Introduction
Spondyloarthropathy (SpA), including ankylosing
spondylitis (AS), is a type of inflammatory disorder, which is
characterized by uveitis and inflammation of the axial skeleton,
and associated to human leukocyte antigen-B27 (1). Initial symptoms of SpA/AS appear in
the late teen and early adult years; however, due to a lack of
signatures for early diagnosis, treatment is frequently delayed,
ultimately leading to disability (2). There is 0.3% incidence rate of AS in
people of Asian descent (3). More
importantly, the molecular mechanisms driving disease progression
are very poorly understood. Therefore, elucidating the pathogenesis
of SpA/AS is urgently warranted.
Previously, microarray analyses have become a
standard approach for finding the alterations underlying the onset
and progression of disease and identifying signatures for diagnosis
and response to treatment (4,5).
According to literature, numerous microarray studies have been
conducted on SpA/AS (6–8). Though these analyses have
successfully identified a number of gene biomarkers distinguishing
subjects with SpA/AS from healthy subjects, the differentially
expressed genes (DEGs) listed in each study have little overlap.
Due to the limited performance ability of DEGs, discovering
potential pathogenic pathways is crucial, as the pathway biomarkers
may enhance the accuracy of detection, relative to individual genes
(9,10). Furthermore, long non-coding
(lnc)RNAs were demonstrated to competitively regulate biological
pathways and exert key functions during the development of
bone-associated disease, for example, AS (11,12).
Therefore, discovering the pathways competitively regulated by
lncRNA may reveal disease pathogenesis and is helpful to expound
the biological roles of lncRNAs in disease. In addition, searching
for sub-pathways instead of the complete pathways may uncover more
meaningful pathways and identify the functions of lncRNAs. The
concept of key local subregion was created (13), which was used to successfully
identify a number of important sub-pathways. So far, no data on
lncRNA-regulated sub-pathways associated with SpA/AS has been
reported.
In the present study, to further reveal the
mechanisms of the initiation and progression of SpA/AS, a
systematical tracking of sub-pathways from the lncRNA competitively
regulated pathways (LCRP) based on the combination of lncRNA data
and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways was
conducted. This method may be beneficial for expounding the
functional roles of lncRNAs in SpA/AS.
Materials and methods
Data collection
Microarray data of E-GEOD-41038 (14) were obtained from the ArrayExpress
at the European Bioinformatics Institute (www.ebi.ac.uk/arrayexpress/) using the terms
‘ankylosing spondylitis’, ‘spondyloarthritis’ and ‘normal control’
on May 29, 2017. In the E-GEOD-41038, there were 15 knee synovial
biopsy tissue samples, including six seronegative SpA, two AS,
three osteoarthritis and four normal control biopsies. The platform
of E-GEOD-41038 was A-MEXP-1172-Illumina HumanRef-8 v 3.0
Expression BeadChip (www.ebi.ac.uk/arrayexpress/experiments/E-GEOD-41038/).
In order to reveal the molecular mechanisms of SpA/AS, we selected
6 seronegative SpA, two AS, and four normal control biopsies for
identifying important signatures between SpA/AS and control. The
other independent published AS microarray data set (E-GEOD-25101)
(15) was used to conduct in
silico validation.
Data preprocessing
EXPRESSO function of Affy package (16) was employed to pre-treat the gene
expression profile. Specific steps included background adjustment
using the robust multiarray average method, normalization via
quartile method, perfect match/mismatch match probe correction by
means of MAS5.0 and MEDIANPOLISH used to summarize the expression
values. Ultimately, 15,593 genes were obtained.
Candidate lncRNA-mRNA
interactions
Firstly, lncRNA-micro (mi)RNA interactions were
collected from StarBase version 2.0 (17), and the proved mRNA-miRNA
interactions were downloaded from the public databases of
mirTarBase (18), miRecords
(19), TarBase (20) and mir2Disease (21). According to the shared miRNAs of
lncRNAs and mRNAs, the candidate lncRNA-mRNA regulated interactions
were obtained. For removing unreliable data, the candidate
competing mRNAs for each lncRNA were filtered using the following
two criteria (22). Criterion one:
A hypergeometric test was used to assess the significance of the
shared miRNAs, and false discovery rate (FDR) <0.05 was selected
as the cut-off threshold. Criterion two: The Jaccard Coefficient of
lncRNA-mRNA interactions was calculated and ordered, and the top
20% lncRNA-mRNA interactions were reserved.
