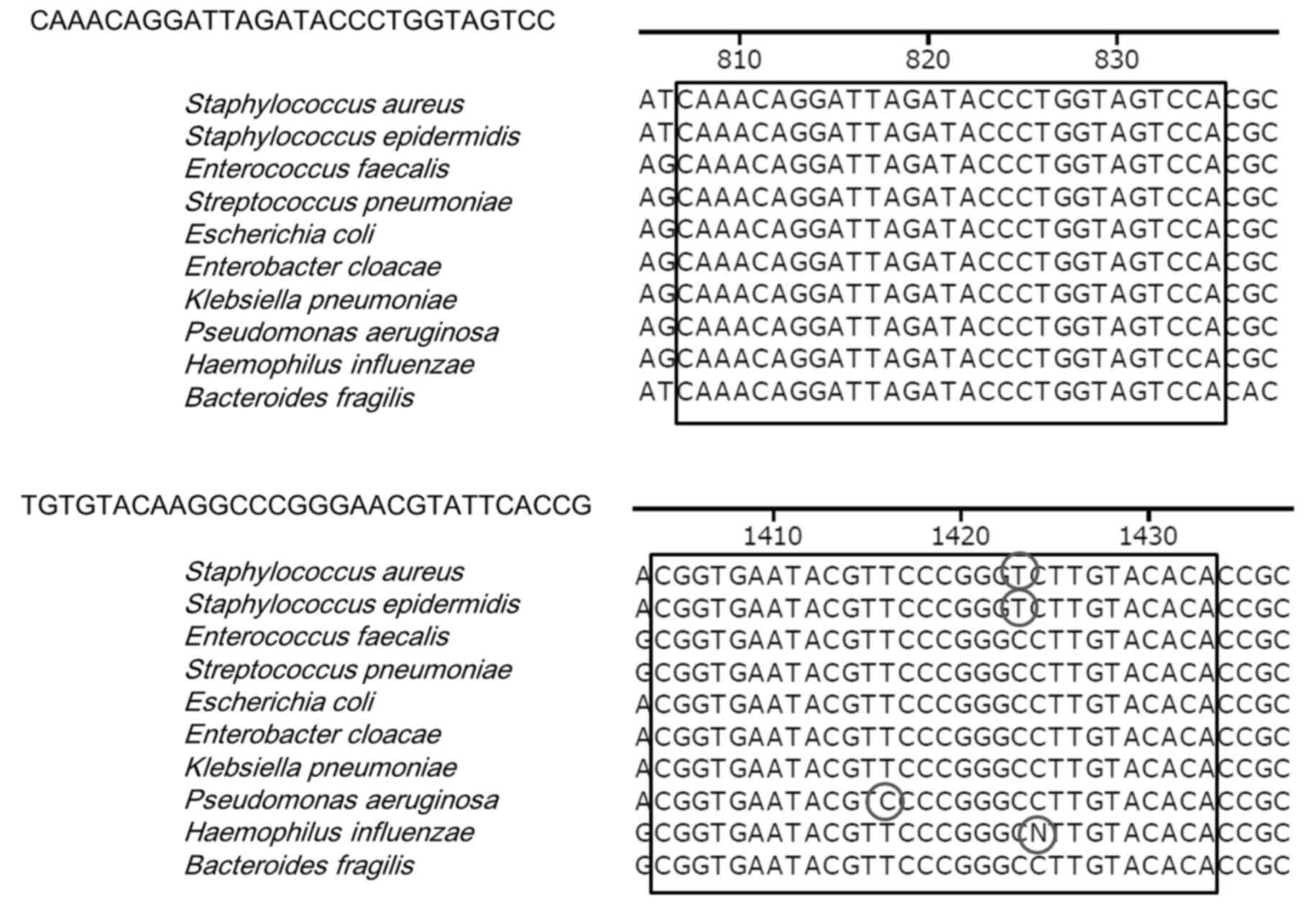
To establish a new PCR protocol, the highly
conserved sequences of the 16S rRNA gene were analyzed. In the 16S
rRNA gene (1,500 bp), it was identified that the sequences at
positions 9, 350, 500, 800, 1,100 and 1,400 were highly conserved.
To increase the sensitivity of the PCR, various primer candidates
whose sequences were GC-rich in the 3′-position were evaluated.
These DNA sequence-related analyses were performed with
commercially available software (DNASTAR Lasergene, Ver.7.1;
DNASTAR, Inc., Madison, WI, USA). The primer pair
5′-CAAACAGGATTAGATACCCTGGTAGTCC-3′ and
5′-TGTGTACAAGGCCCGGGAACGTATTCACCG-3′ was designed on the basis of
its specific amplification of the 16S rRNA gene (800F-1400R;
Fig. 1). Since two additional
potential primer pairs [9F-500R:
(5′-AGAGTTTGATCCTGGCTCAGGATGAACGCT-3′ and
5′-TATTACCGCGGCTGCTGGCACGGAGTTAGC-3′) and 350F-1100R:
(5′-AGAGTTTGATCCTGGCTCAGGATGAACGCT-3′ and
5′-TATTACCGCGGCTGCTGGCACGGAGTTAGC-3′)] failed in providing a
specific amplification of 16S rRNA gene (data not shown), we used
the primer pair shown in Fig. 1.
The fragments of the 16S rRNA gene were amplified with a Gene Amp
PCR system 9700 (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) using the primer pair with the following
conditions: 94°C for 30 sec, 71°C or 55°C for 30 sec and 72°C for
30 sec for genomic DNA samples derived from bacterial strains; and
94°C for 30 sec, 55°C for 30 sec, and 72°C for 30 for
ascites-derived DNA. PCR conditions, including tested DNA templets
and reaction cycles were determined according to the methods
described by Such et al (10). The lower limit of detection of
bacterial DNA was then determined. Bacterial genomic DNA was used
at various concentrations (10, 1 and 0.1 pg) as templates for the
PCR.
