Introduction
Liver injury has been recognized as a serious health
problem worldwide (1) and can be
caused by toxic chemicals, drugs or alcohol abuse, viral infection
and other factors (2,3). However, the mechanisms underlying
liver injury remain to be fully elucidated (4) and few effective therapies are
available for acute liver injury at present. Therefore, it is
necessary to elucidate the possible molecular mechanism underlying
liver injury for developing effective drugs. The collagen β (1-O)
galactosyltransferase 1 (Glt25d1) gene encodes Hyl-specific
galactosyltransferase enzymes, which catalyze the transfer of
β-linked Gal to Hyl residues on collagen (5,6),
which initiates collagen glycosylation during its
post-translational modification. Despite the identification of
Glt25d1, its functional role in collagen glycosylation remains to
be fully elucidated. The biological significance of collagen
glycosylation also remains to be elucidated as it has neither been
associated with a disease nor has a model organism harboring
defective collagen glycosylation been described to date (7,8).
Previous studies have indicated that transgenic mice
with mutations that caused cytoskeletal keratin lacking of
glycosylation modifications in the extracellular matrix (ECM) are
susceptible to liver injury (8,9) and
that glycosyltransferase activities in the extracellular space are
important for cell growth and viability (10). Accordingly, in the present study,
Glt25d1+/− mice, lacking one allele of Glt25d1,
were established, and their susceptibility to liver injury induced
by carbon tetrachloride (CCl4) was compared with that of
wild-type (WT) mice.
Materials and methods
Animals and treatments
To examine the functional roles of Glt25d1 in
vivo, a Glt25d1 conventional knockout mouse model was
established (Glt25d1flox/flox) with loxP sites flanking
exons 2 and 3 of the Glt25d1 gene. The Glt25d1flox/flox
mice were crossed with CMV-Cre transgenic mice, both of which were
purchased from the Model Animal Research Center Of Nanjing
University (Nanjing, China), to facilitate the deletion of exons 2
and 3 of Glt25d1 and generate heterozygous Glt25d1 mice
(Glt25d1+/−). This targeted disruption results in a
frame shift mutation, which leads to the early termination of
GLT25D1 protein translation. However, no homozygous
Glt25d1−/− mice were recovered from the intercrossed
Glt25d1+/− mice, which suggested that a lack of
functional Glt25d1 alleles resulted in embryonic lethality.
A total of 14 female Glt25d1+/− mice, 7–8
weeks of age and 18–20 g in weight, were fed in a specific
pathogen-free laboratory environment, with free access to standard
pellet chow and sterile water. Mice were housed at the Department
of Laboratory Animal Science, Peking University Health Science
Center (Beijing, China), and maintained under a 12/12 h light/dark
cycle, an ambient temperature of 21±2°C and a constant humility of
50±10%. C57BL/6J WT mice of the same age, gender and weight served
as controls. The mice were divided into four groups (n=6–8/group).
Acute liver injury model groups (Glt25d1+/− and WT) were
injected intraperitoneally with a single dose of CCl4
[10 ml/kg body weight dissolved in corn oil (1:6)] as reported
previously (11). The WT control
and Glt25d1+/− control mice were administered the same
dose of corn oil without CCl4. Subsequently, all mice
were sacrificed 48 h following CCl4 injection. Blood and
liver samples were collected for further analyses. All experimental
procedures were performed according to the Animal Care Committee
guidelines and the experimental protocol was approved by the Ethics
Committee of Peking University Health Science Center.
Alanine aminotransferase (ALT) and
aspartate aminotransferase (AST) assessment
Serum samples were separated from the blood by
centrifugation at 3,000 × g for 10 min at 4°C. Serum AST and ALT
levels were determined using a biochemical kit (Sichuan Maccura
Biotechnology, Sichuan, China).
Hematoxylin/eosin (HE) staining
The liver tissues were excised, fixed in 10%
formalin, paraffin-embedded, cut into 4-µm sections, and stained
with HE according to a standard protocol. All the images were
acquired and analyzed using an Axio Observer A1 Fluorescence
Microscope (ZeissAG, Oberkochen, Germany). Six randomly selected
fields (magnification, ×100) were assessed for necrosis according
to standard morphological criteria, including loss of architecture
and morphological changes in the cells, and the necrotic area
percentage was determined as reported previously (12).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the liver tissue using
the TRIzol reagent protocol (Takara Biotechnology Co., Ltd.,
Dalian, China). The concentration and quantity of isolated total
RNA was determined using NanoDrop One Microvolume UV-Vis
Spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Reverse transcription was performed using a PrimeScript RT
Reagent kit (Takara Biotechnology Co., Ltd.) under the following
conditions: 37°C for 15 min, 85°C for 5 sec and then maintained at
4°C. RT-qPCR was performed in a 20 µl reaction volume [diluted cDNA
(2 µl), primers (0.4 µl per primer), SYBR Premix EX Taq (10 µl;
Promega Corporation, Madison, WI, USA) and nuclease-free water (7.2
µl)]. qPCR was performed using a Tetrad2 Peltier Thermal Cycler
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and the reaction
was run on an Applied Biosystems 7500 Real-Time PCR system under
the following conditions: i) holding stage: 95°C for 2 min; ii)
cycling stage: 95°C for 15 sec, 60°C for 1 min, (40 cycles); iii)
melt curve stage: 95°C for 15 sec, 60°C for 1 min, 95°C for 30 sec,
60°C for 15 sec. Gapdh was amplified as a reference gene and
differences between the relative expression of the target genes
were calculated using the 2−ΔΔCq method (13). The sequences of the primers used
for PCR are shown in Table I.
 | Table I.Primer sequences for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I.