Based on the aforementioned two criteria,
informative lncRNA-mRNA competitive interactions were identified,
which constituted 1,749 mRNAs, 7,693 lncRNA-mRNA associations and
835 lncRNAs.
Constructing the co-expressed
lncRNAs-mRNA interactions
In the present study, the Pearson correlation
coefficient (PCC) was used to measure the co-expression possibility
for any pair of informative lncRNA-mRNA interactions using the
matched lncRNA and mRNA expression data, which is reported to
measure the correlation between two variables (23). Relying on Fisher's r-to-Z
transformation (24), the
interaction with r value reaching a significant positive threshold
(P<0.05) were kept.
Selecting important sub-pathways
Detecting seed pathways
All KEGG reference pathways were retrieved from the
KEGG database. Subsequently, the genes of the co-expressed
lncRNAs-mRNA interactions were entered into the reference pathways,
which was utilized to correct the P-values using the
Benjamini-Hochberg procedure (25). Seed pathways were identified based
on the criteria of FDR <0.05.
Establishment of condition-specific
LCRP
R packages were used to convert the seed pathways to
undirected graphs which held the structure of the original pathways
(26). The lncRNAs within the
co-expressed lncRNAs-mRNA interactions were entered into the
pathway graphs, in which lncRNAs associated with their
mediated-mRNAs. Subsequently, the condition-specific LCRP was
constructed, which included lncRNA nodes and lncRNA-mRNA regulated
edges.
Locating sub-pathways competing
regulated by lncRNAs
lncRNAs have been implicated to serve as signature
nodes, as they competitively regulate the interested genes.
Therefore, the combination of lncRNAs and the topology properties
of LCRP is beneficial to effectively locate lncRNA-mediated
subregions. Specifically, the shortest path between any two
signature nodes was analyzed, on condition that the molecule number
between each pair of signature nodes was smaller than the
controlled the strength of regulated signals (n), and these
signature nodes were combined into one. The molecule number
involved in a given pathway more than controlled the sub-pathway
size (s) was regarded as candidate sub-pathways mediated by
lncRNAs s. Herein, n=1 and s=8 in the present
study were utilized to extract the candidate sub-pathways.
Detection of significant sub-pathways
using the attract method
To assess whether the candidate sub-pathways were
competitively regulated by lncRNAs, these candidate sub-pathways
were used to identify the significant sub-pathways using the
attract method (27). On the basis
of the analysis of variance model, Fisher's test was performed for
genes in the candidate sub-pathways and the F-statistic value for
gene ‘a’ was counted as follows:
F(a)=1K-1∑k=1Krk[y·k(a)-y··(a)]21N-K∑k=1K∑b=1rb[ybk(a)-y··(a)]2
In this formula, N was the total number of
sub-pathways; rk represented each cell type; k =1, …, K;
y was the mixed effect model; and b stood for the corresponding
expression value in each replicate sub-pathway. Subsequently, a
t-test was utilized to examine the F-statistics values, and the
P-values were obtained. The FDR was applied to adjust the P-values
using the Benjamini-Hochberg approach. The significant sub-pathways
were identified based on the threshold of FDR <0.05.
Selecting hub lncRNAs in LCRP
network
As reported, hub nodes constantly reflect the
crucial functions of the network. In a biological network, the
degree index was determined as the total count of edges connecting
all nodes. Hence, in the present study, the degree distribution of
the nodes in the LCRP network were measured and the top 10% lncRNAs
with the highest degrees were selected to serve as hub nodes.
In silico validation in the other
independent AS microarray data
To predict these important sub-pathways, further AS
data were downloaded from the publicly available microarray dataset
E-GEOD-25101, which represented 16 patients with AS and 16 normal
patients. For verification, all steps and the defined criteria were
the same as the aforementioned analysis.