The present study used two types of DNA polymerases:
AmpliTaq Gold LD (Applied Biosystems; Thermo Fisher Scientific,
Inc.) and Prime STAR HS (Takara Bio, Inc., Otsu, Japan). All PCRs
were performed according to the manufacturers' protocols.
Commercially available RNase-free water (Takara Bio, Inc.) was used
in all reactions. To obtain the DNA sequences of PCR products, the
amplified products were purified and sequenced according to
standard direct-sequencing techniques. Each sequence was subjected
to analysis with the Basic Local Alignment Search Tool (BLAST) of
GenBank to investigate the homology of the 16S rRNA gene sequences
(https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Results
PCR primers for highly sensitive
amplification of the 16S rRNA gene
Among the three potential primer pairs that
corresponded to the conserved sequences of the bacterial 16S rRNA
gene, the primer pair described in the ‘Patients and methods’
section was successfully used for the amplification of DNA
fragments from multiple bacterial strains and whether the amplified
PCR products were consistent with the fragments of 16S rRNA gene
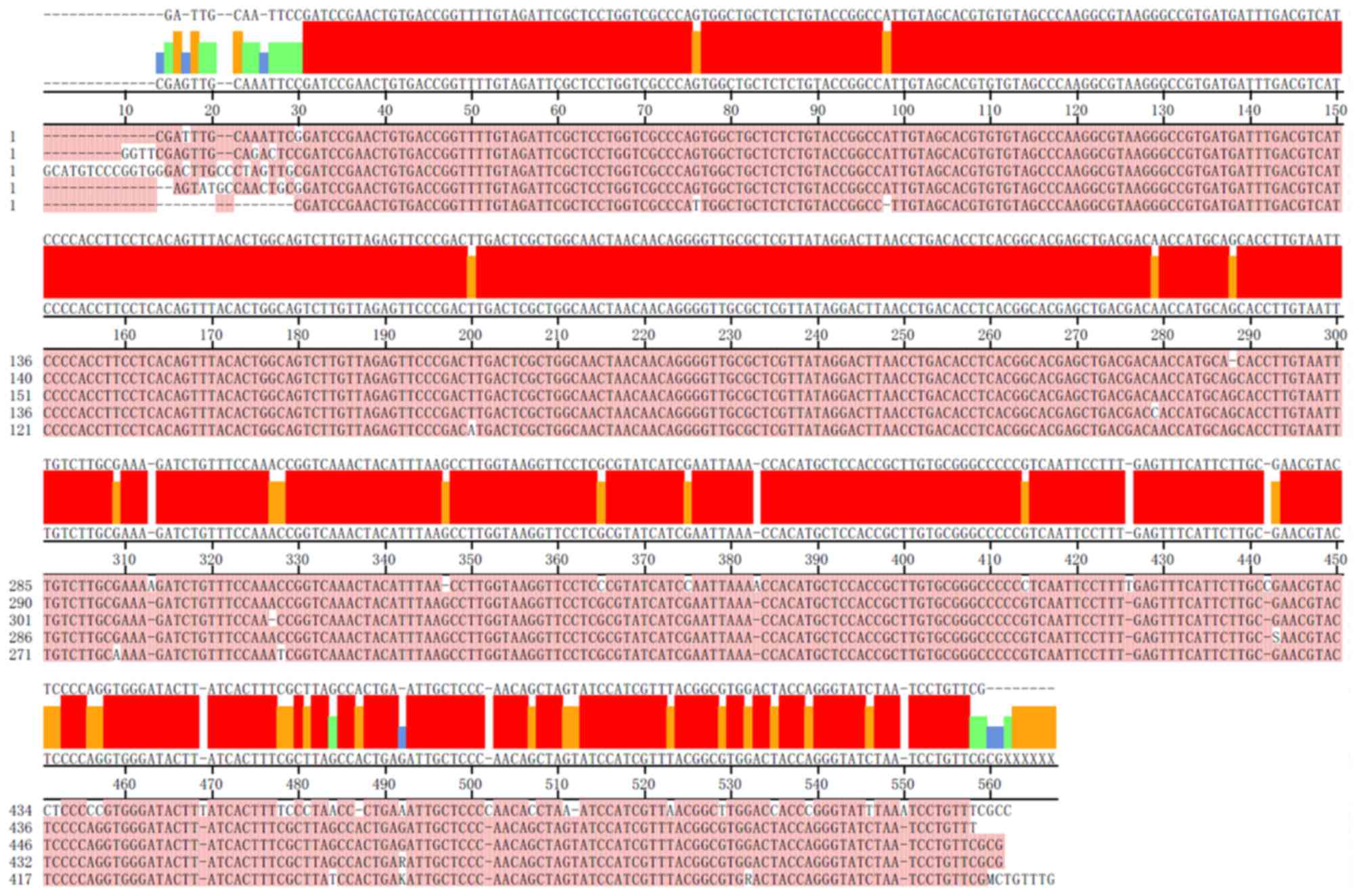
was investigated. Although the DNA sequences were slightly
different from each other, the amplified PCR products had sequences
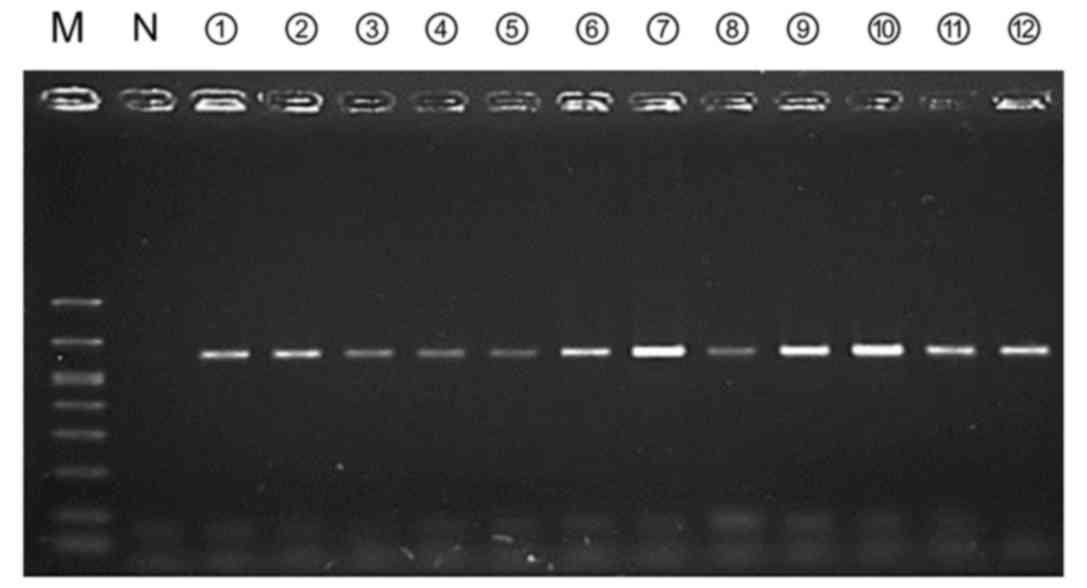
that were highly homologous to the 16S rRNA gene (Fig. 2). When 10 pg of bacterial DNA,
which has been reported as the lower limit of detection for the
conventional PCR method (10), was
used, the 16S rRNA genes of major disease-causing bacterial strains
were amplified (Fig. 3). In
addition, amplification of the 16S rRNA target gene was confirmed
for 59 bacterial strains that were used in our previous study
(5) (data not shown).
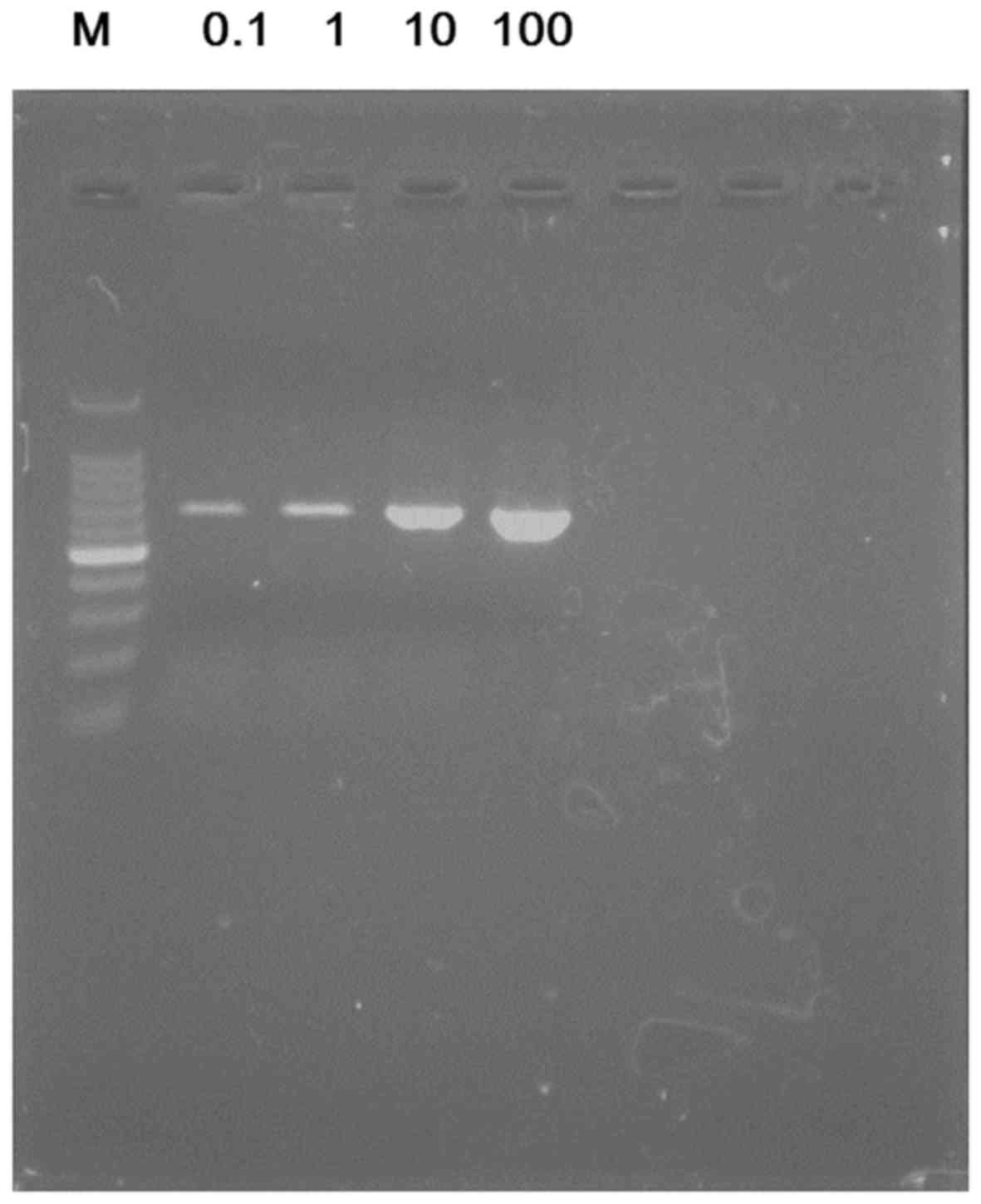
Subsequently, serially diluted bacterial DNA
solutions were used and the lower limit of bacterial DNA
concentration for detection of the 16S rRNA gene with PCR. Evident
bands were obtained with just 0.1 pg of each bacterial DNA template
(Fig. 4).