Primer sequences for reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene (ID) | Primer sequence
(5′-3′) |
|---|
| Tnf-α | Forward:
TGGGCCTCTCATGCACCACC |
|
| Reverse:
GAGGCAACCTGACCACTCTCCCT |
| Il-6 | Forward:
AGACAAAGCCAGAGTCCTTCAG |
|
| Forward:
GCCACCTTTTGACAGTGATGAG |
| Il-1β | Forward:
GCCACCTTTTGACAGTGATGAG |
|
| Reverse:
GACAGCCCAGGTCAAAGGT |
| Gapdh | Forward:
CAATGACCCCTTCATTGAC |
|
| Reverse:
GATCTCGCTCCTGGAAGATG |
Western blot analysis
Total proteins were extracted from the liver using
T-PER™ Tissue Protein Extraction reagent (Thermo Fisher Scientific,
Inc., Waltham, MA, USA; cat. no. 78510) and the protein
concentration in samples was measured using the Pierce™ BCA Protein
Assay kit (Thermo Fisher Scientific, Inc.; cat. no. 23227). An
equivalent quantity of protein (80 µg) was loaded onto
SDS-polyacrylamide gels (10%), electrophoresed and then transferred
onto Immobilon® PVDF membranes (Sigma; EMD Millipore,
Billerica, MA, USA; cat. no. P3313). Following this, the membranes
were blocked for 2 h at room temperature using Tris-buffered saline
containing 0.1% Tween 20 and 5% skimmed milk at room temperature.
The following primary antibodies were used: Monoclonal rabbit
anti-mouse GAPDH (Cell Signaling Technology, Inc., Danvers, MA,
USA; cat. no. 5174, 1:1,000), polyclonal rabbit anti-mouse
caspase-3 (Cell Signaling Technology, Inc.; cat. no. 9662,
1:1,000), polyclonal rabbit anti-mouse Glt25d1 (ProteinTech Group,
Inc., Chicago, IL, USA; cat. no. 16768-1-AP, 1:1,000), monoclonal
rabbit anti-mouse caspase9 (Abcam, Cambridge, UK; cat. no.
ab202068, 1:500), polyclonal rabbit anti-mouse TGF-β1 (ProteinTech
Group, Inc.; cat. no. 21898-1-AP, 1:1,000), monoclonal rabbit
anti-mouse phosphorylated (p)-Smad2/3 (Cell Signaling Technology,
Inc.; cat. no. 8828, 1:1,000) and polyclonal rabbit anti-mouse
Smad2 (ProteinTech Group, Inc.; cat. no. 12570-1-AP, 1:1,000) at
4°C overnight. This was followed by incubation with the goat
anti-mouse (Abcam; cat. no. ab6789, 1:5,000) and anti-rabbit
secondary antibodies (Abcam; cat. no. ab191866, 1:5,000) for 2 h at
room temperature. The sample bands were revealed using the Tanon
Imaging system (Tanon Science and Technology Co., Ltd., Shanghai,
China).
Isolation and culture of mouse primary
hepatocytes
Mouse primary hepatocytes were isolated from the
female WT or Glt25d1+/− mice using a two-step
collagenase perfusion procedure as described previously (14). The hepatocytes were purified
further using 60% percoll (1.076 g/ml) (15,16).
The mean viability and purity of mouse primary hepatocytes
following isolation were identified by trypan blue exclusion and
periodic acid Schiff (PAS) staining, respectively. All the images
were acquired and analyzed using an Axio Observer A1 Fluorescence
Microscope (Zeiss AG). Low activity or dead hepatocytes were
stained blue by trypan blue staining. Six fields (original
magnification, ×100) were selected randomly and the ratio of
hepatocellular viability was calculated according to the following
formula: Cell viability=1-(number of blue cells/total number of
cells) ×100%. The hepatocytes were subjected to PAS staining, which
specifically stains the hepatocyte cytoplasm red, in order to
assess their purity. Six fields (original magnification, ×100) were
selected randomly and the ratio of hepatocellular purity was
calculated according to the following formula: Hepatocellular
purity=(number of red cells/total number of cells) ×100%.
Induction of hepatocyte
death/apoptosis
The cells were plated in 96-well plates at a density
of 30,000 cells/well and cultured in high-glucose Dulbecco's
Modified Eagle Medium (cat. no. 10569010; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (cat.
no. 10100l47; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (cat. no. 1037806; Gibco; Thermo Fisher
Scientific, Inc.) under a humidified atmosphere of 5%
CO2 at 37°C for 24 h. Following this, the cells were
used for the experiments and divided into the following groups:
Groups 1 and 2 (normal control): Culture media were removed and
cells were treated with 100 µl of serum-free medium for 2 h. A
further 100 µl of serum-free medium was then added for the
following 24 h; groups 3 and 4 (CCl4 treatment): Culture
media were removed, and the cells were treated with 100 µl of
serum-free medium for 2 h. A further 100 µl of serum-free medium
containing CCl4 at the selected concentration (1% v/v)
(17) was added for the following
24 h. CCl4 cytotoxicity was determined using a CCK-8
assay kit. The cells were treated with CCK-8 reagent (10 µl/well)
and incubated for a further 3–4 h. The optical density (OD) was
recorded at 450 nm in a microplate reader (Thermo Fisher
Scientific, Inc.). The ratio of cell survival (%) was determined
using the following equation: Cell survival
(%)=[OD(s)-OD(b)/OD(c)-OD(b)] ×100%. OD(s), OD(b) and OD(c)
represent the absorbance values of the sample, blank and negative
control, respectively.