Results
Identifying co-expressed lncRNA-mRNA
interactions and seed pathways
In the present study, PCC was used to determine the
co-expression possibility for any pair of informative lncRNA-mRNA
interactions. Compared with SpA/AS-control, a total of 35 lncRNAs,
131 mRNAs and 145 co-expressed interactions were identified (data
not shown). Subsequently, these 131 mRNAs were respectively aligned
to the reference pathways to further detect the seed pathways. A
total of 82 seed pathways were respectively identified between
SpA/AS and control with the FDR set as <0.05 (Table I). Significantly, the top five
pathways included pathways in cancer, hepatitis B, prostate cancer,
pancreatic cancer and the phosphoinositide 3-kinase (PI3K)-RAC-α
serine/threonine-protein kinase (Akt) signaling pathway.
 | Table I.List of seed pathways between AS and
control. |
Table I.
List of seed pathways between AS and
control.
| Pathways | False discovery
rate |
|---|
| hsa05200: Pathways
in cancer |
1.48×10−23 |
| hsa05161: Hepatitis
B |
1.26×10−14 |
| hsa05215: Prostate
cancer |
1.93×10−14 |
| hsa05212:
Pancreatic cancer |
5.37×10−13 |
| hsa04151: PI3K-Akt
signaling pathway |
3.25×10−12 |
| hsa05220: Chronic
myeloid leukemia |
3.94×10−11 |
| hsa05214:
Glioma |
1.17×10−10 |