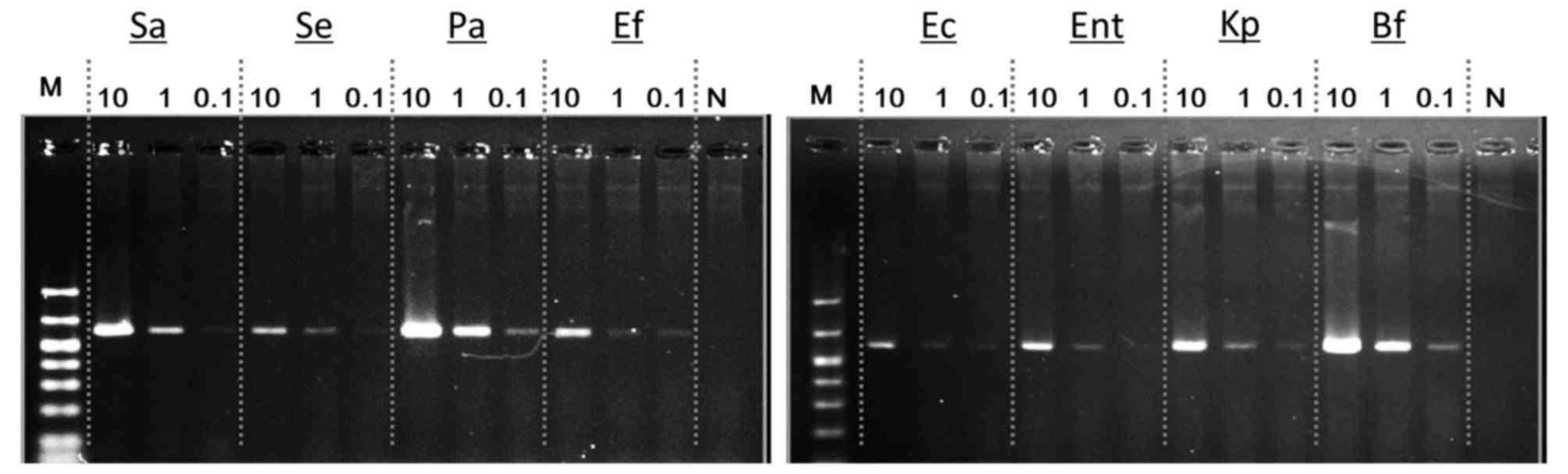 | Figure 4.Amplification of the 16S rRNA gene
using a small amount of bacterial DNA templates. With our PCR
conditions, evidently detectable bands were obtained with only 0.1
pg of bacterial DNA template. rRNA, ribosomal RNA; Sa,
Staphylococcus aureus; Se, Staphylococcus
epidermidis; Pa, Pseudomonas aeruginosa; Ef,
Enterococcus faecalis; Ec, Escherichia coli; Ent,
Enterobacter cloacae subsp. cloacae; Kp, Klebsiella
pneumoniae subsp. pneumoniae; Bf, Bacteroides fragilis;
N, Negative control; M, Marker. |
Detection of bacterial DNA from
non-SBP ascitic fluids
Sensitive PCR amplification of the 16S rRNA gene was
established and then it was investigated whether the gene could be
detected in non-infectious ascites. A total of 24 cirrhotic
patients had sterile (non-infectious) ascites as defined in the
‘Patients and methods’ section. The baseline clinical
characteristics of the patients with non-infectious acsitic fluids
are shown in Table I. All patients
had cirrhotic livers, and the Child-Pugh score was B in 7 patients
and C in 17 patients. A total of 12 patients were diagnosed with
non-viral cirrhosis and 12 with hepatitis B or C viral infection.
The remaining 12 patients had non-viral cirrhosis that was
associated with various liver diseases, including alcoholism,
autoimmune hepatitis, primary biliary cholangitis and cryptogenic
hepatitis (Table I).
 | Table I.Characteristics of the patients. |
Table I.
Characteristics of the patients.
