Introduction
Idiopathic congenital nystagmus is one of the most
common oculomotor disorders, characterized by involuntary,
periodic, and predominantly horizontal oscillations of both eyes
(1). The symptoms usually appear
at birth or during the first few months of life. To date, the
pathogenesis of idiopathic congenital nystagmus remains unclear.
Abnormal brain control of the ocular motor system possibly
contributes to this disease. At present, there is no effective
treatment for idiopathic congenital nystagmus, and only very
limited surgical, optical and pharmaceutical therapies are
available improve the symptoms (2,3).
Idiopathic congenital nystagmus exhibits extreme genetic and
clinical heterogeneity. There are three different inheritance
patterns that have been reported, including X-linked idiopathic
congenital nystagmus [Online Mendelian Inheritance in Man (OMIM)
nos: 300628, 300589], autosomal recessive (OMIM no: 257400) and
autosomal dominant (OMIM nos: 164100, 608345 and 193003) (4,5).
Although the inheritance mode is heterogeneous, X-linked idiopathic
congenital nystagmus is the most commonly reported. Three disease
loci for X-linked idiopathic congenital nystagmus have been mapped
to Xp11.4-p11.3 [Nystagmus (NYS)5, OMIM no. 300589] (6), Xp22.3 (NYS6, OMIM no. 300814)
(7) and Xq26-q27 (NYS1, OMIM no.
310700) (8). The four-point-one,
ezrin, radixin, moesin (FERM) domain-containing 7 gene (FRMD7; OMIM
no. 300628), containing 12 exons and encoding a member of the
protein 4.1 superfamily, is credited for the disease loci at
Xq26-q27, which is the main cause of X-linked idiopathic congenital
nystagmus in Asian populations (1,9). As
demonstrated in Table I, >73
mutations responsible for idiopathic congenital nystagmus have been
identified to date. In the present study, two brothers from a
Chinese family who had been diagnosed with idiopathic congenital
nystagmus were evaluated. To identify the causative gene mutations,
the affected patients were first genotyped with microsatellite
markers flanking the FRMD7 locus. Direct sequencing using gene
specific primers then identified the mutations. The aim of this
study was to identify a causative mutation responsible for
idiopathic congenital nystagmus in a Chinese family, which may be
important for the future genetic diagnosis and treatment of
idiopathic congenital nystagmus.
 | Table I.Genetic mutations in the FRMD7 gene
responsible for X-idiopathic congenital nystagmus, identified since
2008. |
Table I.
Genetic mutations in the FRMD7 gene
responsible for X-idiopathic congenital nystagmus, identified since
2008.
| Author, year | Origin | Mutation type | Nucleotide
change | Protein change | (Refs.) |
|---|
| Bai et al,
2017 | China | Missense | c.284G>T | p.R95M | (10) |
| Jia et al,
2017 | China | Missense | c.473T>A | p.I158N | (11) |
| Bai et al,
2017 | China | Missense | c.521A>T | p.D174V | (10) |
| Gupta et al,
2015 | India | Missense | c.556A>G | p.M186V | (12) |
| Jia et al,
2017 | China | Missense | c.580G>T | p.A194S | (11) |
| Bai et al,
2017 | China | Missense | c.586G>A | p.D196N | (10) |
| Jia et al,
2017 | China | Missense | c.605T>A | p.I202N | (11) |
| Li et al,
2011 | China | Missense | c. 623A>G | p. H208R | (13) |
| Liu et al,
2013 | China | Missense | c.635T>C | – | (14) |
| Bai et al,
2017 | China | Missense | c.T766A | p.F2561 | (10) |
| Al-Moallem et
al, 2015 | Belgium | Missense | c.801C>A | – | (15) |
| Thomas et
al, 2008 | China | Missense | c.811T>C | p.C271R | (16) |
| Jia et al,
2017 | China | Missense | c.811T>A | p.C271S | (11) |
| Al-Moallem et
al and Kohmoto et al, 2015 | Belgium, Japan | Missense | c.875T>C | p.L292P | (15,17) |
| Radhakrishna et
al, 2012 | India | Missense | c.A917G | Q305R | (18) |
| Bai et al,
2017 | China | Missense | c.A973G | p.R325G | (10) |
| Choi et al,
2015 | Korean | Missense | c.A>G | p.M1>V | (19) |
| Zhao et al,
2016 | China | Nonsense | c.1090C>T | p.Q364X | (20) |
| AlMoallem et
al, 2015 | Belgium | Frameshift | c.2036del | – | (15) |
| He et al,
2008 | China | Frameshift |
c.1274-1275delTG | – | (21) |
| Bai et al,
2017 | China | Frameshift | c.999delT | p.H333fs | (10) |
| Jia et al,
2017 | China | Slice mutation | c.57+1G>A | – | (11) |
| Hu et al,
2012 | China | Splicing
mutation | c.163-1G>T | – | (22) |
| AlMoallem et
al, 2015 | Belgium | Splice site
mutation | c.497þ5G>A | – | (15) |
| Hu et al,
2011 | China | Splicing
mutation | c.658+1g>t | – | (23) |
| Al-Moallem et
al, 2015 | Belgium | Copy number