| hsa05211: Renal
cell carcinoma |
1.43×10−10 |
| hsa05218:
Melanoma |
3.77×10−10 |
| hsa05219: Bladder
cancer |
1.01×10−9 |
| hsa05222: Small
cell lung cancer |
4.47×10−9 |
| hsa04510: Focal
adhesion |
3.68×10−8 |
| hsa04066: HIF-1
signaling pathway |
4.21×10−8 |
| hsa04520: Adherens
junction |
7.60×10−8 |
| hsa05203: Viral
carcinogenesis |
1.96×10−7 |
| hsa04110: Cell
cycle |
3.88×10−7 |
| hsa04540: Gap
junction |
6.07×10−7 |
| hsa05223: Non-small
cell lung cancer |
6.97×10−7 |
| hsa04012: ErbB
signaling pathway |
3.96×10−6 |
| hsa05169:
Epstein-Barr virus infection |
4.10×10−6 |
| hsa04010: MAPK
signaling pathway |
4.37×10−6 |
| hsa04912: GnRH
signaling pathway |
6.60×10−6 |
| hsa04320:
Dorso-ventral axis formation |
7.68×10−6 |
| hsa04730: Long-term
depression |
1.26×10−5 |
| hsa05131:
Shigellosis |
2.05×10−5 |
| hsa05166: HTLV-I
infection |
2.12×10−5 |
| hsa04115: p53
signaling pathway |
3.21×10−5 |
| hsa05120:
Epithelial cell signaling in Helicobacter pylori
infection |
3.21×10−5 |
| hsa05213:
Endometrial cancer |
4.22×10−5 |
| hsa04910: Insulin
signaling pathway |
5.07×10−5 |
| hsa05145:
Toxoplasmosis |
6.38×10−5 |
| hsa04722:
Neurotrophin signaling pathway |
6.86×10−5 |
| hsa04916:
Melanogenesis |
9.53×10−5 |
| hsa04350: TGF-β
