Introduction
Coronary artery disease is a major problem worldwide
account for the greatest proportion of cardiovascular diseases
which cause 31.5% global deaths (1,2).
With advances in the prevention, diagnosis and management of
cardiovascular diseases, the mortality rate of acute myocardial
infarction (AMI) has decreased. However, chronic heart failure
(CHF) remains a major cause of morbidity and mortality in patients
with post-infarcted hearts (3,4). In
addition, treatments for CHF impose a heavy burden on society and
monopolize numerous health care resources in industrialized
countries (5). Therefore, the
search for novel pharmacological approaches in the prevention and
treatment of CHF is urgently needed.
Alcohol dehydrogenase 2 (ALDH2) is a member of the
ALDH gene family and is a key enzyme in the metabolism of
acetaldehyde and other toxic aldehydes (6). The cardioprotective role of ALDH2 in
cardiac ischemic events was first reported by Chen et al
(7). Subsequent studies have
demonstrated that ALDH2 provides beneficial effects in alcoholic
cardiomyopathy, ischemia-reperfusion (I/R) injury and heart failure
(8–10). Knockout of ALDH2 was reported to
exacerbate cardiac contractile dysfunction and promote apoptosis
induced by endoplasmic reticulum stress induction, as manifested by
the alterations in the ejection fraction and fractional shortening
(11). Activation or
overexpression of ALDH2 was demonstrated to protect against cardiac
injury by diminishing AMI size, ameliorating cardiac dysfunction
and preventing reperfusion arrhythmias (6,12,13).
Alda-1 is a selective agonist of ALDH2 (14), which increases productive
substrate-enzyme interactions and protects ALDH2 from
substrate-induced inactivation by binding near the exit of the
substrate-binding tunnel (15).
Previous studies have demonstrated that ALDH2 activation exhibits
beneficial effects on I/R injury (9). In rats pre-treated with Alda-1 for 5
min in the left ventricle prior to ischemia, infarction damage was
reduced by ~60% by clearing the toxic reactive aldehydes, such as
4-hydroxynonenal (4-HNE) the key mediator leading to oxidative
stress (7). Additionally, in a rat
model of MI, sustained treatment with Alda-1, either for 4 weeks
starting at 24 h post-MI or for 6 weeks starting at 4 weeks
following permanent MI, maintained mitochondrial bioenergetic
status, prevented excessive oxidative stress and improved
ventricular function and remodeling (10,13).
Improvement of long-term survival is the key goal of
CHF drug therapy. Although certain therapeutic agents exhibited
favorable effects on ventricular function and remodelling, they
were associated with increased mortality rates. For example, the
tumor necrosis factor antagonist etanercept is a cytokine inhibitor
that is able to reverse ventricular remodeling over 3-months
treatment; however, etanercept failed to demonstrate any long-term
benefit in a 6-month long-term study (16,17).
Peroxisome proliferator-activated receptor-γ (PPAR-γ) serves a
prominent role in cardiac function, but the effects of PPAR-γ
agonists in cardiac diseases remain controversial, as chronic
PPAR-γ therapy may be deleterious (18). Systolic improvement therapy with
digoxin, which reversed remodelling in dilated cardiomyopathy, was
associated with a significant increase in mortality from all
cardiac disease-associated causes among patients with atrial
fibrillation as well as heart failure (19). The effects of long-term treatment
with Alda-1 on CHF post-MI remain unclear. In the present study,
Alda-1 treatment began 1 week following AMI and was sustained for
20 weeks. The effects were determined by investigating the
mortality rate, cell apoptosis and collagen fiber formation, as
well as toxic aldehyde clearance.
Materials and methods
Animals and surgical procedures
All experimental procedures involving animals were
approved by the Animal Care and Use Committee at Southern Medical
University (Guangzhou, China). A total of 66 specific-pathogen-free
male Wistar rats (200–250 g, 7 weeks old) were obtained from the
Experimental Animal Center of Southern Medical University after
animal ethic approval (Guangzhou, China; Animal Quarantine
Conformity Certificate no. 4402102052). The rats were maintained in
a room with a 12-h light/dark cycle, constant temperature of
22–26°C, constant humidity of 40–60% and free access to tap water
and food. To induce MI, left coronary artery ligation was performed
as previously described (20). A
total of ~20% of the rats (n=9) failed to survive the first week
following ligation procedure owing to acute heart failure or
malignant arrhythmia. All rats were examined by echocardiography
1-week post-MI surgery to eliminate unqualified rats that either
did not develop sufficient MI (i.e. left ventricular ejection
fraction >50% compared with the Sham group) or with severe
complications. The rest of the successfully ligated rats were
randomly assigned into two experimental groups: The MI group, in
which MI was induced and rats were treated orally with 0.9% normal
saline (1 ml/100 g/day), and the Alda-1-treated group, in which
MI-induced rats were treated orally with Alda-1 (D43490, Merch
Millipore, Darmstadt, Germany; 16 mg/kg/day) started from 1 week
after MI surgery (14).
Sham-operated rats served as the control group; they underwent
surgery, but not left coronary artery ligation, and were treated
orally with 0.9% normal saline (1 ml/100 g/day). Each group
consisted of 18 rats and all animals underwent gavage
administration for 20 weeks. All the procedures in this animal
study were performed in accordance with the approval by the
Institute of Animal Care and Use Committee of Southern Medical
University. All the rats were weighed (body weight, BW), performed
echocardiography and then euthanized with pentobarbitone sodium
after 20 weeks treatment. Heart tissue was harvested, weighted
(Heart weight, HW) and separated into two parts, which were snap
froze in liquid nitrogen (stored at −80°C, for protein, enzyme and
biomarker assay), and fixed in 10% neutral buffered formalin,
embedded and cut into paraffin section afterwards (stored at room
temperature for histopathology, immunohistochemistry and TUNEL
apoptosis assay).
Transthoracic echocardiographic
measurements
A non-invasive transthoracic echocardiography method
was used to assess heart function at 1 week following surgery to
exclude rats that failed in developing MI, and evaluate the
morphology and function of the left ventricle at 20 week before
rats' sacrifice as previously described (20).