variation | 1.4 Mb
deletion | – | (15) |
| Jia et al,
2017 | China | Deletion | c.41_43delAG | – | (11) |
| Al-Moallem et
al, 2015 | Belgium | Deletion | c.660del | – | (15) |
| Jia et al,
2017 | China | Deletion | c.689-690delAG | p.Ser232del | (11) |
| Du et al,
2011 | China | Deletion |
c.1486-1489delTTTT | p.F497fs26X | (24) |
| Bai et al,
2017 | China | Shear mutation | c.162+2T>C | – | (10) |
| Bai et al,
2017 | China | Shear mutation | c.206-1G>A | – | (10) |
| Jia et al,
2017 | China | Insert |
c.1492-1493insA | p.Y498X | (11) |
| Schorderet et
al, 2007 | Switzerland | Variant | c.383-11G>A | – | (25) |
| Schorderet et
al, 2007 | Switzerland | Variant | c.206-20T>C | – | (25) |
Materials and methods
Clinical evaluation and DNA
sampling
The present study was approved by the ethics
committee of Xiamen Eye Center Affiliated Xiamen University
(Xiamen, China) and adhered to the guidelines of the Declaration of
Helsinki. The study participants included the two affected brothers
and their parents, as well as 100 unaffected control subjects (45
male, 55 female; age range 18–56, recruited from 1st January-29th
February 2016); written informed consent was received from all
participants prior to enrolment. All subjects underwent
comprehensive ophthalmologic evaluation, including eye movement
recording using the Metrovision MonPack 3 vision monitoring system
(Metrovision, Lille, France), fundus examination, and retinal
optical coherence tomography (OCT) imaging. Peripheral venous blood
samples (2 ml) were obtained from the affected brothers, their
unaffected parents, and the 100 controls.
Mutational analysis and
multiple-sequence alignment
The coding exons and splice junctions of the FRMD7
gene were amplified via polymerase chain reaction (PCR) using PCR
primers (Table III) that have
been previously published (14).
PCR products were purified and directly sequenced using an ABI
A3730 Automated Sequencer (Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The suspected mutation
identified in the two affected brothers was further confirmed as a
mutation by sequencing of other family members and control males
from the same ethnic background. Mutation descriptions followed the
nomenclature recommended by the Human Genomic Variation Society
(www.hgvs.org/). The amino acid sequences of the
FRMD7 protein from various species were obtained from the National
Center for Biotechnology Information website (www.ncbi.nlm.nih.gov/). The amino acid sequences of
the FRMD7 gene from different species were aligned by the CLC
Sequence Viewer 6 (CLC bio A/S; www.clcbio.com). The potential impact of amino acid
substitution on the function of the FRMD7 protein was predicted
using online tools, including the PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/)
and SIFT (http://sift.jcvi.org/) programs.
| Table III.Primers and PCR conditions for
FRMD7. |
Table III.
Primers and PCR conditions for
FRMD7.
| Exon | Sequence
(5′>3′) | Annealing temp | Length |
|---|
| FRMD7 E1 |
CCCTTGGGTGTGCATTACTT | 58°C | 308 bp |
|
|
TTGCTCTCTAATGGGCTGTTC |
|
|
| FRMD7 E2 |
CAGAGAGTCTTGTGGCTTCTAC | 58°C | 380 bp |
|
|
TGGCCAAGGTGACTGTTAAT |
|
|
| FRMD7 E3 |
CCACCTATTTGACATTGCTGTTT | 58°C | 399 bp |
|
|
CCAGCAGCATGATTTCTTTCATC |
|
|
| FRMD7 E4 |
AGTCTGTAGGAGGGAGTGATG | 58°C | 418 bp |
|
|
TCCTGTAGTCTCCACCCTTT |
|
|
| FRMD7 E5 |
GCCTGCCAAAGTGTTCAATC | 58°C | 455 bp |
|
|
GCCATGCTGTTTCTCTCTATCT |
|
|
| FRMD7 E6+7 |
TTTGATGGAGGACAAGGGTATG | 58°C | 660 bp |
|
|
CCCTTTCTGGCTGGTGATAAT |
|
|
| FRMD7 E8 |
CATCCTTGTATCCCACCTGAAATG | 58°C | 590 bp |
|
|
CTGGTCACTCCCATTTGACTTA |
|
|
| FRMD7 E9 |
GTTTCTGGCAGAGAAGAGTGAGT | 58°C | 638 bp |
|
|
CATCTTCCTCCCTCCTAGTTAG |
|
|
| FRMD7 E10+11 |
CTCTGCCTGGTCCTTGAATAAG | 58°C | 562 bp |
|
|
CCAGGAAGCTAACCTACTCAAA |
|
|
| FRMD7 E12a |
CTGGAAGTAGGATGGCATTGAG | 58°C | 845 bp |
|
|
TTGGAGTACTTGCAGGTCTTG |
|
|
| FRMD7 E12b |
CAGGTCAGCAGGTTGGTATTAT | 58°C | 916 bp |
|
|
GAGGAGAGGGCATGTTCTAAAG |
|
|
Protein structural modeling
The secondary structure of the human wild type was
predicted by the Phyre2 program (www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index,
2.0) and 3D structures of the human wild type and mutant FRMD7
proteins were predicted using Iterative Threading ASSEmbly
Refinement (I-TASSER; zhanglab.ccmb.med.umich.edu/I-TASSER), and the results
saved in a Protein Data Bank (PDB) file format. The PDB files
obtained for the two proteins were then analyzed using PyMOL1.7.0.0
(pymol.org/2/) to visualize the structural
variations.