| Parameter | Median value |
|---|
| Age (years) | 59 (50–82) |
| Sex |
|
|
Male | 17 |
|
Female | 7 |
| Etiology |
|
|
HBV | 4 |
|
HCV | 8 |
|
Non-viral | 12 |
| Child-Pugh
classification |
|
| A | 0 |
| B | 7 |
| C | 17 |
| Total bilirubin
(mg/dl) | 2.3
(0.5–28.3) |
| Albumin (g/dl) | 2.7 (2.0–4.3) |
| PT-INR | 1.35
(1.02–2.30) |
| Hepatocellular
carcinoma |
|
|
Present | 6 |
|
Absent | 18 |
| PMN count of
ascites (cells/µl) | 10 (1–168) |
Although the conditions used for PCR in the present
study allowed for highly sensitive amplification of the 16S rRNA
gene, it was further investigated if conditions could be
established for obtaining complete amplification of the gene from
non-infectious ascites. When the results of the PCRs were carefully
analyzed, the possibility that a PCR product was present in the
negative control sample was noted, although the signal was very
weak and uncertain (Fig. 3, lane
N). To increase the sensitivity of the PCR, another DNA polymerase
with higher efficacy of amplification was used (Prime STAR HS) and
the annealing temperature selected as 55°C. It was confirmed that
the 16S rRNA target gene was more evidently amplified with the 0.1
pg of the bacterial DNA template under this condition (Fig. 5) compared with the results in
Fig. 4. It was then attempted to
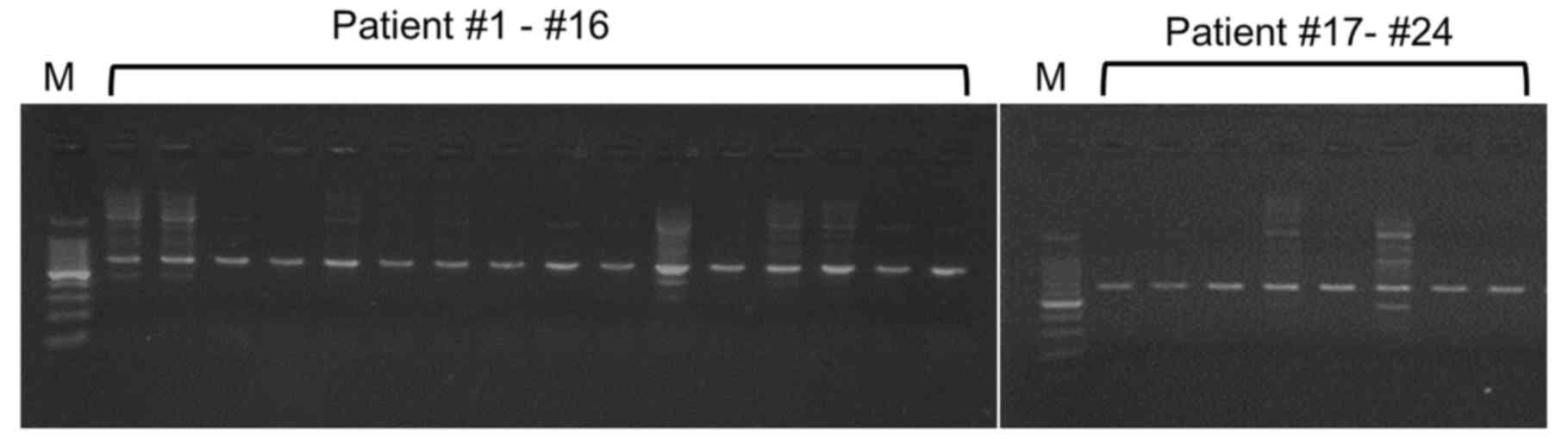
detect the 16S rRNA target gene from non-infectious ascites. On
doing so, it was identified that the PCR products were amplified
from all 24 non-SBP samples (Fig.
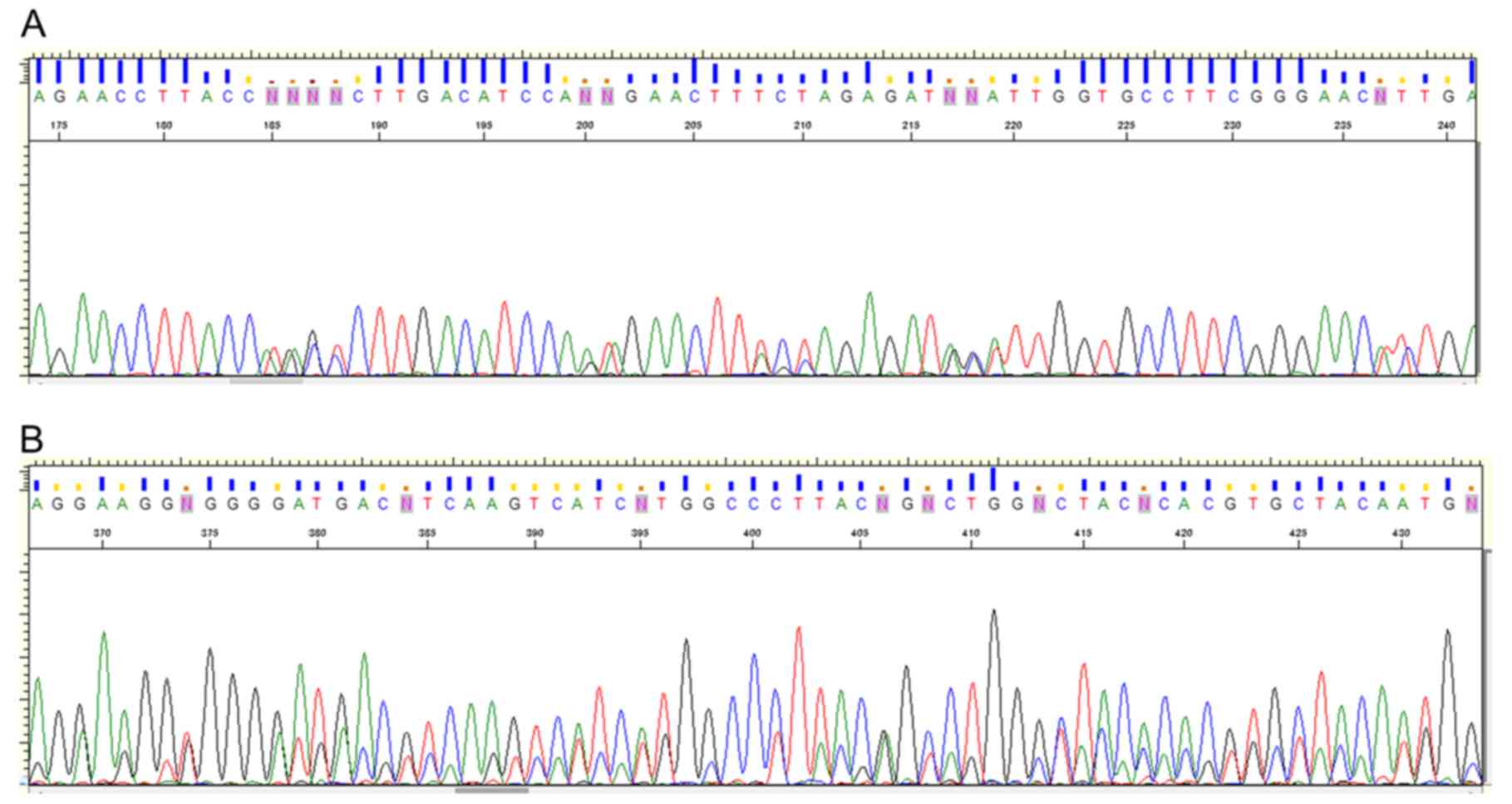
6). All PCR products had homologous sequences to the 16S rRNA
gene; however, multi-peak signals were observed at numerous
sequence points, and identification of specific pathogens was
difficult, suggesting that each PCR product contained the genomic
DNA fragments of polymicrobacteria (Fig. 7). These results suggested that
sensitive amplification of the 16S rRNA gene was achieved with our
PCR protocol and that bacterial DNA can be amplified from all
samples independently of spontaneous bacterial infection or
bacterial translocation in the abdominal cavity, since these
conditions are considered to be a monobacterial infection in
ascitic fluid (1–4).
Discussion
SBP is an infectious disease that develops in
cirrhotic patients with ascites. Bacterial culture often fails to
detect the pathogen. It is known that the 16S rRNA gene is present
in multiple copies in the genomes of bacterial pathogens and
numerous bacterial species contain up to 7 copies of the gene
(19). The presence of multiple
copies can increase the possibility of detecting small numbers of
pathogens, compared with assays performed for a single-copy gene.
Therefore, PCR amplification of the bacterium-specific 16S rRNA
gene is a useful method for investigating a broad range of
bacterial species. However, it is unclear whether the PCR-based
detection of the 16S rRNA gene is useful for determining the
causative pathogen. A major problem is that the 16S rRNA gene can
be amplified not only in SBP ascites but also in non-SBP sterile
ascites, which makes it difficult to determine the clinical
significance of this method.
Numerous commercially available recombinant DNA
polymerases are generated in bacterial cells, and concerns about
the presence of bacterial DNA in experimental items for PCR have
been reported (15–17). Although several trials have been
carried out to eliminate or reduce the amount of contaminating DNA
(20–23), no method for absolute purification
has been established (16).