Statistical analysis
Quantitative data are expressed as the mean ±
standard deviation, and statistical evaluation was performed using
SPSS 18.0 statistical software (SPSS, Inc., Chicago, IL, USA).
Significance was determined by Student's t-test or two-way analysis
of variance followed by Tukey's test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Downregulation of Glt25d1 aggravates
CCl4-induced hepatic injury
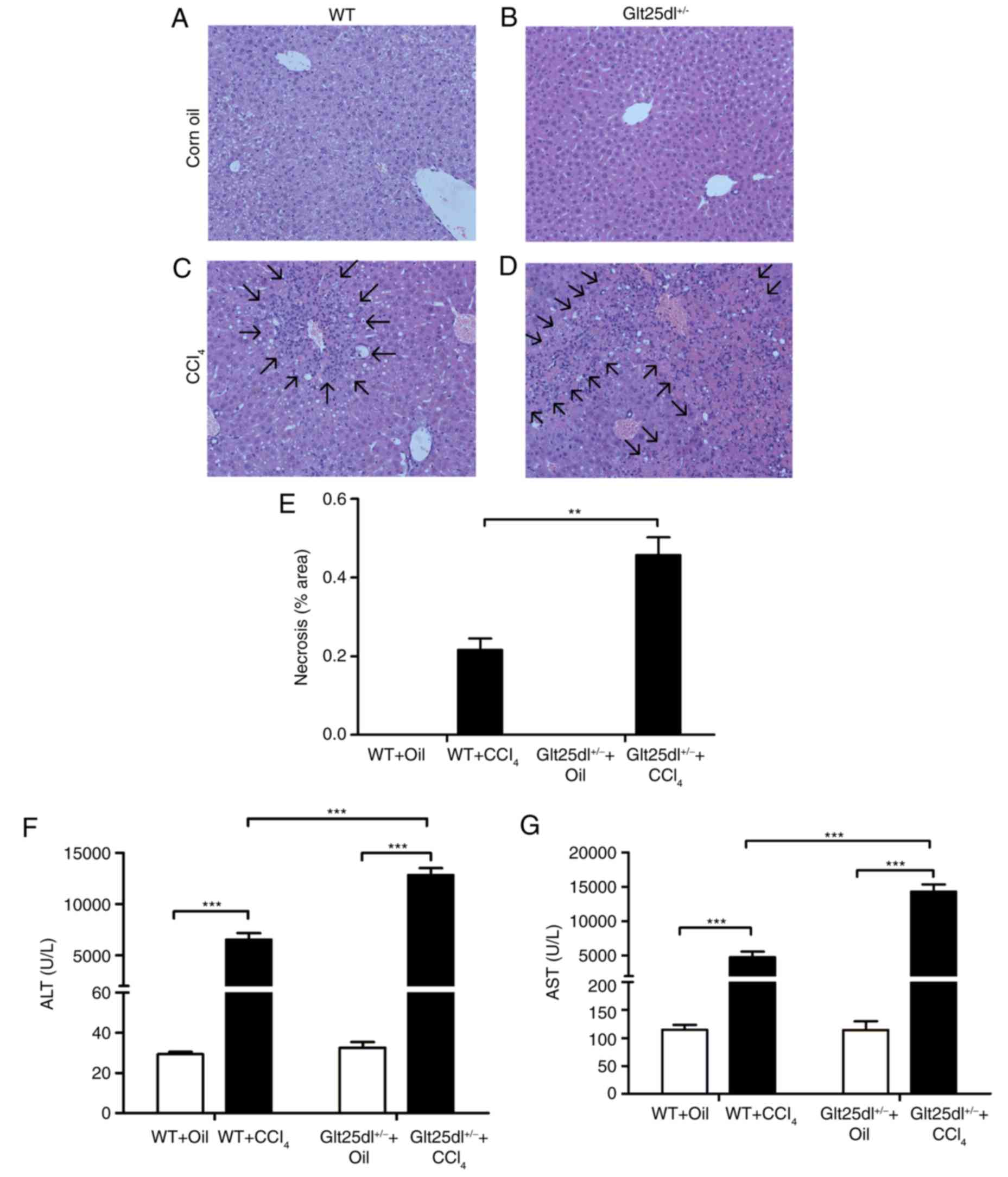
As shown in Fig.