Histological examination
The heart was fixed in 10% neutral-buffered formalin
at room temperature for 48 h, embedded in paraffin and cut into 5
µm sections. The sections were subjected to hematoxylin & eosin
(H&E) and Mallory's Trichrome staining, following standard
procedures (21). All
histopathological alterations were evaluated by two investigators
that were blinded to the study. Five randomly selected fields from
each section were examined at magnification, ×400 and analyzed
using NIS-Elements F 3.2 software accompanied with microscope
(Nikon ECLIPS Ti-S, Japan). In H&E staining paraffin sections,
myocardial size was measured on the cross-section profile with
idealized outline. In the Mallory's Trichrome staining paraffin
sections, collagen volume fractions were semi-quantified with the
quotient of the area of collagen divided by the total area occupied
by heart tissue in each field.
ALDH2 enzymatic activity
ALDH2 Enzymatic Activity Assay kit (GenMed
Scientifics Inc., Wilmington, DE, USA) was used to measure ALDH2
activity in tissue lysates, according to the manufacturer's
protocol. Previously deep-frozen heart tissues (10 mg) were ground
to a powder under liquid nitrogen and homogenized. The homogenates
were sonicated on ice and then centrifuged at 10,000 × g for 30 min
at 4°C. ALDH2 enzymatic activity was measured at 25°C in 1 ml
reaction system containing 33 mM sodium pyrophosphate (pH 8.8), 0.8
mM NAD+, 15 µM propionaldehyde and 0.1 ml tissue
homogenate. Reduced nicotinamide-adenine dinucleotide phosphate
(NADH) production was determined spectrophotometrically by
monitoring the alterations in absorbance intensity at 340 nm every
30 sec for 5 min. The ALDH2 reaction rates were expressed as µmol
NADH/min/mg protein.
Immunohistochemistry
Immunohistochemical staining of 4-HNE and collagen
types I and III was performed using the streptavidin peroxidase
method. Polyclonal immunoglobulin G antibodies against 4-HNE
(catalog no. ab46545, Abcam, Cambridge, UK), collagen type I
(catalog no. ab34710, Abcam, Cambridge, UK) and collagen type III
(catalog no. ab7778, Abcam, Cambridge, UK) were used at 1:100
dilution and incubated at 4°C overnight. Staining procedures were
using StreptAvidin-Biotin Complex (SABC) Kit (SA1022, Boster
Corporation, Wuhan, China) and DAB kit (AR1022, Boster Corporation,
Wuhan, China) following the manufacturer's protocol. Nuclei were
counterstained with H&E staining. Five randomly selected fields
from each section were examined at magnification, ×400 and analyzed
using NIS-Elements F 3.2 software accompanied with microscope
(Nikon ECLIPS Ti-S, Japan). The positive contents were presented by
the percentage of immunoreactive stained area, which were stained
into brown. 4-HNE accumulation was calculated as the percentage of
4-HNE-positive stained cells/total cells.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) apoptosis assay
Apoptosis was detected using the TUNEL Detection kit
(Roche Diagnostics GmbH, Mannheim, Germany), according to the
manufacturer's protocol. The myocardium was stained with myosin-7
(MYH7; 1:200, cat. no. sc-168678, Santa Cruz Biotechnology, Inc.,
Dallas, Texas, USA) overnight at 4°C, and nuclei were stained with
DAPI (0.5 µg/ml, cat. no. 10236276001, Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) for 5 min at room temperature. Fluorescence was
detected by confocal microscopy. Whole cells with blue-colored
nuclei were considered apoptotic. The results were
semi-quantitatively scored by taking an average of the number of
apoptotic cells per field at ×400 magnification; five fields were
evaluated per tissue sample and cardiomyocyte apoptosis was
represented as apoptosis index (AI), and calculated as follows:
AI=number of TUNEL-positive cells/total number of cells.
Western blotting
Heart tissue (20 mg) protein samples were extracted
and quantified, according to methods reported previously (22). The protein lysates (30 µg) were
separated by 10% SDS-PAGE and electrotransferred onto
polyvinylidene fluoride membranes and blocked with 5% nonfat milk
at room temperature for 1 h. The membranes were incubated overnight
with primary antibodies including anti-ALDH2 antibody (catalog no.
3221-1, Epitomics; Abcam), anti-4-HNE antibody (catalog no.
ab46545; Abcam), anti-GAPDH catalog no. 5174, Cell Signaling
Technology, Inc., Danvers, MA, USA) and anti-cleaved-caspase-3
(catalog no. 9661, Cell Signaling Technology, Inc., Danvers, MA,
USA); all with dilution 1:2,000 at 4°C, followed by HRP-conjugated
secondary antibodies (catalog no. 7074, Cell Signaling Technology,
Inc., Danvers, MA, USA) with dilution 1:2,000 at room temperature
for 1 h. Protein bands were visualized by Enhanced
Chemiluminescence detection (GE Healthcare Bio-Sciences,
Pittsburgh, PA, USA). Images were captured and documented with the
Image Station 2000MM CCD system (Kodak, Rochester, NY, USA).
Caspase-3 activity assay
A caspase-3 activity assay was performed using a
colorimetric Caspase-3 Activity Assay kit (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China), according to the
manufacturer's protocol. Briefly, 200 µg tissue lysate was combined
with 100 µl reaction buffer containing 5 µl caspase-3 substrate
DEVD-pNA (4 mM), 1% NP-40, 20 mM Tris-HCl (pH 7.5), 137 mM
n-acetyl-cysteine and 10% glycerol. The lysates were incubated at
37°C for 2 h in the dark and the absorbance was measured at a
wavelength of 405 nm using a microplate reader, as previously
described (23).
Serum B-type natriuretic peptide (BNP)
concentration assay
The Rat BNP ELISA kit (cat. no. Ab108816, Abcam) was
used to measure serum BNP, according to the manufacturer's
protocol. Blood samples were collected from the abdominal aorta
into plain blood collection tubes prior to the rats being
sacrificed. Serum was separated from whole blood following clot
formation and stored at −80°C. Briefly, serum samples and BNP
standards were captured by a BNP specific antibody, which had been
pre-coated onto the bottom of 96-well plates, and subsequently
detected with a BNP specific biotinylated detection antibody, which
was linked with a streptavidin-peroxidase conjugate. The chromogen
substrate was catalysed by streptavidin-peroxidase to visualize
colours into blue. The reactions were stopped and the absorbance
was read on microplate reader at 450 nm wavelength to determine the
final concentration based on the standard curve. Each sample was
assayed with duplicates.