Results
Phenotypic characterization of the
affected brothers
There were two affected individuals in this
two-generation family. The probands (II:1 and II:2) presented with
nystagmus as the first symptom. The clinical findings are
summarized in Table II. Mental
retardation, night blindness and photophobia were not observed in
either of the two affected individuals. No head nodding was
observed in the two affected patients, although one exhibited a
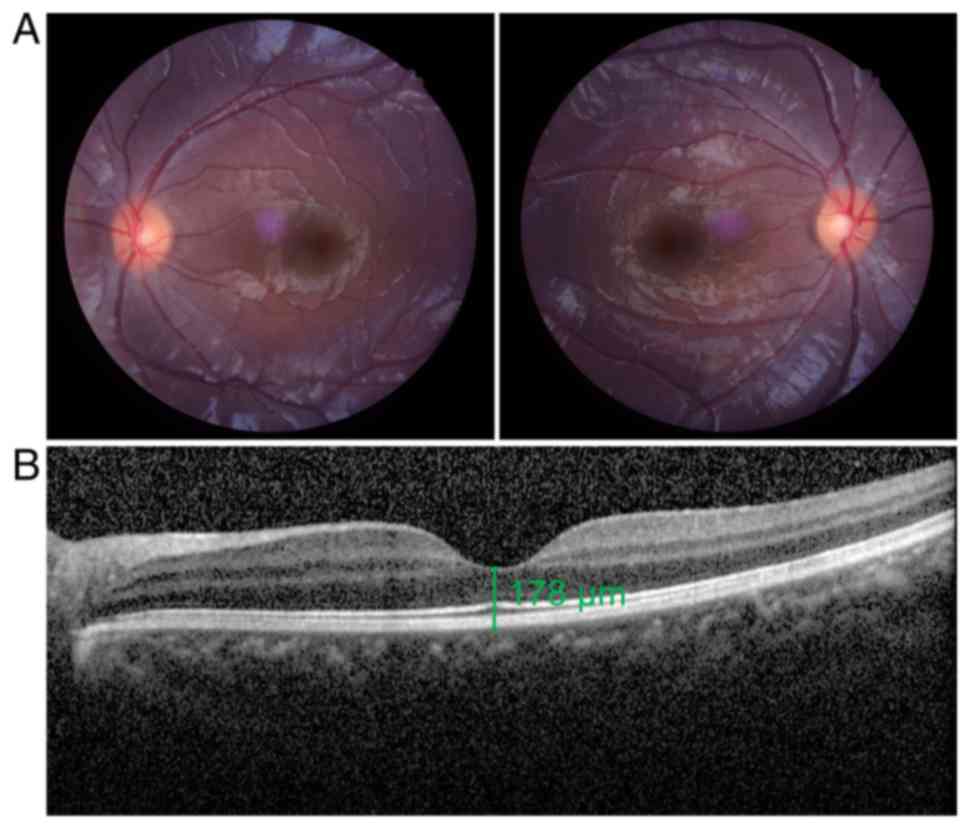
clear head tilt. As demonstrated in Fig. 1, a representative ophthalmological
examination results of both probands suggested that there was no
indication of other ocular anomalies. OCT imaging demonstrated that
the retinal nerve fiber layer thickness was within the normal
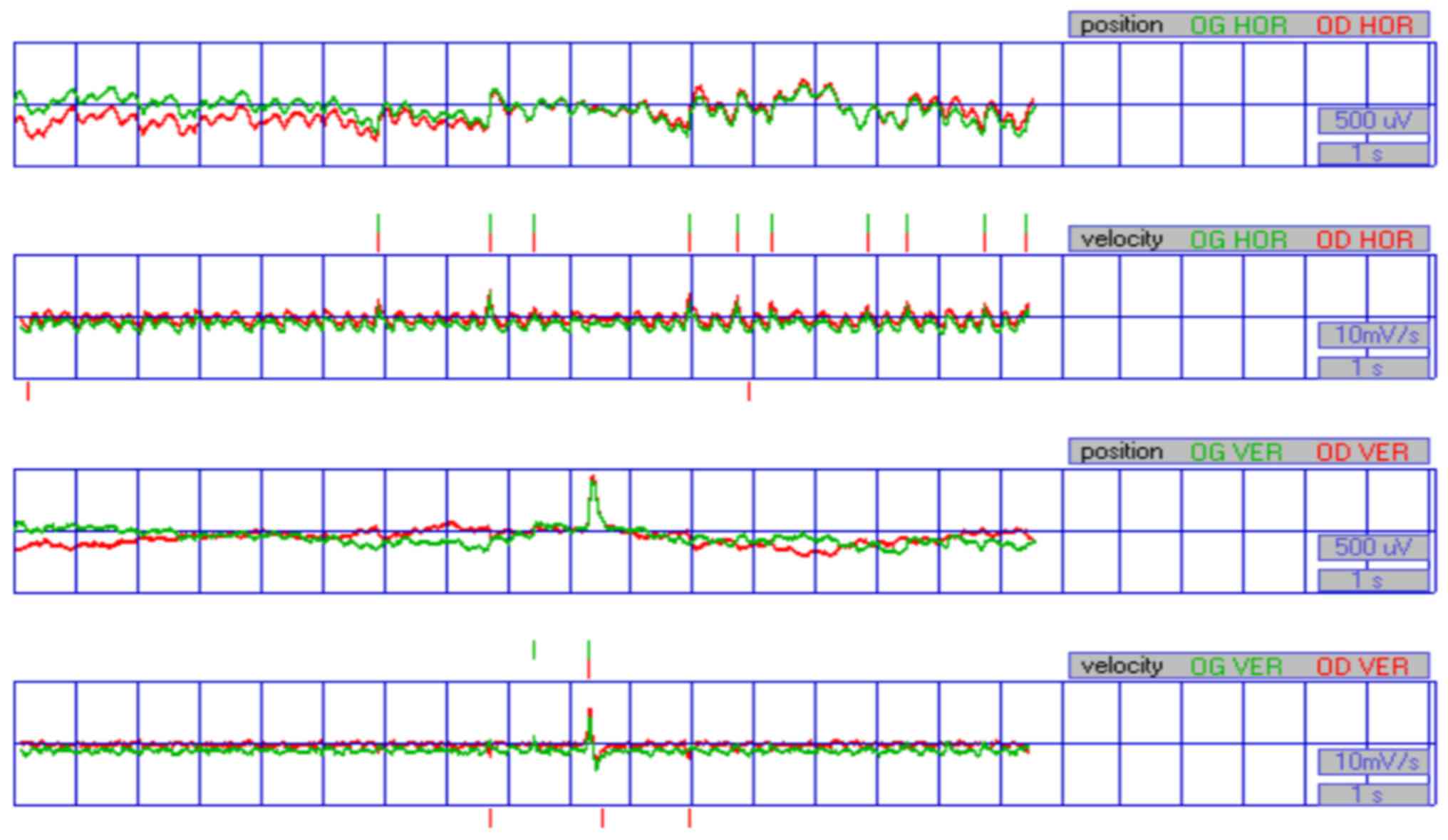
range. Eye movement recordings revealed that nystagmus of the
proband 1 was predominantly a horizontal waveform (Fig. 2). The ocular oscillation could be
slightly slowed at a right gaze of 5°. Patients in this family were
confirmed to have X-linked congenital nystagmus based on the
comprehensive ophthalmologic examination and family history.
| Table II.Clinical features of individuals with
idiopathic congenital nystagmus in the present study. |
Table II.
Clinical features of individuals with
idiopathic congenital nystagmus in the present study.
| Phenotype | I:1 | II:2 |
|---|
| Sex | Male | Male |
| Age (years) | 7 | 6 |
| Age at diagnosis
(years) | 4 | 3 |
| Visual
activity | OD20/100 | OD20/50 |
| at
presentation | OS20/80 | OS20/50 |
| Nystagmus | Horizontal | Horizontal |
| Abnormal head
movement | Negative | Clear head
tilt |
| Neurological