1A-D, the HE staining showed severe hepatocyte degeneration and
necrosis at the centrilobular zones at 48 h post-CCl4
administration. The Glt25d1+/− mice exhibited larger
annular necrotic lesion areas around the hepatic lobule portal area
compared with the WT mice (0.48±0.10, vs. 0.22±0.06, P<0.01) as
shown in Fig. 1E (arrows indicate
lesions) and exhibited bridging necrosis. The above results were
supported by a higher increase of serum ALT and AST levels in the
Glt25d1+/− mice compared with the WT mice (P<0.001;
Fig. 1F and G). For further
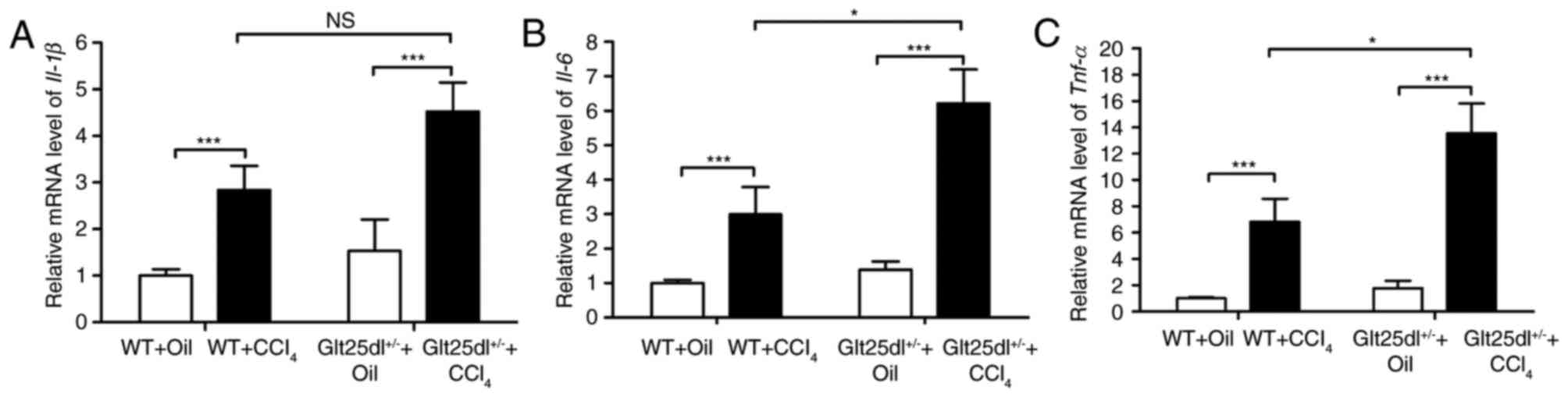
confirmation of these results, the mRNA expression of inflammatory
cytokines was examined. CCl4 treatment caused elevated
expression of Tnf-α, Il-6 and Il-1β in the livers of
the WT mice, and the expression levels of Tnf-α and
Il-6 were increased further in the Glt25d1+/−
mice (all P<0.05; Fig.
2A-C).
Glt25d1 deficiency increases
CCl4-induced hepatocellular apoptosis
The levels of the cleaved forms of caspase-3 and −9
in the Glt25d1+/− liver were significantly higher
compared with those in the WT liver (P<0.001 and P<0.01,
respectively; Fig. 3A-C).
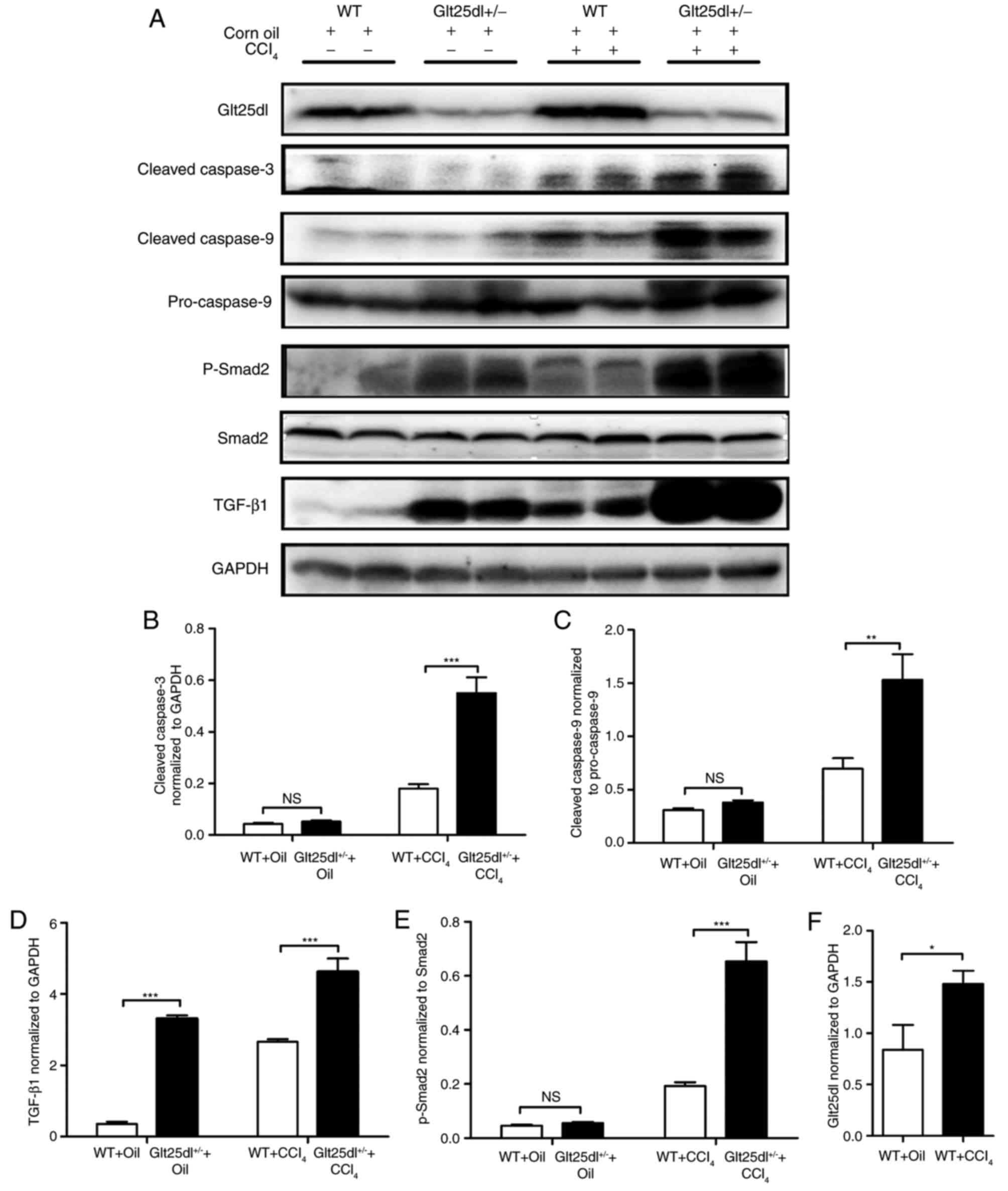 | Figure 3.Apoptosis is severe and the
TGF-β1/Smad2 signaling pathway is significantly activated in
Glt25d1+/− mice. (A) Representative western blots of
TGF-β, Glt25d1, cleaved caspase-3, cleaved caspase-9, caspase-9,
p-Smad2/3 and Smad2 in the liver. Quantification of individual
protein bands of (B) cleaved caspase-3, (C) cleaved caspase-9, (D)
TGF-β, (E) p-Smad2 and (F) Glt25d1 were normalized to the
corresponding GAPDH levels (n=6–8). *P<0.05, **P<0.01,