signaling pathway |
1.05×10−4 |
| hsa05142: Chagas
disease (American trypanosomiasis) |
1.20×10−4 |
| hsa05210:
Colorectal cancer |
1.33×10−4 |
| hsa04810:
Regulation of actin cytoskeleton |
1.58×10−4 |
| hsa05216: Thyroid
cancer |
1.67×10−4 |
| hsa04062: Chemokine
signaling pathway |
1.81×10−4 |
| hsa04725:
Cholinergic synapse |
2.26×10−4 |
| hsa04726:
Serotonergic synapse |
2.42×10−4 |
| hsa04664: Fc
epsilon RI signaling pathway |
2.87×10−4 |
| hsa04360: Axon
guidance |
5.40×10−4 |
| hsa05221: Acute
myeloid leukemia |
5.98×10−4 |
| hsa04660: T cell
receptor signaling pathway |
.6.42×10−4 |
| hsa04728:
Dopaminergic synapse |
6.77×10−4 |
| hsa05160: Hepatitis
C |
7.56×10−4 |
| hsa04150: mTOR
signaling pathway |
7.88×10−4 |
| hsa05132:
Salmonella infection |
1.01×10−3 |
| hsa04114: Oocyte
meiosis |
1.11×10−3 |
| hsa04064: NF-κB
signaling pathway |
1.41×10−3 |
| hsa04144:
Endocytosis |
1.88×10−3 |
| hsa04662: B cell
receptor signaling pathway |
2.05×10−3 |
| hsa04380:
Osteoclast differentiation |
2.85×10−3 |
| hsa05100: Bacterial
invasion of epithelial cells |
2.89×10−3 |
| hsa05016:
Huntington's disease |
2.95×10−3 |
| hsa04620: Toll-like
receptor signaling pathway |
3.37×10−3 |
| hsa05010:
Alzheimer's disease |
3.82×10−3 |
| hsa04621: NOD-like