findings | Normal | Normal |
Mutation analysis and protein
structure modeling
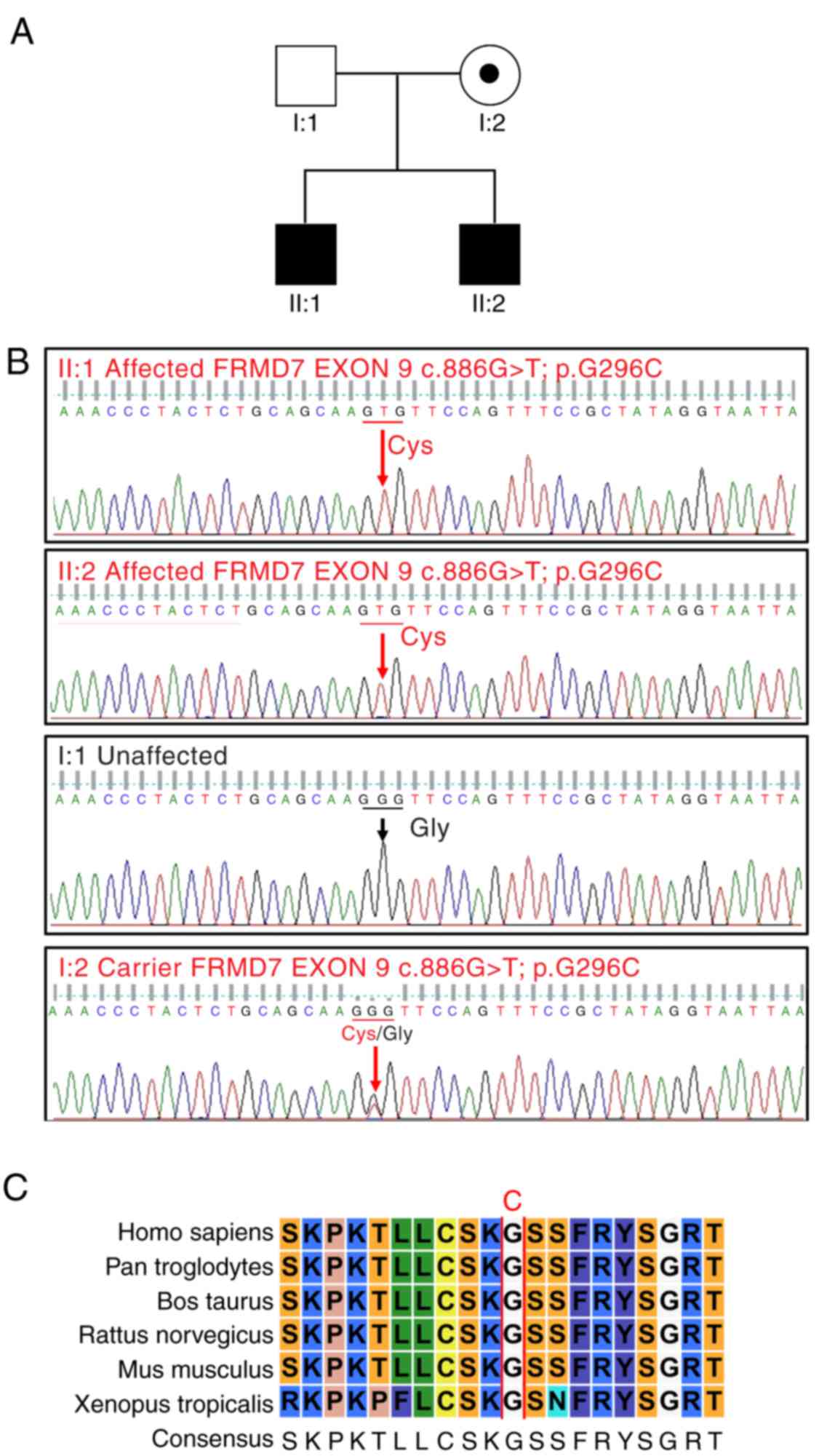
A pedigree analysis of the patients is presented in
Fig. 3A. Sequencing the FRMD7 gene
revealed a G to T transition (c.886G>T) in exon 9 (Fig. 3B), which caused a conservative
substitution of a glycine to a cysteine at codon 296 (p.G296C).
This mutation was identified in both affected individuals and was
present in the mother, but was absent in the father and in all
control subjects. The FRMD7 p.G296C mutation is not present in any
single nucleotide polymorphism database. In addition, this mutation
was predicted with high confidence to be ‘probably damaging’ and
‘damaging’ by Polyphen-2 (score, 0) and SIFT (score, 2.54),
respectively. Furthermore, multiple sequence alignment of the FRMD7
gene from different species was performed. The results demonstrated
that codon 296, where the mutation (p.G296C) occurred, was located
within a phylogenetically conserved region (Fig. 3C). Considering the above, it is
reasonable to conclude that the p.G296C substitution is a causative
mutation with the disease.
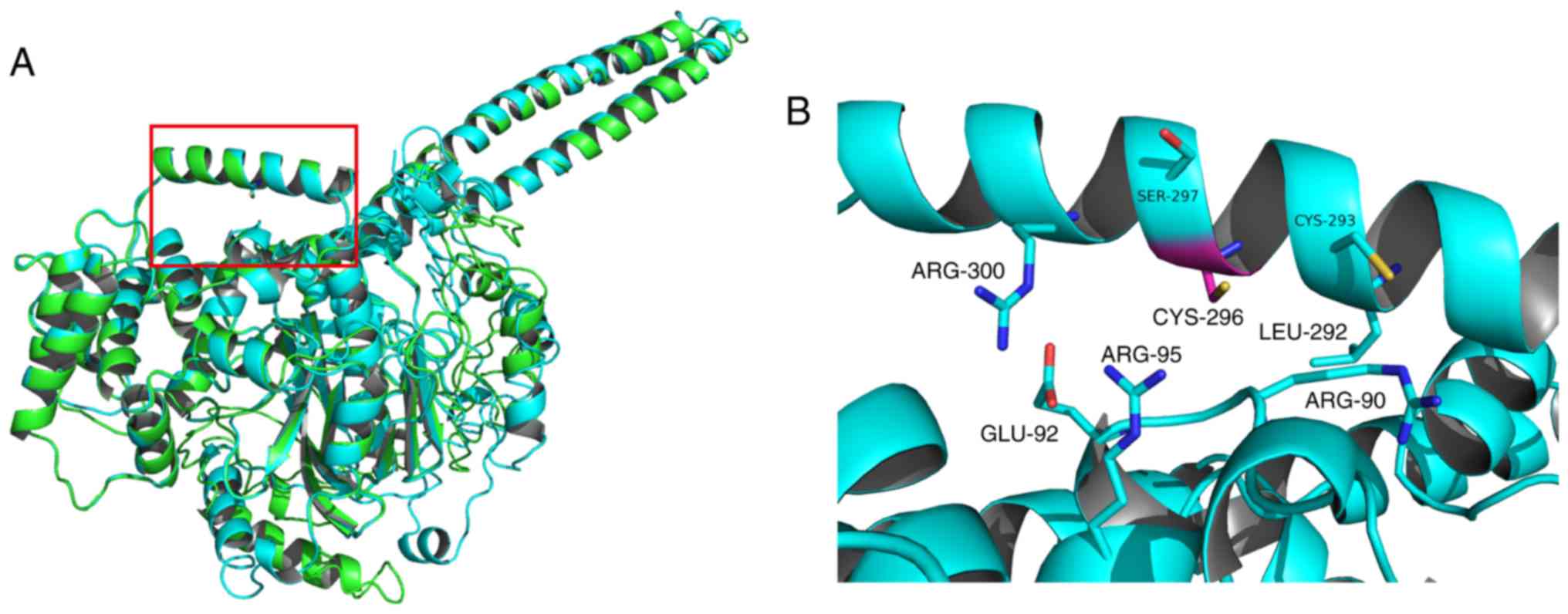
Using the Phyre2 program (www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index,
2.0), the secondary structure of the wild type FRMD7 protein was
predicted, the three-dimensional structure of the wild type FRMD7
protein (1–336) was modeled, and p.G296C was mapped within the
structure (Fig. 4). The results
demonstrated that, with the p.G296C mutation, the amino acid
residues at 296 were largely surrounded by hydrophilic polar amino
acid residues. Substitution of glycine to cysteine at codon 296