***P<0.001; significance determined by Student's t-test or
two-way analysis of variance followed by Tukey's test. Glt25d1,
collagen β (1-O) galactosyltransferase 1; CCl4, carbon
tetrachloride; WT, wild-type; TGF-β, transforming growth factor-β;
Smad, small mothers against decapentaplegic; p-, phosphorylated;
NS, not significant. |
Activation of the TGF-β1/Smad2
signaling pathway in Glt25d1+/− mice
Compared with the control mice, the model mice
exhibited elevated levels of TGF-β1 and p-Smad2/Smad2. The
upregulation of the TGF-β1/Smad2 signaling pathway in the
Glt25d1+/− liver was more marked compared with that in
the WT liver (P<0.001, Fig. 3A, D
and E). CCl4 treatment increased the protein level
of Glt25d1 in the WT and Glt25d1+/− mice (P<0.05;
Fig. 3A and F).
Downregulation of Glt25d1 enhances
CCl4-induced cytotoxicity in mouse primary hepatocytes
in vitro
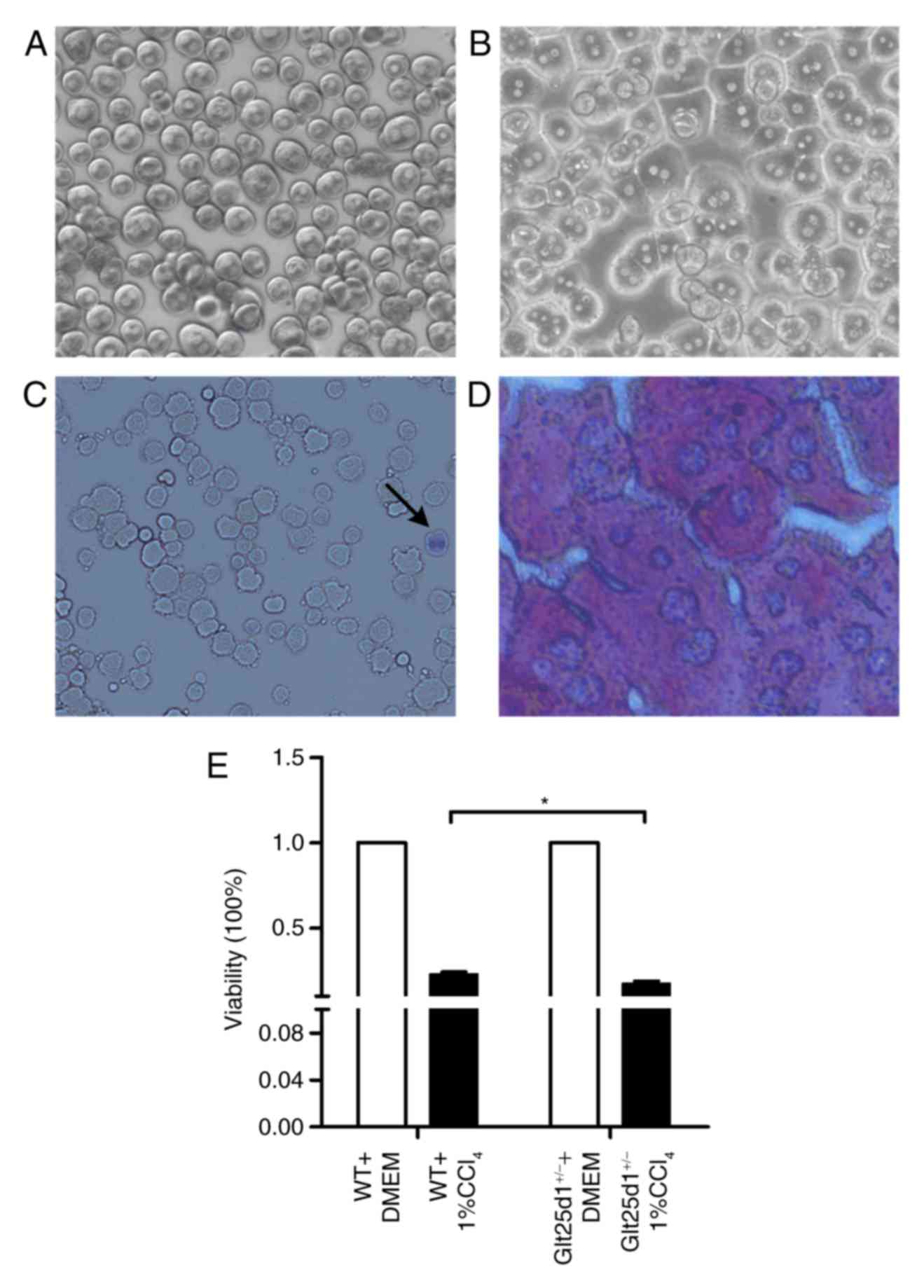
The morphology of mouse primary hepatocytes
following initial plating and incubation for 4 h is shown
respectively in Fig. 4A and B. Low
activity or dead hepatocytes were positively stained by trypan blue
(Fig. 4C). Six randomly selected
fields (magnification, ×100) were subsequently investigated, and
the mean viability of primary hepatocytes was revealed to be
>94.0% (data not shown). As presented in Fig. 4D, the purity of cultured
hepatocytes was investigated by PAS staining, which specifically
stains the hepatocyte cytoplasm red. Following this, six randomly
selected fields (magnification, ×100) were investigated, and the
mean purity of primary hepatocytes was revealed to be 96.25±1.20%
(data not shown).