Statistical analysis
Data were presented as the mean ± standard
deviation. Statistical analysis was performed using GraphPad Prism
7.0 (GraphPad Software, Inc., La Jolla, CA, USA). The statistical
significance of each variable was estimated using one-way analysis
of variance followed by a Bonferroni post hoc correction between
all groups. Comparison of survival curves was performed by
Kaplan-Meier and the Mantel-Cox log-rank test. P<0.05 was
considered to indicate a statistically significant difference. Data
of protein expression, enzyme activity, biomarker level and
paraffin section staining are from a minimum of 3 independent
experiments.
Results
Alda-1 treatment reduces CHF mortality
in post-MI rats
ALDH2 enzymatic activity was reduced in MI group
(P<0.01; Fig. 1A), as well as
ALDH2 expression (Fig. 1B). Alda-1
treatment significantly upregulated ALDH2 enzymatic activity
(P<0.001; Fig. 1A), but did not
affect ALDH2 protein expression (Fig.
1B) compared with the MI group. Survival curve analysis
indicated that the mortality rates of the Sham group and MI group
were 5.6 and 50%, respectively (Fig.
1C). Treatment with Alda-1 significantly improved the survival
rate of chronic MI rats (16.6%; P=0.0297). In addition, results
from the ELISA assay demonstrated that Alda-1 treatment
significantly reduced BNP levels in the serum of MI rats
(P<0.001; Fig. 1D).
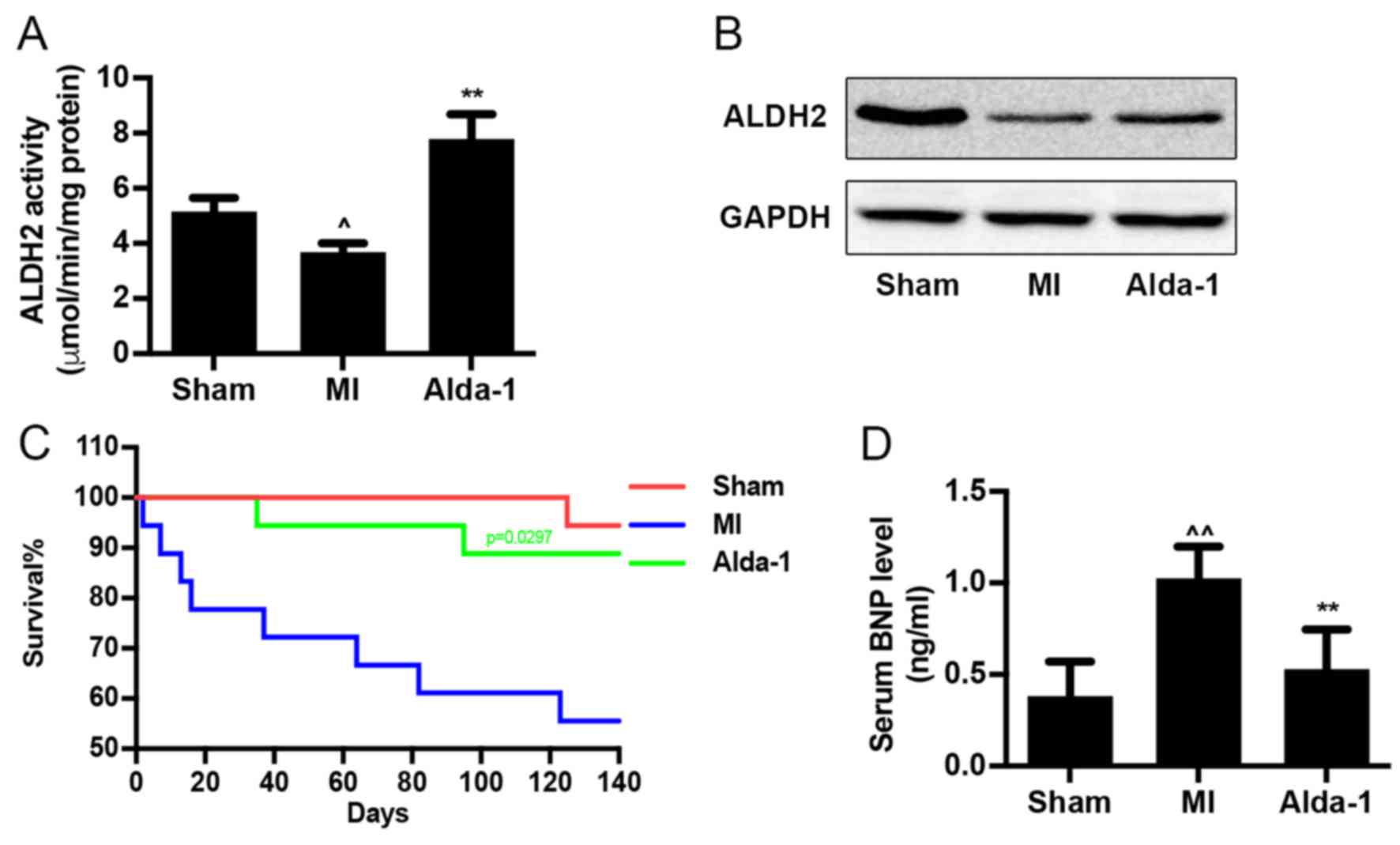 | Figure 1.Long-term treatment with Alda-1
increases survival rate of CHF in post-MI rats. (A) ALDH2 activity
in heart tissue from each group; 3 replicates of each sample,
n=4/group. (B) ALDH2 protein expression levels were determined by
western blot analysis using ALDH2 specific antibody; GAPDH was used
as the loading control. (C) Kaplan-Meier survival curves and
Mantel-Cox log rank test were used to examine the survival rates
for rats in the Sham, MI and Alda-1 groups. Alda-1 treatment
significantly improved 20-week survival rates; P=0.0297, Alda-1 vs.
MI. Sham, n=17; MI, n=9; Alda-1, n=15. (D) BNP level in serum from
each group; 3 replicates of each sample, n=4/group.