increased the regional polarity of the amino acids. Furthermore,
the thiol group of cysteine is a reducing group, able to change the
local electron density. Changes in hydrophobicity and electron
density may cause structural instability of the FRMD7 protein
leading to protein dysfunction, thus causing idiopathic congenital
nystagmus, but this requires further investigation.
Discussion
Idiopathic congenital nystagmus is an inherited
ocular disorder that develops at birth or shortly after and
persists throughout the patient's life (26). Nystagmus has an estimated
prevalence of 6.72 per 100,000 for individuals younger than 19
years (27). Idiopathic congenital
nystagmus is associated with significant negative psychosocial and
functional consequences, including negative social stigma and poor
visual function scores (28,29).
There is a great interest in the treatment and early diagnosis of
idiopathic congenital nystagmus. However, there is currently no
effective treatment for idiopathic congenital nystagmus, and only
limited surgical, optical and pharmaceutical therapies have been
tested for the improvement of nystagmus waveforms and visual acuity
(2,3). The early diagnosis of congenital
nystagmus is critical for facilitating habilitation, providing
genetic counseling, and potential gene therapies in the future
(30). The diagnosis of idiopathic
congenital nystagmus remains being a challenge and potential
patients are usually required to undergo numerous examinations
(31). Nystagmus is currently
clinically described in terms of its peak-to peak amplitude,
frequency, mean velocity and waveform (32). In the present study, idiopathic
congenital nystagmus in two brothers from a Chinese family was