The Glt25d1+/− primary hepatocytes
exhibited decreased viability upon treatment with 1%
CCl4. Compared with the WT cells, the
Glt25d1+/− primary hepatocytes were more sensitive to
CCl4-induced toxicity and showed greater loss of
viability (P<0.05; Fig.
4E).
Discussion
The present study demonstrated that
Glt25d1+/− mice, which exhibited downregulated Glt25d1
synthesis, developed aggravated CCl4-induced liver
injury compared with WT mice in vivo. It also provided
evidence suggesting that Glt25d1 deficiency may aggravate
CCl4-induced liver injury via TGF-β1/Smad2 signal
pathway activation.
The ECM is known to have multiple biological
functions, including cell self-renewal, adhesion, differentiation,
proliferation and survival (18,19).
Despite the well-regulated homeostasis of ECM components,
particularly the level of collagen, which is important in tissue
repair and remodeling (20,21),
few studies have investigated the role of collagen glycosylation in
the pathogenesis or resolution of acute liver injury. In 2009,
Schegg et al (5) confirmed
that collagen glycosylation during post-translational modification
was catalyzed by Glt25d1 and Glt25d2 in vitro. Subsequent
investigations have demonstrated that Glt25d1 is required for the
secretion of high molecular weight adiponectin (6) and that Glt25d1 deletion induces
collagen accumulation in osteosarcoma cells in vitro
(7). The present study is the
first, to the best of our knowledge, to examine the effect of
Glt25d1 on acute liver injury in vivo.
The present study utilized CCl4, a
chemical hepatotoxin, to generate a classical animal model of acute
hepatic injury (22,23) in Glt25d1+/− and WT mice.
The results demonstrated that the Glt25d1+/− mice
developed aggravated CCl4-induced liver injury compared
with the WT mice. HE staining of the Glt25d1+/− liver
tissues showed severe hepatocyte degeneration and necrosis
(Fig. 1D), which was supported by
a higher increase in the levels of serum ALT and AST (Fig. 1E and G). These data also provided
evidence that Glt25d1 had beneficial effects in reducing hepatic
necrosis. Apoptosis or programmed cell death, particularly
hepatocyte apoptosis, is a common feature of various liver diseases
(24). Previous studies have
reported severe hepatocyte apoptosis in CCl4-induced
acute liver injury (25,26). In order to evaluate the extent of
liver tissue apoptosis in the present study, western blot analysis
for cleaved caspase-3, cleaved caspase-9, and pro-caspase-9 was
performed, which revealed that the apoptosis was more severe in the
livers of the Glt25d1+/− than in the livers of the WT
mice (Fig. 3A). It is well known
that hepatocyte necrosis can recruit numerous inflammatory cells,
which secrete cytokines, including TNF-α, IL-6 and IL-1β, and
trigger an inflammatory response (27,28).
In addition, evidence suggests that apoptosis is a pro-inflammatory
and fibrogenic stimulus (29).
Consistent with the above results, the mRNA levels of Tnf-α
and Il-6 were markedly increased in the livers of
Glt25d1+/− mice in the present study (Fig. 2B and C), which indicated that
normal collagen glycosylation may alleviate CCl4-induced
liver dysfunction. Hyaluronan (HA), a ubiquitous glycosaminoglycan,
is another common component of the ECM (30). Consistent with the results of the
present study, HA-knockout mice exhibited increased hepatic injury
in CCl4-induced acute liver injury (31). These results demonstrated that
integrity and repair of the ECM is important in ameliorating liver
injury.
Under the same CCl4 treatment,
Glt25d1+/− primary hepatocytes showed increased loss of
viability in vitro (Fig.
4D), which was consistent with the results of Baumann and
Hennet, who suggested that the complete loss of collagen
glycosylation catalyzed by GLT25D1 and GLT25D2 impairs osteosarcoma
cell proliferation and viability (7). This suggests that GLT25D1 deficiency
may directly influence CCl4-induced cytotoxicity in
primary hepatocytes.