receptor signaling pathway |
3.89×10−3 |
| hsa04210:
Apoptosis |
5.01×10−3 |
| hsa04370: VEGF
signaling pathway |
5.21×10−3 |
| hsa04512:
ECM-receptor interaction |
5.30×10−3 |
| hsa05034:
Alcoholism |
5.99×10−3 |
| hsa04666: Fc γ
R-mediated phagocytosis |
6.59×10−3 |
| hsa04720: Long-term
potentiation |
7.75×10−3 |
| hsa04330: Notch
signaling pathway |
1.12×10−2 |
| hsa04961: Endocrine
and other factor-regulated calcium reabsorption |
1.16×10−2 |
| hsa05162:
Measles |
1.18×10−2 |
| hsa04310: Wnt
signaling pathway |
1.48×10−2 |
| hsa05164: Influenza
A |
1.61×10−2 |
| hsa05152:
Tuberculosis |
1.71×10−2 |
| hsa05130:
Pathogenic Escherichia coli infection |
1.84×10−2 |
| hsa05168: Herpes
simplex infection |
2.10×10−2 |
| hsa04914:
Progesterone-mediated oocyte maturation |
2.32×10−2 |
| hsa05020: Prion
diseases |
2.83×10−2 |
| hsa04713: Circadian
entrainment |
3.34×10−2 |
| hsa04650: Natural
killer cell mediated cytotoxicity |
4.11×10−2 |
| hsa04723:
Retrograde endocannabinoid signaling |
4.16×10−2 |
| hsa04530: Tight
junction |
4.24×10−2 |
| hsa05202:
Transcriptional misregulation in cancer |
4.77×10−2 |
| hsa04971: Gastric
acid secretion |
4.96×10−2 |
| hsa05133:
Pertussis |
4.97×10−2 |
Constructing the condition-specific
LCRP and identifying sub-pathways