^P<0.01 vs. Sham, ^^P<0.001 vs. Sham,
**P<0.001 vs. MI. ALDH2, alcohol dehydrogenase 2; BNP, B-type
natriuretic protein; MI, myocardial infarction. |
Alda-1 reduces heart size and improves
cardiac function
H&E staining of heart tissues demonstrated that
the size of myocardial cells was increased (P<0.001; Fig. 2A and B) in the MI group. Compared
with the Sham group, the heart weight/body weight (HW/BW) ratio of
the MI group was significantly increased (P<0.001; Fig. 2C). The heart size of the chronic MI
rats was detected by echocardiography in two-dimensional-guided
M-mode of left ventricle. In MI group, visualization of wall
movement weakening and dilated chamber; while in Alda-1 treatment
group, changes induced by MI were reversed (Fig. 2D). The left ventricular dimension
at the end of diastole (LVIDd) and left ventricular dimension at
the end of systole (LVIDs) were significantly increased in MI group
compared with the Sham group (P<0.001; Fig. 2E and F, respectively). In addition,
the MI group exhibited significantly reduced left ventricular
ejection fraction (LVEF) and left ventricular fractional shortening
(LVFS) compared with the Sham group (P<0.001; Fig. 2G and H, respectively). These
results indicated that the ventricular remodeling model was
successfully established, as previously reported (20). MI rats in the Alda-1 treatment
group demonstrated a significantly lower HW/BW ratio and smaller
cardiac myocytes compared with the MI group (P<0.001; Fig. 2C and D, respectively). Alda-1
treatment also significantly reduced LVIDd and LVIDs (P<0.001;
Fig. 2E and F, respectively),
whereas LVEF and LVFS were elevated (P<0.001; Fig. 2G and H, respectively) compared with
the MI group.
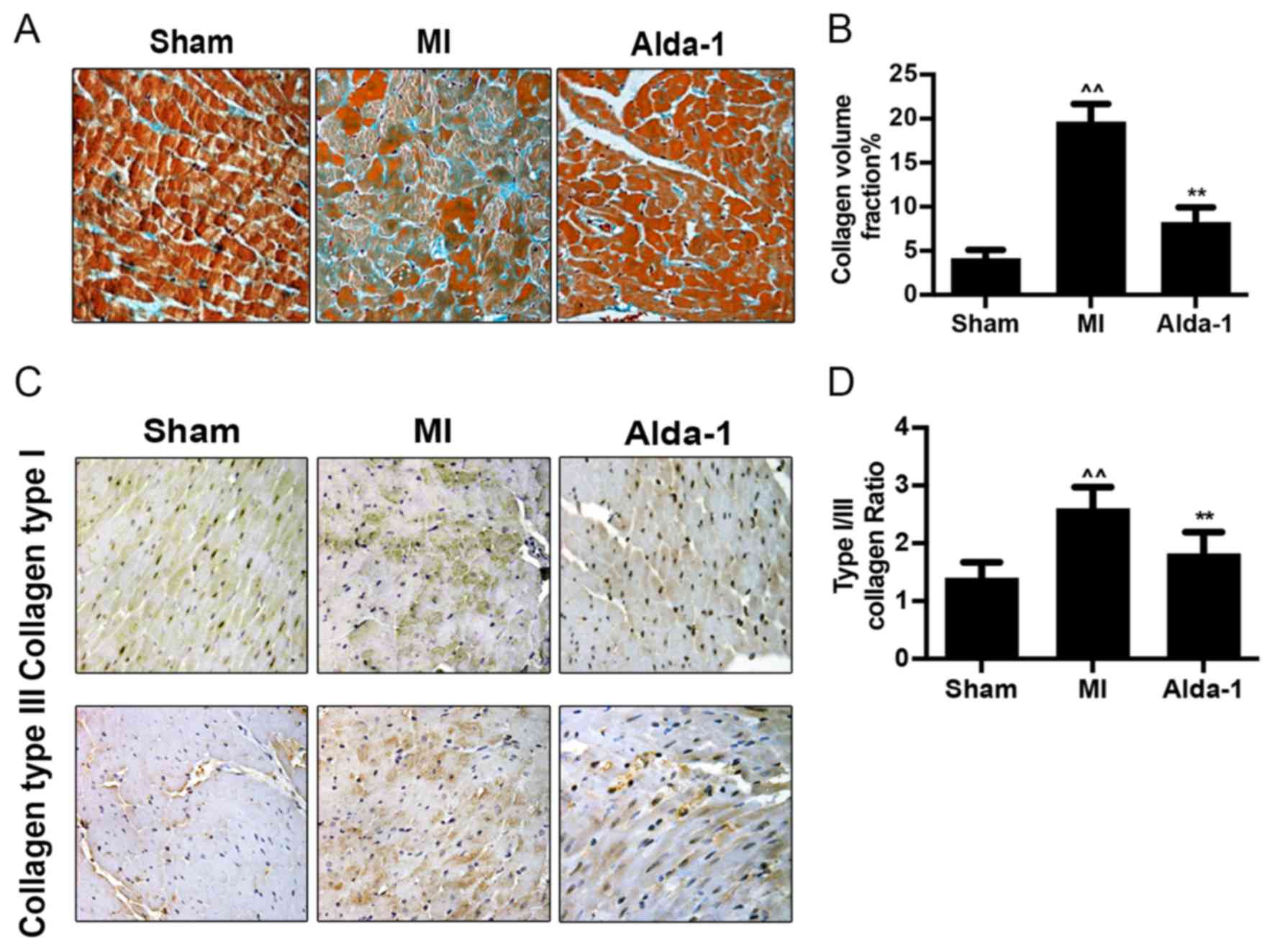
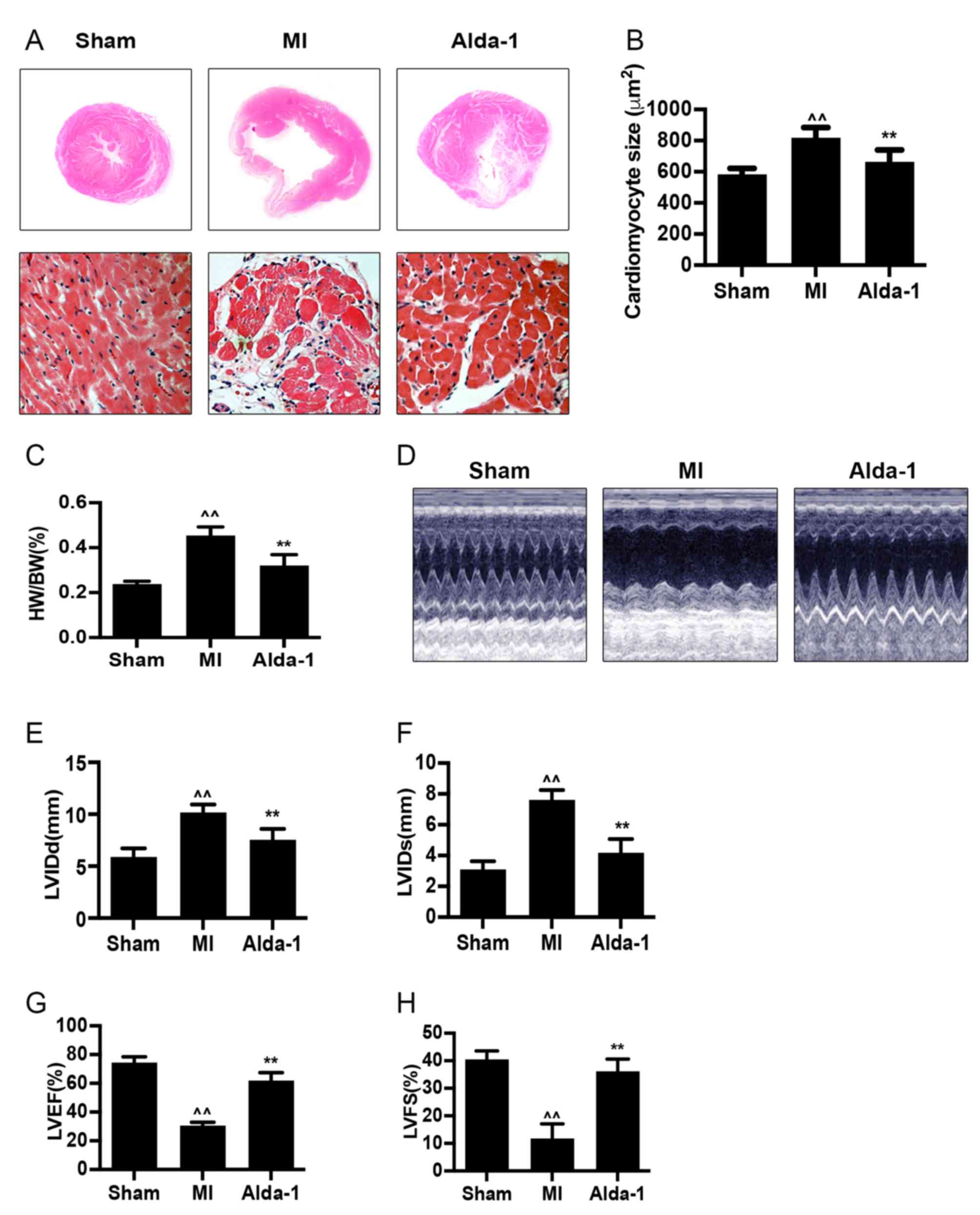 | Figure 2.Long-term treatment with Alda-1