Therefore, a sensitive method may detect small amounts of
contaminating bacterial genomic DNA. However, if the amplified 16S
rRNA gene from genomic DNA products of the clinical samples really
represents the contaminated DNA, all PCRs should demonstrate
positive results independent of bacterial infection. Nevertheless,
previous studies have demonstrated that the PCR method can amplify
16S rRNA gene in fewer than 60% of sterile ascites samples
(10–14). Therefore, it has been also
suggested that amplification of the 16S rRNA gene is associated
with early detection of bacterial translocation in cirrhotic
ascites, since a small number of bacteria are presumed to invade
the intraperitoneal cavity of cirrhotic patients with ascites via
several pathways (24,25). Conversely, it has also been
reported that PCR detection of bacterial DNA in non-infectious
ascites is not directly associated with the development of SBP
(26), and the clinical
implications of detecting the 16S rRNA gene with PCR remain
unclear.
The present study attempted to amplify the 16S rRNA
gene in non-infectious ascites with newly-established conditions
for PCR. Using this PCR protocol, a positive band could be obtained
with 0.1 pg of bacterial DNA templates. This limit is 100 times
more sensitive than the previously reported PCR protocols, whose
lower limits of bacterial DNA templates were ~10 pg (10,27,28).
However, difficulty was experienced in determining the bacterial
species with DNA sequencing due to the possible presence of DNA
fragments corresponding to multiple microbial species (Fig. 7). Soriano et al (14) studied 20 non-infectious ascites
samples and could amplify the 16S rRNA gene in 12 samples. However,
they succeeded in definitive bacterial identification only in 6
cases. They mentioned that the reason for the failure of the
sequencing reaction could be low initial DNA concentration or the
use of a mixture of amplification products that corresponded to
multiple bacterial species. Tilburg et al (18) reported the probable contamination
of a commercially available PCR Master Mix with bacterial DNA. They
mentioned that the contamination was most likely caused by the use
of compounds of animal origin due to the asymptomatic presence of
several microorganisms in animals. As described above, although
several studies have aimed to avoid bacterial DNA contamination of
DNA polymerase (20–23), complete eradication of bacterial
DNA is thought to be very difficult (16). The present study consistently
demonstrated that a simple conventional PCR targeting the 16S rRNA
gene invites criticism with respect to the identification of the
causative pathogen.
To confirm the results of the present study, PCR was
repeatedly performed, including using different batch numbers of
PCR reagents, 16S rRNA genes were amplified from all non-SBP
samples. The results of the present study may reflect contamination
of commercially available PCR systems with multiple bacterial
species. The possibility that the PCR products reflect the presence
of bacterial DNA due to bacterial translocation cannot be
dismissed, but, taking into account previous studies that
demonstrated the risk of contamination, the contaminating bacterial
DNA fragments would be considered to be mainly responsible for the
reproducible and complete amplification of bacterial DNA. It is
therefore suggested that contamination with bacterial DNA would be
a commonly observed inevitable problem when using highly sensitive
conventional PCR to identify the pathogen.