Following the extraction of seed pathways of the two
groups, the seed pathways were respectively transformed into
undirected graphs, and the 35 lncRNAs in the co-expressed
lncRNA-mRNA interactions of SpA/AS were embedded into pathway
graphs as nodes by associating with their regulated-mRNAs. An
SpA/AS-specific LCRP was established, which covered lncRNA nodes in
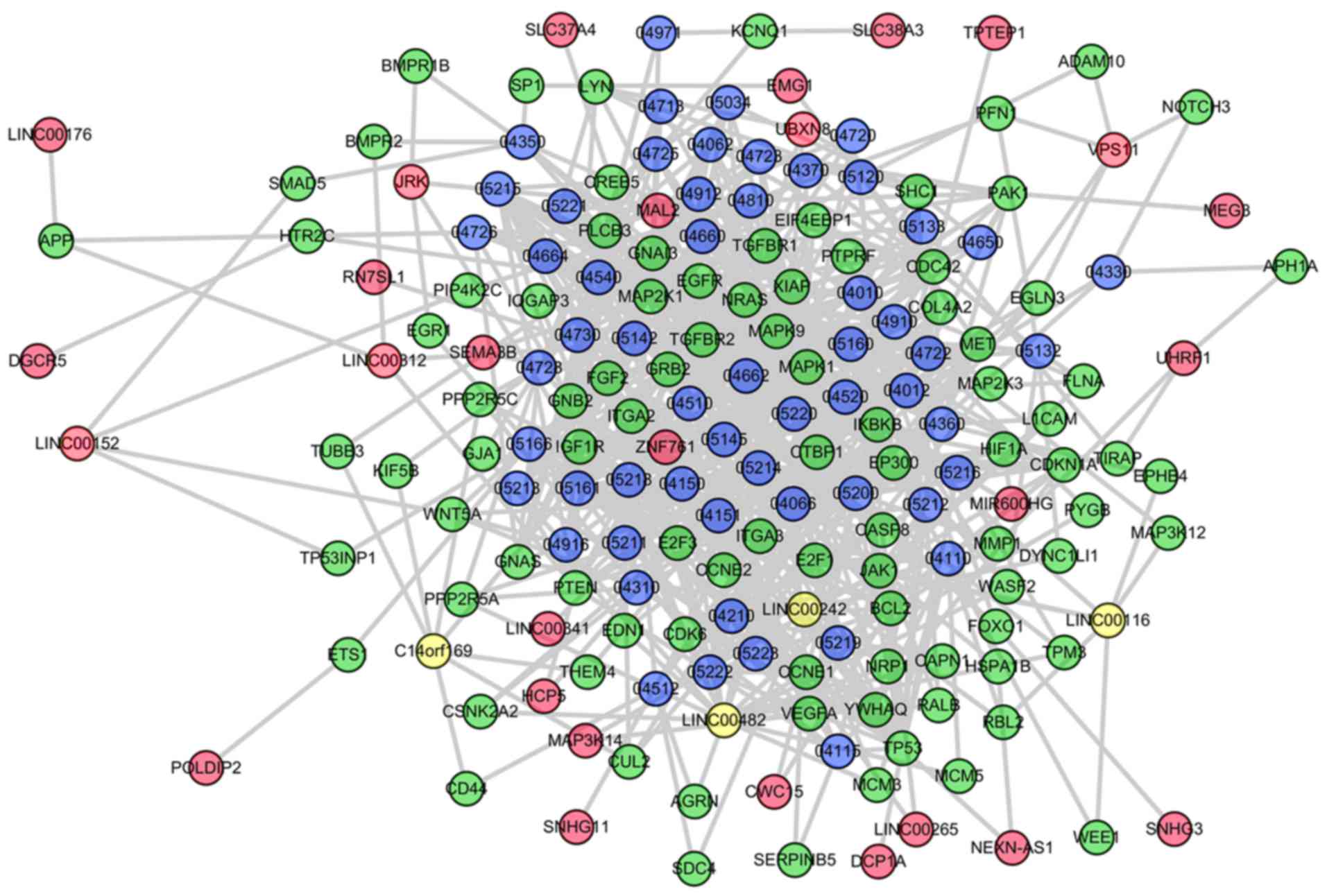
addition to lncRNA-mRNA edges. Specific LCRPs are presented in
Fig. 1. In the SpA/AS-specific
network, it was identified that overall, 35 significant lncRNAs
competitively regulated sub-pathways involved in 56 complete
pathways.
The top three sub-pathways that are competitively
regulated by lncRNAs in the comparison between AS and control
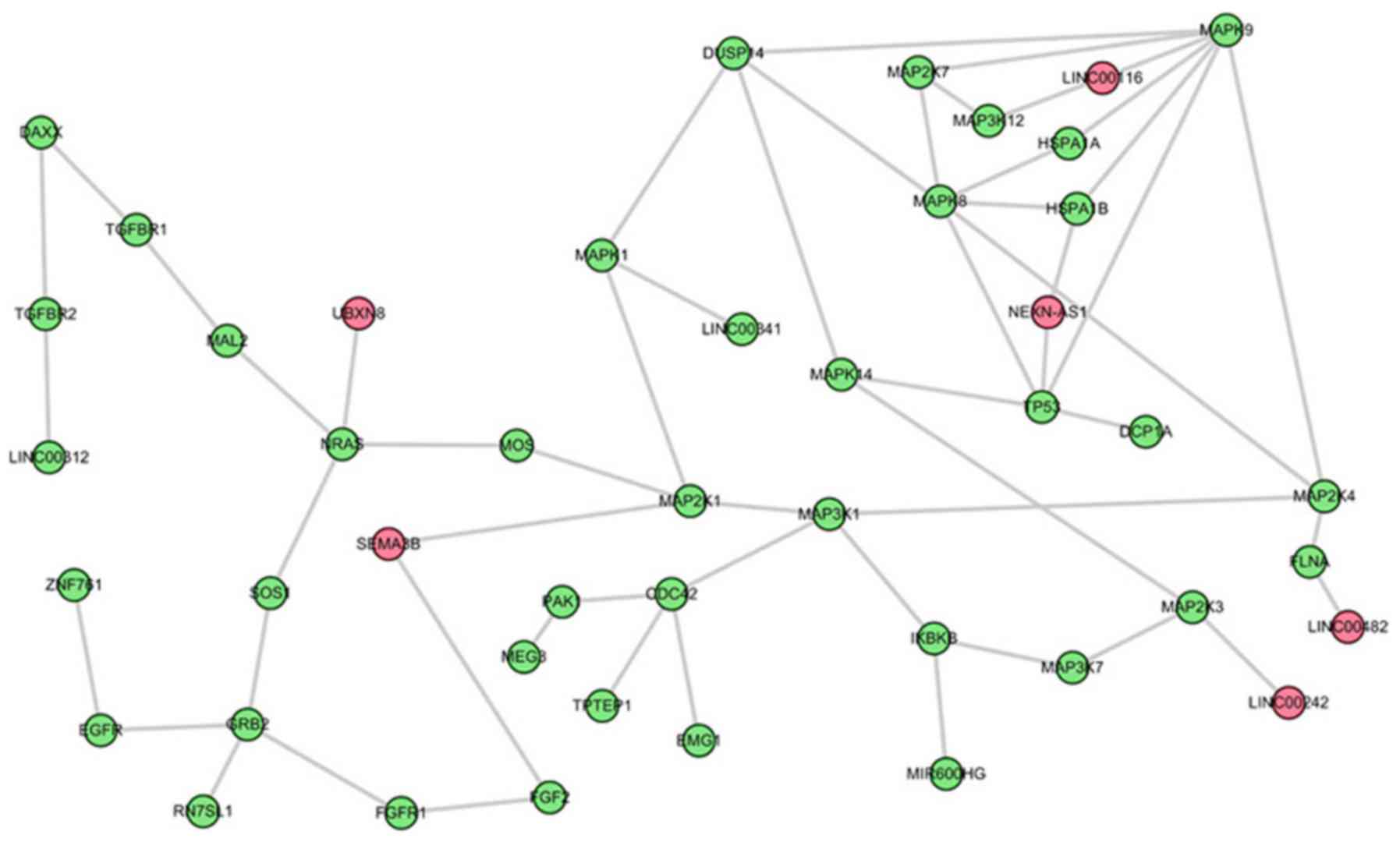
groups were further analyzed. The first is the most significant
sub-pathway path: 04010_1, which was a subregion of
mitogen-activated protein kinase (MAPK) signaling pathway (Fig. 2). Based on this module composition,
it was observed that this subregion was competitively regulated by
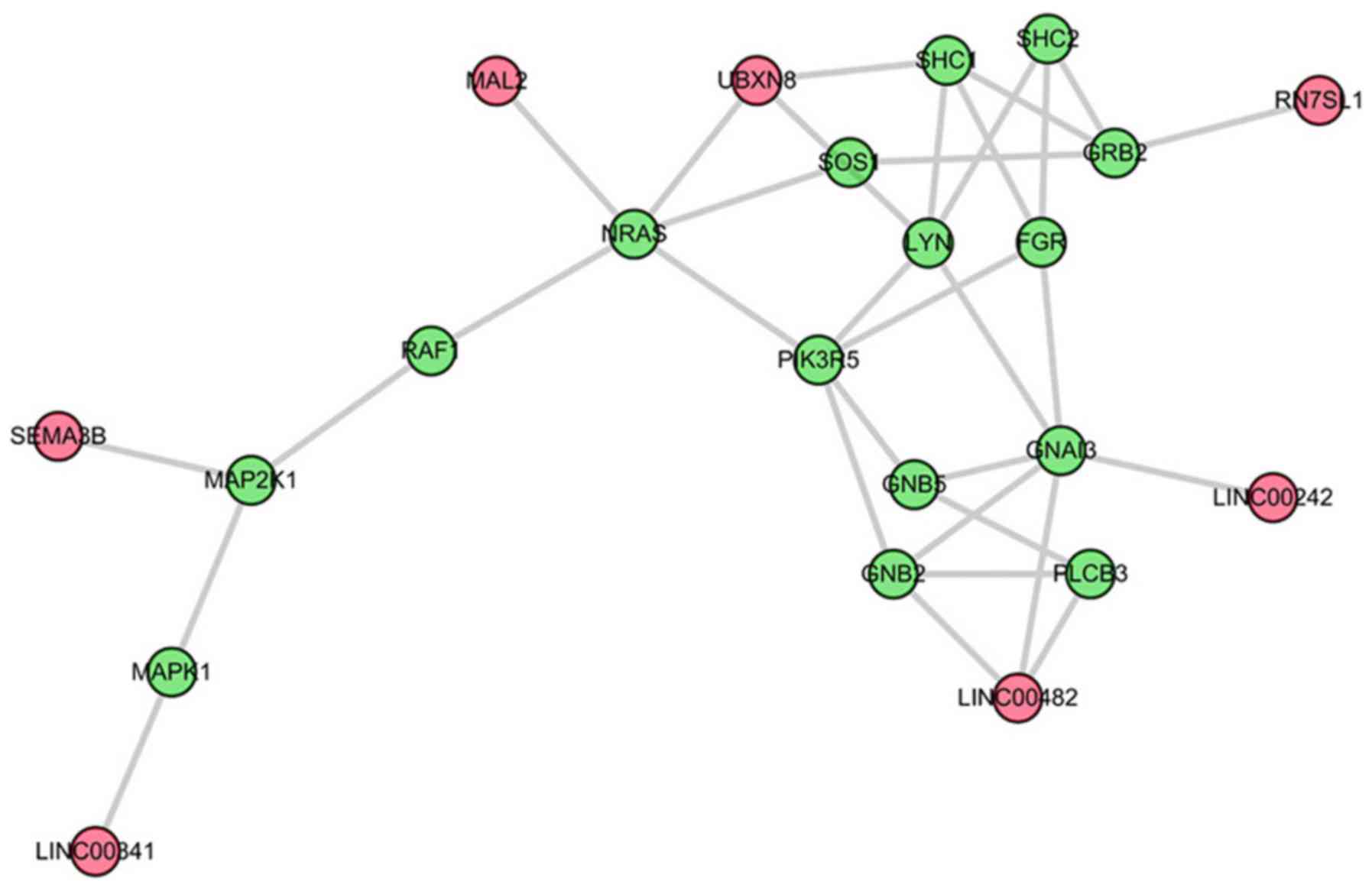
six lncRNAs. The second significant sub-pathway was path: 04062-1,
an important sub region in the chemokine signaling pathway
(Fig. 3). This sub-pathway was