attenuates ventricular remodeling and improves cardiac function.
(A) Representative H&E stained cross-section images of heart
(top); representative H&E staining micrograph images of left
ventricles in non-infarcted area in each group (bottom);
magnification, ×400. (B) Myocardial size is summarized and
demonstrated; n=4/group. (C) HW/BW ratios in each group. (D)
Echocardiography was performed 20 weeks following treatment.
Representative images of 2D-guided M-mode echocardiographic of the
left ventricle in each group are exhibited. (E) Quantitative
analysis of LVIDd. (F) Quantitative analysis of LVIDs. (G)
Quantitative analysis of LVEF. (H) Quantitative analysis of LVFS.
Sham, n=17; MI, n=9; Alda-1, n=15; ^^P<0.001 vs.
Sham, **P<0.001 vs. MI. H&E, hematoxylin and eosin; HW/BW,
heart weight/body weight; LVEF, left ventricular ejection fraction;
LVFS, left ventricular fractional shortening; LVIDd, left
ventricular dimension at end diastole; LVIDs, left ventricular
dimension at end systole. |
Alda-1 inhibits collagen formation in
post-MI rats with CHF
Mallory's trichrome staining demonstrated that
collagen formation and collagen volume fraction were significantly
increased in the MI group compared with the Sham group (P<0.001;
Fig. 3A and B).
Immunohistochemistry staining images illustrated that the
expression of collagen types I and III in the MI group was elevated
compared with expression in the Sham group (Fig. 3C). The increase of type I collagen
exceeded that of type III, with an increased collagen type I/III
ratio (P<0.001; Fig. 3D).
Alda-1 treatment reduced collagen volume (P<0.001; Fig. 3B), collagen type I and type III
expression (Fig. 3C) and collagen
type I/III ratio (P<0.001; Fig.
3D), compared with untreated MI rats.