Recently, in addition to the efforts on eliminating
contaminating bacterial DNA, several new approaches have been
reported to succeed in providing a clinical significance of 16S
rRNA gene amplification (29–32).
For instance, the amount of 16S rRNA gene has been shown to be
associated with the prognosis of cirrhotic patients (29,30).
Additionally, advanced PCR-based methods for identifying bacterial
pathogens, which cause SBP, have been also reported; excellent
results were obtained with these assays, which should provide an
improved approach for detecting pathogens (31,32).
To the best of the authors' knowledge, the present study is the
first report regarding the 100% amplification of 16S rRNA gene from
non-infectious ascitic samples by a conventional PCR method. Its
results suggest limitations of the simple PCR amplification and
support the importance of the abovementioned recent superior
techniques (29–32).
In conclusion, although recent advanced methods
should demonstrate a clinical relevance, it is difficult to
accurately detect the bacterial translocation in cirrhotic ascites
with only a simple conventional PCR targeting the 16S rRNA gene.
Careful attention is required to interpret the results based on
simple amplification of 16S rRNA gene with conventional PCR.
Acknowledgements
The authors thank Ms. Kanazawa N., Ms. Deguchi N.,
Ms. Matsushita Y. and Ms. Fujii S. (Hyogo College of Medicine,
Nishinomiya, Japan) for their technical assistance.
Funding
No funding was received.
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
HE contributed to the study design, performed the
experiments, analyzed the data and drafted the manuscript. SI-I and
AM performed the experiments and edited the manuscript. NA, TT and
HN contributed to the sample collection, data acquisition and data
analysis. YI, YS, RT, NIk, KH, CN, TN, KY, YM, NIs, YY, AI and HI
contributed to sample collection and data acquisition. SN
contributed to the study design, analyzed the data and edited the
manuscript. All authors were involved in the manuscript revision
and approved the final version of the manuscript.
Ethics approval and consent to
participate
The study protocol conformed to the ethical
guidelines of the 1975 Helsinki declaration and patients who agreed
to the research use of ascites were enrolled following their
informed consent. The present study was approved by the Ethics
Committee/Institutional Review Board of Hyogo College of
Medicine.
Consent for publication
Patients gave informed written consent.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SBP
|
spontaneous bacterial peritonitis
|
|
PMN
|
polymorphonuclear neutrophil
|
|
ISH
|
in-situ hybridization
|
|
PCR
|
polymerase chain reaction
|
|
rRNA
|
ribosomal RNA
|
References
|
1
|
Runyon BA: AASLD Practice Guidelines
Committee: Management of adult patients with ascites due to
cirrhosis: An update. Hepatology. 49:2087–2107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Garcia-Tsao G and Lim JK: Members of
Veterans Affairs Hepatitis C Resource Center Program: Management
and treatment of patients with cirrhosis and portal hypertension:
recommendations from the Department of Veterans Affairs Hepatitis C
Resource Center Program and the National Hepatitis C Program. Am J
Gastroenterol. 104:1802–1829. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
European Association for the Study of the
Liver: EASL clinical practice guidelines on the management of
ascites, spontaneous bacterial peritonitis, and hepatorenal
syndrome in cirrhosis. J Hepatol. 53:397–417. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wiest R, Krag A and Gerbes A: Spontaneous
bacterial peritonitis: Recent guidelines and beyond. Gut.
61:297–310. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Enomoto H, Inoue S, Matsuhisa A, Aizawa N,
Imanishi H, Saito M, Iwata Y, Tanaka H, Ikeda N, Sakai Y, et al:
Development of a new in situ hybridization method for the detection
of global bacterial DNA to provide early evidence of a bacterial
infection in spontaneous bacterial peritonitis. J Hepatol.
56:85–94. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Picard FJ and Bergeron MG: Rapid molecular
theranostics in infectious diseases. Drug Discov Today.
7:1092–1101. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baron EJ: Implications of new technology
for infectious diseases practice. Clin Infect Dis. 43:1318–1323.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Woo PC, Lau SK, Teng JL, Tse H and Yuen
KY: Then and now: Use of 16S rDNA gene sequencing for bacterial
identification and discovery of novel bacteria in clinical
microbiology laboratories. Clin Microbiol Infect. 14:908–934. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sontakke S, Cadenas MB, Maggi RG, Diniz PP
and Breitschwerdt EB: Use of broad range16S rDNA PCR in clinical
microbiology. J Microbiol Methods. 76:217–225. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Such J, Francés R, Muñoz C, Zapater P,
Casellas JA, Cifuentes A, Rodríguez-Valera F, Pascual S, Sola-Vera
J, Carnicer F, et al: Detection and identification of bacterial DNA
in patients with cirrhosis and culture-negative, nonneutrocytic
ascites. Hepatology. 36:135–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Francés R, Zapater P, González-Navajas JM,
Muñoz C, Caño R, Moreu R, Pascual S, Bellot P, Pérez-Mateo M and
Such J: Bacterial DNA in patients with cirrhosis and noninfected
ascites mimics the soluble immune response established in patients
with spontaneous bacterial peritonitis. Hepatology. 47:978–985.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sugihara T, Koda M, Maeda Y, Matono T,
Nagahara T, Mandai M, Ueki M and Murawaki Y: Rapid identification
of bacterial species with bacterial DNA microarray in cirrhotic
patients with spontaneous bacterial peritonitis. Inter Med.