A number of studies have shown that the TGF-β1
signaling pathway is involved in the development of various
diseases (32–34) and that activation of the
TGF-β1/Smad signaling pathway may aggravate the severity of
CCl4-induced liver injury (35). To determine whether the
glycosylation effect of GLT25D1 alleviates acute liver injury
through TGF-β1 signaling pathways, the present study evaluated the
protein levels of p-Smad2, Smad2 and GLT25D1 in the liver tissues.
The results indicated that, upon CCl4 administration,
the protein expression levels of GLT25D1 and p-Smad2, and the ratio
of p-Smad2/Smad2 were upregulated, compared with the control mice.
In addition, activation of the TGF-β1/Smad2 signaling pathway was
more marked in the Glt25d1+/−mice. These data
demonstrated that GLT25D1 deficiency may aggravate
CCl4-induced liver injury through TGF-β1/Smad2 signaling
pathway activation.
There were several limitations of the present study.
Firstly, due to the embryonic lethality induced by conventional
knockout, it was not possible to obtain Glt25d1−/−
homozygous mice. In follow-up investigations on embryonic
lethality, the Glt25d1−/− mouse embryos exhibited severe
developmental arrest and usually died prior to embryonic day 13.5.
To better understand the function of Glt25d1, the generation of
liver-specific Glt25d1 mutant mice is underway.
Secondly, in order to evaluate
CCl4-induced hepatotoxicity in the WT and
Glt25d1+/− primary hepatocytes in vitro, ALT, AST
and lactate dehydrogenase require identification in the culture
supernatants. These associated experiments are also underway.
In conclusion, using Glt25d1+/− mice, the
present study demonstrated that Glt25d1 deficiency aggravated
CCl4-induced acute liver injury through activation of
the TGF-β1/Smad2 signaling pathway. Although further investigation
is required to elucidate the detailed mechanisms responsible for
these observations, the role of Glt25d1 requires consideration for
examining novel therapeutic strategies against acute liver
injury.
Acknowledgements
The authors would like to thank Spandidos
Publications for their English language editing.
Funding
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81271901 and
30671875), the Beijing Natural Science Foundation (grant no.
7152073) and the Science Foundation of Capital Medicine Development
(grant no. 2014-2-2171) to Professor Hongshan Wei.
Availability of data and materials
Not applicable.
Authors' contribution
XY and LH performed the experiments and wrote the
manuscript. JM, YL and MZ performed the experiments. JY and JZ
helped to raise animals and established the animal model of acute
liver injury. FX analyzed the data. HW designed the study and
critically revised the manuscript for important intellectual
content.
Ethics approval and consent to
participate
All experimental procedures were performed according
to the Animal Care Committee guidelines and the experimental
protocol was approved by the Ethics Committee of Peking University
Health Science Center.
Patient consent for publication
Not applicable.
Competing interests
The authors confirm that they have no competing
interests.
References
|
1
|
Wang FS, Fan JG, Zhang Z, Gao B and Wang
HY: The global burden of liver disease: The major impact of China.
Hepatology. 60:2099–2108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen Q, Zhan Q, Li Y, Sun S, Zhao L, Zhang
H and Zhang G: Schisandra lignan extract protects against carbon
tetrachloride-induced liver injury in mice by inhibiting oxidative
stress and regulating the nf-kb and Jnk signaling pathways. Evid
Based Complement Alternat Med. 2017:51402972017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi H, Han W, Ren F, Chen D, Chen Y and
Duan Z: Augmenter of liver regeneration protects against carbon
tetrachloride-induced liver injury by promoting autophagy in mice.
Oncotarget. 8:12637–12648. 2017.PubMed/NCBI
|
|
4
|
Ren F, Zhang L, Zhang X, Shi H, Wen T, Bai
L, Zheng S, Chen Y, Chen D, Li L and Duan Z: Inhibition of glycogen
synthase kinase 3β promotes autophagy to protect mice from acute
liver failure mediated by peroxisome proliferator-activated
receptor α. Cell Death Dis. 7:e21512016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schegg B, Hulsmeier AJ, Rutschmann C, Maag
C and Hennet T: Core glycosylation of collagen is initiated by two
beta(1-O) galactosyltransferases. Mol Cell Biol. 29:943–952. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Webster JA, Yang Z, Kim YH, Loo D, Mosa
RM, Li H and Chen C: Collagen beta (1-O) galactosyltransferase 1
(GLT25D1) is required for the secretion of high molecular weight
adiponectin and affects lipid accumulation. Biosci Rep. 37(pii):
BSR201701052017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baumann S and Hennet T: Collagen
accumulation in osteosarcoma cells lacking GLT25D1 collagen
galactosyltransferase. J Biol Chem. 291:18514–18524. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ku NO, Strnad P, Zhong BH, Tao GZ and
Omary MB: Keratins let liver live: Mutations predispose to liver
disease and crosslinking generates Mallory-Denk bodies. Hepatology.