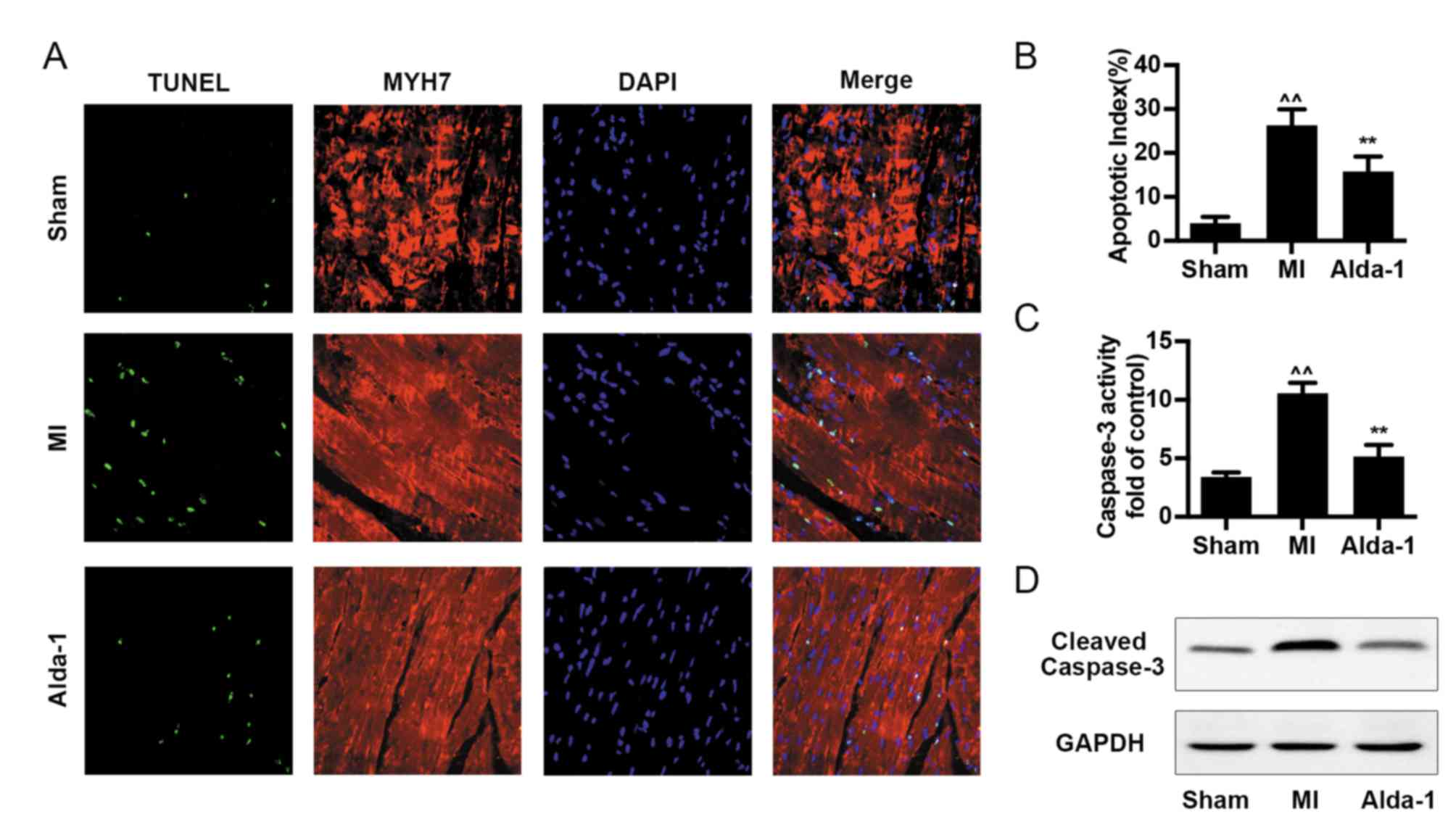
Alda-1 reduces myocyte apoptosis in
post-MI rats with CHF
Cell apoptosis was analyzed by TUNEL assay,
caspase-3 activity and cleaved-caspase-3 expression. The number of
TUNEL-positive cells and the apoptotic index were increased in the
MI group compared with the Sham group (P<0.001; Fig. 4A and B). Alda-1 treatment
significantly reduced the levels cell apoptosis, including
TUNEL-positive cell number and apoptotic index (P<0.001;
Fig. 4A and B), caspase-3 activity
(P<0.001; Fig. 4C) and
cleaved-caspase-3 expression (Fig.
4D).
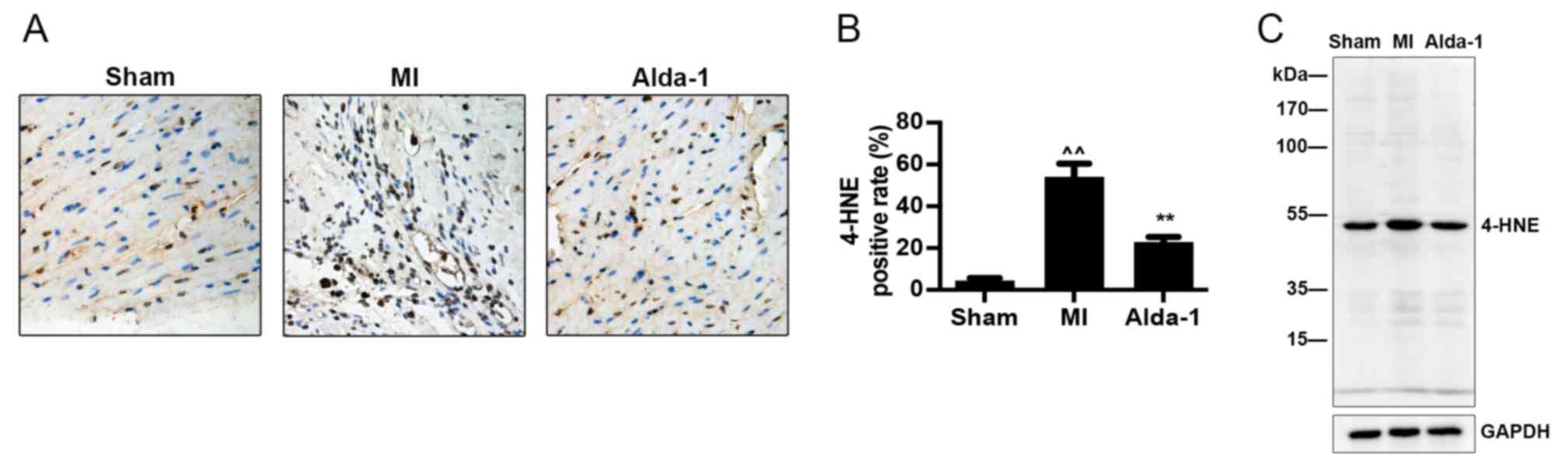
Alda-1 increases clearance of 4-HNE
toxic aldehydes
Immunohistochemistry and western blotting
demonstrated that 4-HNE modified protein expression was
significantly increased in the MI group compared with the Sham
group (P<0.001; Fig. 5A and B,
respectively). The results also demonstrated that Alda-1 treatment
reduced 4-HNE modified protein expression compared with the MI
group (P<0.001; Fig. 5).
Discussion
In the present study, the effects of oral Alda-1
treatment on long-term survival and ventricular remodeling in
post-MI model rats with CHF were investigated. The results
demonstrated that Alda-1 treatment improved the long-term survival
and ventricular remodeling in chronic MI rats. Ventricular
remodelling following MI and the subsequent development into heart
failure is a long pathological process. However, the association
between ventricular remodeling and long-term survival is not
consistent. Contemporary clinical drugs such as etanercept, digoxin
and rosiglitazone exert preventive effects on ventricular
remodelling without improvement of long-term survival (16–19).