first diagnosed using eye movement recordings and a comprehensive
ophthalmologic examination.
Recently, a next-generation sequencing panel has
been used for the early diagnosis of idiopathic congenital
nystagmus (31). Comprehensive
understanding of the mutation spectrum is the foundation for
efficient early genetic diagnosis. To identify the causative
mutations in the two affected brothers, direct sequencing of
genomic DNA samples from the affected brothers was conducted. The
results revealed a G to T transition (c.886G>T) in exon 9 of the
FRMD7 gene, which resulted in a conservative substitution of a
glycine to a cysteine at codon 296 (p.G296C), leading to idiopathic
congenital nystagmus. So far, >40 mutations causing idiopathic
congenital nystagmus have been identified in the FRMD7 gene,
predominantly concentrated in two key regions: The FERM and FERM
adjacent domains (9).
Identification of the mutations is not only critical for early
diagnosis, but also for understanding the pathogenesis of
idiopathic congenital nystagmus. The genetic etiology of idiopathic
congenital nystagmus is not yet fully understood, especially
regarding sporadic cases. Identifying previously unknown genetic
mutations involved in idiopathic congenital nystagmus will allow
the development of cell and animal-based models to help classify
idiopathic congenital nystagmus and guide future therapies. The
present findings provide further insight into the genetic spectrum
for idiopathic congenital nystagmus, which is useful for the
earlier genetic diagnosis, genetic counseling, and potential future
gene therapy of patients with idiopathic congenital nystagmus.