46:1639–1649. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ku NO, Toivola DM, Strnad P and Omary MB:
Cytoskeletal keratin glycosylation protects epithelial tissue from
injury. Nat Cell Biol. 12:876–885. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang C, Kovanen V, Raudasoja P, Eskelinen
S, Pospiech H and Myllyla R: The glycosyltransferase activities of
lysyl hydroxylase 3 (LH3) in the extracellular space are important
for cell growth and viability. J Cell Mol Med. 13:508–521. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Louis H, Van Laethem JL, Wu W, Quertinmont
E, Degraef C, Van den Berg K, Demols A, Goldman M, Le Moine O,
Geerts A and Devière J: Interleukin-10 controls neutrophilic
infiltration, hepatocyte proliferation, and liver fibrosis induced
by carbon tetrachloride in mice. Hepatology. 28:1607–1615. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li R, Wang Y, Zhao E, Wu K, Li W, Shi L,
Wang D, Xie G, Yin Y, Deng M, et al: Maresin 1, a proresolving
lipid mediator, mitigates carbon tetrachloride-induced liver injury
in mice. Oxid Med Cell Longev. 2016:92037162016.PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nitta T, Kim JS, Mohuczy D and Behrns KE:
Murine cirrhosis induces hepatocyte epithelial mesenchymal
transition and alterations in survival signaling pathways.
Hepatology. 48:909–919. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li P, Liu S, Lu M, Bandyopadhyay G, Oh D,
Imamura T, Johnson AM, Sears D, Shen Z, Cui B, et al:
Hematopoietic-derived galectin-3 causes cellular and systemic
insulin resistance. Cell. 167:973–984 e912. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Goncalves LA, Vigario AM and
Penha-Goncalves C: Improved isolation of murine hepatocytes for in
vitro malaria liver stage studies. Malar J. 6:1692007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tong J, Yao X, Zeng H, Zhou G, Chen Y, Ma
B and Wang Y: Hepatoprotective activity of flavonoids from
Cichorium glandulosum seeds in vitro and in vivo carbon
tetrachloride-induced hepatotoxicity. J Ethnopharmacol.
174:355–363. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lucendo-Villarin B, Khan F, Pernagallo S,
Bradley M, Iredale JP and Hay DC: Maintaining hepatic stem cell
gene expression on biological and synthetic substrata. BioRes Open
Access. 1:50–53. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Arriazu E, Ruiz de Galarreta M, Cubero FJ,
Varela-Rey M, Perez de Obanos MP, Leung TM, Lopategi A, Benedicto
A, Abraham-Enachescu I and Nieto N: Extracellular matrix and liver
disease. Antioxid Redox Signal. 21:1078–1097. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Duarte S, Baber J, Fujii T and Coito AJ:
Matrix metalloproteinases in liver injury, repair and fibrosis.
Matrix Biol 44–46. 1–156. 2015.
|
|
21
|
Fausto N, Campbell JS and Riehle KJ: Liver
regeneration. J Hepatol. 57:692–694. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mizuoka H, Shikata N, Yang J, Takasu M,
Inoue K and Tsubura A: Biphasic effect of colchicine on acute liver
injury induced by carbon tetrachloride or by dimethylnitrosamine in
mice. J Hepatol. 31:825–833. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pritchard DJ, Wright MG, Sulsh S and
Butler WH: The assessment of chemically induced liver injury in
rats. J Appl Toxicol. 7:229–236. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guicciardi ME and Gores GJ: Apoptosis: A
mechanism of acute and chronic liver injury. Gut. 54:1024–1033.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang BY, Zhang XY, Guan SW and Hua ZC:
Protective effect of procyanidin B2 against CCl4-induced acute
liver injury in mice. Molecules. 20:12250–12265. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Karakus E, Karadeniz A, Simsek N, Can I,
Kara A, Yildirim S, Kalkan Y and Kisa F: Protective effect of Panax
ginseng against serum biochemical changes and apoptosis in liver of
rats treated with carbon tetrachloride (CCl4). J Hazard Mater.
195:208–213. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang F, Wang X, Qiu X, Wang J, Fang H,
Wang Z, Sun Y and Xia Z: The protective effect of Esculentoside A
on experimental acute liver injury in mice. PLoS One.
9:e1131072014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zeng B, Su M, Chen Q, Chang Q, Wang W and
Li H: Protective effect of a polysaccharide from anoectochilus
roxburghii against carbon tetrachloride-induced acute liver injury
in mice. J Ethnopharmacol. 200:124–135. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo J and Friedman SL: Hepatic
fibrogenesis. Semin Liver Dis. 27:413–426. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chanmee T, Ontong P and Itano N:
Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett.
375:20–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McCracken JM, Jiang L, Deshpande KT,
O'Neil MF and Pritchard MT: Differential effects of hyaluronan
synthase 3 deficiency after acute vs chronic liver injury in mice.
Fibrogenesis Tissue Repair. 9:42016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shirasaki T, Honda M, Shimakami T, Murai
K, Shiomoto T, Okada H, Takabatake R, Tokumaru A, Sakai Y,
Yamashita T, et al: Impaired interferon signaling in chronic
hepatitis C patients with advanced fibrosis via the transforming
growth factor beta signaling pathway. Hepatology. 60:1519–1530.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dooley S and ten Dijke P: TGF-β in
progression of liver disease. Cell Tissue Res. 347:245–256. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Macias-Silva M, Abdollah S, Hoodless PA,
Pirone R, Attisano L and Wrana JL: MADR2 is a substrate of the
TGFbeta receptor and its phosphorylation is required for nuclear
accumulation and signaling. Cell. 87:1215–1224. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Niu L, Cui X, Qi Y, Xie D, Wu Q, Chen X,
Ge J and Liu Z: Involvement of TGF-β1/smad3 signaling in carbon
tetrachloride-induced acute liver injury in mice. PLoS One.
11:e01560902016. View Article : Google Scholar : PubMed/NCBI
|