Therefore, the observations of long-term effect and its end point
of mortality are also required in the pharmacological study of
heart failure, other than general mechanism studies. Currently, the
maximum length of Alda-1 treatment demonstrating improved
ventricular function and remodelling is 6 weeks (10). The results of the present study
showed the involvement of ALDH2 in a longer period (20 weeks), gave
more powerful evidence of cardio protective effect of Alda-1.
Left ventricular hypertrophy, dilation and cavity
distortion are the main features of ventricular remodeling
(24). In the present study,
cardiac structure was observed by echocardiography. MI model rats
exhibited increased HW/BW ratio, LVIDs and LVIDd, and decreased
LVEF and LVFS, which indicated that alteration of cardiac function
in chronic MI promoted cardiac remodeling. MI rats treated with
Alda-1 exhibited higher LVEF and LVFS and lower HW/BW ratio, LVIDd
and LVIDs compared with untreated MI rats. These results indicated
that the early activation of ALDH2, prior to pathological
remodeling occurs, and extension of Alda-1 treatment course for 20
weeks or longer, may effectively prevent cardiac remodeling. In
addition, the level of circulating BNP, a biomarker of cardiac
hypertrophy and heart failure, was measured and the levels were
consistent with the echocardiographic data, which suggested a
favorable prognosis in MI rats receiving Alda-1 treatment.
Cardiac fibrosis, including fibroblast proliferation
and the accumulation of extracellular matrix, serve a central and
dynamic role in ventricular remodelling processes (25,26).
Results from the present demonstrated increased collagen production
in rats in the MI group was upregulated, along with an increased
collagen type I to type III ratio; these effects were attenuated by
Alda-1 treatment. These data demonstrated that long-term treatment
with Alda-1 attenuated the processes mediating cardiac fibrosis in
CHF post-MI model rats, which were in agreement with a previous
study that reported Alda-1 treatment for 6 weeks in MI rats
attenuated cardiac fibrosis (10).
Apoptosis serves a crucial role in I/R injury,
cardiac remodeling and heart failure. Apoptosis progressively
occurs from the first day following infarction (27,28).
The loss of myocardial cells may lead to a decrease in cardiac
function reserves in the surviving myocardium and results in heart
failure (29). In the present
study, it was demonstrated that myocardial apoptosis was at a high
level at 20 weeks following MI. Alda-1 treatment decreased the
apoptosis of myocardial cells, which indicated potential
anti-apoptotic action is a key protective mechanism of Alda-1
against CHF.
In the present study, it was demonstrated that ALDH2
enzymatic activity was suppressed in the MI group and the mortality
rate was increased with the elevated level of the reactive
aldehydes 4-HNE, and these effects were dramatically attenuated by
treatment with Alda-1. 4-HNE staining positive rates and TUNEL
staining positive cells could be demonstrated in the heart
following MI, which suggested that the overload of aldehydes may
damage cardiac function and aggravate cardiac remodeling, as the
toxic aldehydes may induce apoptosis in remote areas of the
myocardium. ALDH2 enzymatic activity was suppressed when the heart
was in ischemic conditions and this low ALDH2 activity accelerated
4-HNE accumulation and cardiac remodeling. The results coincided
with previous study which indicated 4-HNE also directly inhibits
ALDH2, forming a feedback loop (30). Alda-1 protects the heart by
increasing enzymatic activity and indirectly clearing aldehydes. In
addition, a previous study demonstrated that Alda-1 may prevent
4-HNE-induced inactivation of ALDH2 (7). However, in the present results, the
activator of ALDH2 only affected ALDH2 enzymatic activity, but not
ALDH2 expression.
In summary, to the best of our knowledge, the
present study demonstrated for the first time that Alda-1 improves
long-term survival and cardiac function, attenuates heart
remodeling, apoptosis and fibrosis in rats with permanent MI. The
results of the present suggest that Alda-1 may be a novel
therapeutic agent for CHF post MI.
Acknowledgements
All the authors would like to acknowledge the
contribution of Senior Laboratory Technician Yurao Chen as a valued
mentor of animal study and histology in this research.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant nos. 81673805, 81373575,
81673949 and 81601779), The Guangdong Natural Science Foundation
(grant nos. 2014A030313495 and 2014A030310210), The Science and
Technology Planning Project of Guangdong Province (grant nos.
2014A020221013 and 2014A020221059), The Science Technology and
Innovation Committee of Shenzhen (grant no. JCYJ20150630164505508),
Planned Science Technology Project of Guangzhou (201804010064),
Medical Science and Technology Research Project of Guangdong
Province (grant no. A2016029) and The Traditional Chinese Medicine
Bureau of Guangdong Province (grant no. 20161260 and 20162002).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
YZ and BL provided the concept, administration,
supervision, resources and funding, and validated the data. YH, HC,
XZ, ZT, HF and YT curated the data. YH, HC, XZ, ML, WJ, WY and YW
analyzed the data. LX, WZ and BL analyzed the data with the
software. YH, HC, XZ and BL prepared the figures. YH and HC wrote
the manuscript. All authors reviewed and edited the final
manuscript.
Ethics approval and consent to
participate
All the procedures in this animal study were
performed in accordance with the approval by the Institute of
Animal Care and Use Committee of Southern Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al:
Executive summary: Heart disease and stroke statistics-2014 update:
A report from the American Heart Association. Circulation.
129:399–410. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman
M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C,
et al: Heart disease and stroke statistics-2017 Update: A Report
From the American Heart Association. Circulation. 135:e146–e603.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
O'Meara E, Thibodeau-Jarry N, Ducharme A
and Rouleau JL: The epidemic of heart failure: A lucid approach to
stemming the rising tide. Can J Cardiol. 30 12 Suppl:S442–S454.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al:
Executive summary: Heart disease and stroke statistics-2013 update:
A report from the American Heart Association. Circulation.
127:143–152. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barquera S, Pedroza-Tobías A, Medina C,
Hernández-Barrera L, Bibbins-Domingo K, Lozano R and Moran AE:
Global overview of the epidemiology of atherosclerotic
cardiovascular disease. Arch Med Res. 46:328–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ma H, Guo R, Yu L, Zhang Y and Ren J:
Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial
ischaemia/reperfusion injury: Role of autophagy paradox and toxic
aldehyde. Eur Heart J. 32:1025–1038. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen CH, Budas GR, Churchill EN, Disatnik
MH, Hurley TD and Mochly-Rosen D: Activation of aldehyde
dehydrogenase-2 reduces ischemic damage to the heart. Science.