In summary, a novel mutation in the FRMD7 gene
causing idiopathic congenital nystagmus was identified using direct
sequencing. G to T transition (c.886G>T) in exon 9 that resulted
in the conservative substitution of a glycine to a cysteine at
codon 296 (p.G296C) was identified in two affected brothers from a
Chinese family. The present study expanded the already known gene
mutation spectrum for idiopathic congenital nystagmus, providing
more evidence for the genetic heterogeneity associated with
idiopathic congenital nystagmus. This novel finding is important
for faster diagnosis, prenatal testing, and improving the
understanding of the molecular pathogenesis of idiopathic
congenital nystagmus.
Acknowledgements
Not applicable.
Funding
The present study was financially supported by
grants from the Department of Science and Technology of Fujian
Province (Science and Technology Project, Grant No. 2018D0015), the
Natural Science Foundation of China (grant no. 81270999/H1204;
www.nsfc.gov.cn/), Professor Academic Development
Fund of Fujian Medical University (grant no. JS14019; www.nsfc.gov.cn/), and the Science Technology project
of Zhejiang Province (grant no. 2017C37176; www.zjkjt.gov.cn).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ, FG and YZ designed the experiments. YX, YH, TY,
MP, YY and WF performed the experiments. YX, JZ and YZ analyzed the
results and wrote the manuscript.
Ethics approval and consent to
participate
All procedures performed in the present study
involving human participants were in accordance with the ethical
standards of the institutional and/or national research committee
and with the 1964 Helsinki declaration and its later amendments or
comparable ethical standards. Written informed consent for
participation in the study was obtained from all participants or
their legal guardians prior to their inclusion.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tarpey P, Thomas S, Sarvananthan N, Mallya
U, Lisgo S, Talbot CJ, Roberts EO, Awan M, Surendran M, McLean RJ,
et al: Mutations in FRMD7, a newly identified member of the FERM
family, cause X-linked idiopathic congenital nystagmus. Nat Genet.
38:1242–1244. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Serra A, Dell'Osso LF, Jacobs JB and
Burnstine RA: Combined gaze-angle and vergence variation in
infantile nystagmus: Two therapies that improve the
high-visual-acuity field and methods to measure it. Invest
Ophthalmol Vis Sci. 47:2451–2460. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Balzer BWR, Catt CJ, Bou-Abdou M and
Martin FJ: Visual acuity improves in children and adolescents with
idiopathic infantile nystagmus. Asia Pac J Ophthalmol (Phila).
7:99–101. 2018.PubMed/NCBI
|
|
4
|
Kerrison JB, Arnould VJ, Barmada MM,
Koenekoop RK, Schmeckpeper BJ and Maumenee IH: A gene for autosomal
dominant congenital nystagmus localizes to 6p12. Genomics.
33:523–526. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu Y, Zhuang J, Ge X, Zhang X, Wang Z,
Sun J, Yang J and Gu F: Identifcation of a novel mutation p.I240T
in the FRMD7 gene in a family with congenital nystagmus. Sci Rep.
3:30842013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cabot A, Rozet JM, Gerber S, Perrault I,
Ducroq D, Smahi A, Souied E, Munnich A and Kaplan J: A gene for
X-linked idiopathic congenital nystagmus (NYS1) maps to chromosome
xp11.4-p11.3. Am J Hum Genet. 64:1141–1146. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bassi MT, Schiaffino MV, Renieri A, De
Nigris F, Galli L, Bruttini M, Gebbia M, Bergen AA, Lewis RA and
Ballabio A: Cloning of the gene for ocular albinism type-1 from the
distal short arm of the × chromosome. Nat Genet. 10:13–19. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kerrison JB, Vagefi MR, Barmada MM and
Maumenee LH: Congenital motor nystagmus linked to xq26-q27. Am J
Hum Genet. 64:600–607. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Watkins RJ, Thomas MG, Talbot CJ, Gottlob
I and Shackleton S: The role of FRMD7 in idiopathic infantile
nystagmus. J Ophthalmol. 2012:4609562012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bai D, Shi W, Qi Z, Li W, Wei A, Cui Y, Li
C and Li L: Clinical feature and waveform in infantile nystagmus
syndrome in children with FRMD7 gene mutations. Sci China Life Sci.
60:707–713. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jia X, Zhu X, Li Q, Jia X, Li S and Guo X:
Novel mutations of FRMD7 in chinese patients with congenital motor
nystagmus. Mol Med Rep. 16:1753–1758. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gupta S, Pathak E, Chaudhry VN, Chaudhry
P, Mishra R, Chandra A, Mukherjee A and Mutsuddi M: A novel
mutation in FRMD7 causes X-linked idiopathic congenital nystagmus
in a north indian family. Neurosci Lett. 597:170–175. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li N, Wang X, Wang Y, Wang L, Ying M, Han
R, Liu Y and Zhao K: Investigation of the gene mutations in two
chinese families with X-linked infantile nystagmus. Mol Vis.