48:3–10. 2009. View Article : Google Scholar
|
|
13
|
Bruns T, Sachse S, Straube E, Assefa S,
Herrmann A, Hagel S, Lehmann M and Stallmach A: Identification of
bacterial DNA in neutrocytic and non-neutrocytic cirrhotic ascites
by means of a multiplex polymerase chain reaction. Liver Int.
29:1206–1214. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Soriano G, Esparcia O, Montemayor M,
Guarner-Argente C, Pericas R, Torras X, Calvo N, Román E, Navarro
F, Guarner C and Coll P: Bacterial DNA in the diagnosis of
spontaneous bacterial peritonitis. Aliment Pharmacol Ther.
33:275–284. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Corless CE, Guiver M, Borrow R,
Edwards-Jones V, Kaczmarski EB and Fox AJ: Contamination and
sensitivity issues with a real-time universal 16S rRNA PCR. J Clin
Microbiol. 38:1747–1752. 2000.PubMed/NCBI
|
|
16
|
Philipp S, Huemer HP, Irschick EU and
Gassner C: Obstacles of multiplex real-time PCR for bacterial 16S
rDNA: Primer specifity and DNA decontamination of Taq polymerase.
Transfus Med Hemother. 37:21–28. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Evans GE, Murdoch DR, Anderson TP, Potter
HC, George PM and Chambers ST: Contamination of Qiagen DNA
extraction kits with Legionella DNA. J Clin Microbiol.
41:3452–3453. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tilburg JJ, Nabuurs-Franssen MH, van
Hannen EJ, Horrevorts AM, Melchers WJ and Klaassen CH:
Contamination of commercial PCR master mix with DNA from Coxiella
burnetii. J Clin Microbiol. 48:4634–4635. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brosius J, Dull TJ, Sleeter DD and Noller
HF: Gene organization and primary structure of a ribosomal RNA
operon from Escherichia coli. J Mol Biol. 148:107–127. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Corless CE, Guiver M, Borrow R,
Edwards-Jones V, Fox AJ and Kaczmarski EB: Simultaneous detection
of Neisseria meningitidis, Haemophilus influenzae, and
Streptococcus pneumoniae in suspected cases of meningitis
and septicemia using real-time PCR. J Clin Microbiol. 39:1553–1558.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carroll NM, Adamson P and Okhravi N:
Elimination of bacterial DNA from Taq DNA polymerases by
restriction endonuclease digestion. J Clin Microbiol. 37:3402–3404.
1999.PubMed/NCBI
|
|
22
|
Klaschik S, Lehmann LE, Raadts A, Hoeft A
and Stuber F: Comparison of different decontamination methods for
reagents to detect low concentrations of bacterial 16S DNA by
real-time-PCR. Mol Biotechnol. 22:231–242. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Silkie SS, Tolcher MP and Nelson KL:
Reagent decontamination to eliminate false-positives in
Escherichia coli qPCR. J Microbiol Methods. 72:275–282.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Benjamin J, Singla V, Arora I, Sood S and
Joshi YK: Intestinal permeability and complications in liver
cirrhosis: A prospective cohort study. Hepatol Res. 43:200–207.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aberg F, Helenius-Hietala J, Meurman J and
Isoniemi H: Association between dental infections and the clinical
course of chronic liver disease. Hepatol Res. 44:349–353. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zapater P, Francés R, González-Navajas JM,
de la Hoz MA, Moreu R, Pascual S, Monfort D, Montoliu S, Vila C,
Escudero A, et al: Serum and ascitic fluid bacterial DNA: A new
independent prognostic factor in noninfected patients with
cirrhosis. Hepatology. 48:1924–1931. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hiramatsu K, Harada K, Tsuneyama K, Sasaki
M, Fujita S, Hashimoto T, Kaneko S, Kobayashi K and Nakanuma Y:
Amplification and sequence analysis of partial bacterial 16S
ribosomal RNA gene in gallbladder bile from patients with primary
biliary cirrhosis. J Hepatol. 33:9–18. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Islam D, Bandholtz L, Nilsson J, Wigzell
H, Christensson B, Agerberth B and Gudmundsson G: Downregulation of
bactericidal peptides in enteric infections: A novel immune escape
mechanism with bacterial DNA as a potential regulator. Nat Med.
7:180–185. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hardick J, Won H, Jeng K, Hsieh YH, Gaydos
CA, Rothman RE and Yang S: Identification of bacterial pathogens in
ascitic fluids from patients with suspected spontaneous bacterial
peritonitis by use of broad-range PCR (16S PCR) coupled with
high-resolution melt analysis. J Clin Microbiol. 50:2428–2432.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fagan KJ, Rogers GB, Melino M, Arthur DM,
Costello ME, Morrison M, Powell EE and Irvine KM: Ascites bacterial
burden and immune cell profile are associated with poor clinical
outcomes in the absence of overt infection. PLoS One.
10:e01206422015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rogers GB, van der Gast CJ, Bruce KD,
Marsh P, Collins JE, Sutton J and Wright M: Ascitic microbiota
composition is correlated with clinical severity in cirrhosis with
portal hypertension. PLoS One. 8:e748842013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Krohn S, Böhm S, Engelmann C, Hartmann J,
Brodzinski A, Chatzinotas A, Zeller K, Prywerek D, Fetzer I and
Berg T: Application of qualitative and quantitative real-time PCR,
direct sequencing, and terminal restriction fragment length
polymorphism analysis for detection and identification of
polymicrobial 16S rRNA genes in ascites. J Clin Microbiol.
52:1754–1757. 2014. View Article : Google Scholar : PubMed/NCBI
|