321:1493–1495. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Doser TA, Turdi S, Thomas DP, Epstein PN,
Li SY and Ren J: Transgenic overexpression of aldehyde
dehydrogenase-2 rescues chronic alcohol intake-induced myocardial
hypertrophy and contractile dysfunction. Circulation.
119:1941–1949. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aldi S, Takano K, Tomita K, Koda K, Chan
NY, Marino A, Salazar-Rodriguez M, Thurmond RL and Levi R:
Histamine H4-receptors inhibit mast cell renin release in
ischemia/reperfusion via protein kinase C ε-dependent aldehyde
dehydrogenase type-2 activation. J Pharmacol Exp Ther. 349:508–517.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gomes KM, Campos JC, Bechara LR, Queliconi
B, Lima VM, Disatnik MH, Magno P, Chen CH, Brum PC, Kowaltowski AJ,
et al: Aldehyde dehydrogenase 2 activation in heart failure
restores mitochondrial function and improves ventricular function
and remodelling. Cardiovasc Res. 103:498–508. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liao J, Sun A, Xie Y, Isse T, Kawamoto T,
Zou Y and Ge J: Aldehyde dehydrogenase-2 deficiency aggravates
cardiac dysfunction elicited by endoplasmic reticulum stress
induction. Mol Med. 18:785–793. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koda K, Salazar-Rodriguez M, Corti F, Chan
NY, Estephan R, Silver RB, Mochly-Rosen D and Levi R: Aldehyde
dehydrogenase activation prevents reperfusion arrhythmias by
inhibiting local renin release from cardiac mast cells.
Circulation. 122:771–781. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gomes KM, Bechara LR, Lima VM, Ribeiro MA,
Campos JC, Dourado PM, Kowaltowski AJ, Mochly-Rosen D and Ferreira
JC: Aldehydic load and aldehyde dehydrogenase 2 profile during the
progression of post-myocardial infarction cardiomyopathy: Benefits
of Alda-1. Int J Cardiol. 179:129–138. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun L, Ferreira JC and Mochly-Rosen D:
ALDH2 activator inhibits increased myocardial infarction injury by
nitroglycerin tolerance. Sci Transl Med. 3:107ra1112011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Perez-Miller S, Younus H, Vanam R, Chen
CH, Mochly-Rosen D and Hurley TD: Alda-1 is an agonist and chemical
chaperone for the common human aldehyde dehydrogenase 2 variant.
Nat Struct Mol Biol. 17:159–164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bozkurt B, Torre-Amione G, Warren MS,
Whitmore J, Soran OZ, Feldman AM and Mann DL: Results of targeted
anti-tumor necrosis factor therapy with etanercept (ENBREL) in
patients with advanced heart failure. Circulation. 103:1044–1047.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mann DL, McMurray JJ, Packer M, Swedberg
K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L, et
al: Targeted anticytokine therapy in patients with chronic heart
failure: Results of the Randomized Etanercept Worldwide Evaluation
(RENEWAL). Circulation. 109:1594–1602. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Blasi ER, Heyen J, Hemkens M, McHarg A,
Ecelbarger CM and Tiwari S: Effects of chronic PPAR-agonist
treatment on cardiac structure and function, blood pressure, and
kidney in healthy sprague-dawley rats. PPAR Res. 2009:2378652009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Whitbeck MG, Charnigo RJ, Khairy P, Ziada
K, Bailey AL, Zegarra MM, Shah J, Morales G, Macaulay T, Sorrell
VL, et al: Increased mortality among patients taking
digoxin-analysis from the AFFIRM study. Eur Heart J. 34:1481–1488.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou YC, Liu B, Li YJ, Jing LL, Wen G,
Tang J, Xu X, Lv ZP and Sun XG: Effects of buyang huanwu decoction
on ventricular remodeling and differential protein profile in a rat
model of myocardial infarction. Evid Based Complement Alternat Med.
2012:3852472012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pitarys CJ II, Virmani R, Vildibill HD Jr,
Jackson EK and Forman MB: Reduction of myocardial reperfusion
injury by intravenous adenosine administered during the early
reperfusion period. Circulation. 83:237–247. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu B, Zhang J, Liu W, Liu N, Fu X, Kwan H
and Liu S, Liu B, Zhang S, Yu Z and Liu S: Calycosin inhibits
oxidative stress-induced cardiomyocyte apoptosis via activating
estrogen receptor-α/β. Bioorg Med Chem Lett. 26:181–185. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cai HB, Sun XG, Liu ZF, Liu YW, Tang J,
Liu Q, Ji BM, Song YH, Zhou YC, Yang MH and Lv ZP: Effects of
dahuangzhechong pills on cytokines and mitogen activated protein
kinase activation in rats with hepatic fibrosis. J Ethnopharmacol.
132:157–164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tham YK, Bernardo BC, Ooi JY, Weeks KL and
McMullen JR: Pathophysiology of cardiac hypertrophy and heart
failure: Signaling pathways and novel therapeutic targets. Arch
Toxicol. 89:1401–1438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jain M, Liao R, Ngoy S, Whittaker P,
Apstein CS and Eberli FR: Angiotensin II receptor blockade
attenuates the deleterious effects of exercise training on post-MI
ventricular remodelling in rats. Cardiovasc Res. 46:66–72. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun Y and Weber KT: Infarct scar: A
dynamic tissue. Cardiovasc Res. 46:250–256. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu Y, Yang H, Song L, Li N, Han QY, Tian
C, Gao E, Du J, Xia YL and Li HH: AGGF1 protects from myocardial
ischemia/reperfusion injury by regulating myocardial apoptosis and
angiogenesis. Apoptosis. 19:1254–1268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Takemura G and Fujiwara H: Role of
apoptosis in remodeling after myocardial infarction. Pharmacol
Ther. 104:1–16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mill JG, Stefanon I, dos Santos L and
Baldo MP: Remodeling in the ischemic heart: The stepwise
progression for heart failure. Braz J Med Biol Res. 44:890–898.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Doorn JA, Hurley TD and Petersen DR:
Inhibition of human mitochondrial aldehyde dehydrogenase by
4-hydroxynon-2-enal and 4-oxonon-2-enal. Chem Res Toxicol.
19:102–110. 2006. View Article : Google Scholar : PubMed/NCBI
|