17:461–468. 2011.PubMed/NCBI
|
|
14
|
Liu Z, Mao S, Pu J, Ding Y, Zhang B and
Ding M: A novel missense mutation in the FERM domain containing 7
(FRMD7) gene causing X-linked idiopathic congenital nystagmus in a
chinese family. Mol Vis. 19:1834–1840. 2013.PubMed/NCBI
|
|
15
|
Al-Moallem B, Bauwens M, Walraedt S,
Delbeke P, De Zaeytijd J, Kestelyn P, Meire F, Janssens S, van
Cauwenbergh C, Verdin H, et al: Novel FRMD7 mutations and genomic
rearrangement expand the molecular pathogenesis of X-linked
idiopathic infantile nystagmus. Invest Ophthalmol Vis Sci.
56:1701–1710. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thomas S, Proudlock FA, Sarvananthan N,
Roberts EO, Awan M, McLean R, Surendran M, Kumar AS, Farooq SJ,
Degg C, et al: Phenotypical characteristics of idiopathic infantile
nystagmus with and without mutations in FRMD7. Brain.
131:1259–1267. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kohmoto T, Okamoto N, Satomura S, Naruto
T, Komori T, Hashimoto T and Imoto I: A FRMD7 variant in a japanese
family causes congenital nystagmus. Hum Genome Var. 2:150022015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Radhakrishna U, Ratnamala U, Deutsch S,
Bartoloni L, Kuracha MR, Singh R, Banwait J, Bastola DK, Johar K,
Nath SK and Antonarakis SE: Novel homozygous, heterozygous and
hemizygous FRMD7 gene mutations segregated in the same
consanguineous family with congenital X-linked nystagmus. Eur J Hum
Genet. 20:1032–1036. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Choi JH, Shin JH, Seo JH, Jung JH and Choi
KD: A start codon mutation of the FRMD7 gene in two korean families
with idiopathic infantile nystagmus. Sci Rep. 5:130032015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao H, Huang XF, Zheng ZL, Deng WL, Lei
XL, Xing DJ, Ye L, Xu SZ, Chen J, Zhang F, et al: Molecular genetic
analysis of patients with sporadic and X-linked infantile
nystagmus. BMJ Open. 6:e0106492016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
He X, Gu F, Wang Z, Wang C, Tong Y, Wang
Y, Yang J, Liu W, Zhang M and Ma X: A novel frameshift mutation in
FRMD7 causing X-linked idiopathic congenital nystagmus. Genet Test.
12:607–613. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu Y, Shen J, Zhang S, Yang T, Huang S and
Yuan H: A novel splicing mutation of the FRMD7 gene in a Chinese
family with X-linked congenital nystagmus. Mol Vis. 18:87–91.
2012.PubMed/NCBI
|
|
23
|
Hu J, Liang D, Xue J, Liu J and Wu L: A
novel GPR143 splicing mutation in a Chinese family with X-linked
congenital nystagmus. Mol Vis. 17:715–722. 2011.PubMed/NCBI
|
|
24
|
Du W, Bu J, Dong JM, Jia Y, Li J, Liang C,
Si S and Wang L: A novel frame-shift mutation in FRMD7 causes
X-linked idiopathic congenital nystagmus in a Chinese family. Mol
Vis. 17:2765–2768. 2011.PubMed/NCBI
|
|
25
|
Schorderet DF, Tiab L, Gaillard MC, Lorenz
B, Klainguti G, Kerrison JB, Traboulsi EI and Munier FL: Novel
mutations in FRMD7 in X-linked congenital nystagmus. Mutation in
brief #963. Online. Hum Mutat. 28:5252007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li N, Wang L, Cui L, Zhang L, Dai S, Li H,
Chen X, Zhu L, Hejtmancik JF and Zhao K: Five novel mutations of
the FRMD7 gene in Chinese families with X-linked infantile
nystagmus. Mol Vis. 14:733–738. 2008.PubMed/NCBI
|
|
27
|
Nash DL, Diehl NN and Mohney BG: Incidence
and types of pediatric nystagmus. Am J Ophthalmol. 182:31–34. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sarvananthan N, Surendran M, Roberts EO,
Jain S, Thomas S, Shah N, Proudlock FA, Thompson JR, McLean RJ,
Degg C, et al: The prevalence of nystagmus: The leicestershire
nystagmus survey. Invest Ophthalmol Vis Sci. 50:5201–5206. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Penix K, Swanson MW and DeCarlo DK:
Nystagmus in pediatric patients: Interventions and patient-focused
perspectives. Clin Ophthalmol. 9:1527–1536. 2015.PubMed/NCBI
|
|
30
|
Holmström G, Bondeson ML, Eriksson U,
Åkerblom H and Larsson E: ‘Congenital’ nystagmus may hide various
ophthalmic diagnoses. Acta Ophthalmol. 92:412–416. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thomas MG, Maconachie G, Sheth V, McLean
RJ and Gottlob I: Development and clinical utility of a novel
diagnostic nystagmus gene panel using targeted next-generation
sequencing. Eur J Hum Genet. 25:725–734. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Clement RA, Whittle JP, Muldoon MR, Abadi
RV, Broomhead DS and Akman O: Characterisation of congenital
nystagmus waveforms in terms of periodic orbits. Vision Res.
42:2123–2130. 2002. View Article : Google Scholar : PubMed/NCBI
|