regulated by seven lncRNAs. Notably, LINC00482 and UBXN8 regulated
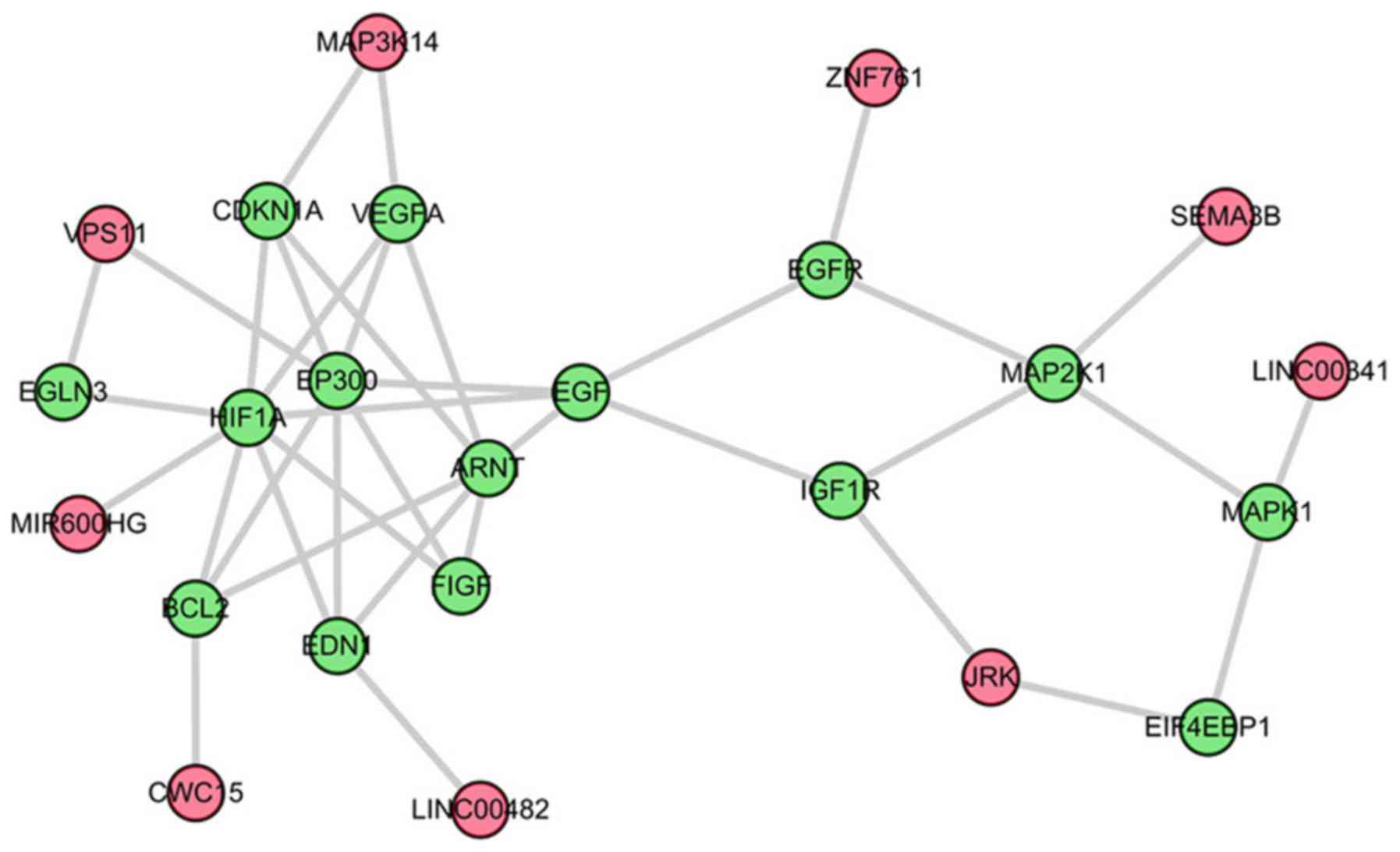
three genes. The third sub-pathway, path: 04066_2, was a part of
the HIF-1 signaling pathway (Fig.
4).
Dissecting hub lncRNAs in the LCRP
network
To dissect key lncRNAs associated with
spondyloarthropathy, degree analysis was conducted for all nodes
within the LCRP. According to the degree distribution, four hub
lncRNAs in SpA/AS-specific LCRP were identified, including
LINC00482 (degree=22), LINC00242 (degree=9), C14orf169 (degree=7)
and LINC00116 (degree=7). The degree distribution of all lncRNAs in
the SpA/AS-specific LCRP network is presented in Table II.
| Table II.Degree distribution of all lncRNAs in
the SpA/AS-specific LCRP network. |
Table II.
Degree distribution of all lncRNAs in
the SpA/AS-specific LCRP network.
| LncRNAs | Degree |
|---|
| LINC00482 | 22 |
| LINC00242 | 9 |
| C14orf169 | 7 |
| LINC00116 | 7 |
| VPS11 | 5 |
| UBXN8 | 4 |
| LINC00312 | 4 |
| JRK | 4 |
| LINC00152 | 4 |
| ZNF761 | 3 |
| UHRF1 | 3 |
| MIR600HG | 2 |
| SEMA3B | 2 |
| MAL2 | 2 |
| NEXN-AS1 | 2 |
| EMG1 | 2 |
| MAP3K14 | 2 |
| CWC15 | 2 |
| LINC00265 | 2 |
| HCP5 | 2 |
| LINC00341 | 1 |
| RN7SL1 | 1 |
| DCP1A | 1 |
| TPTEP1 | 1 |
| MEG3 | 1 |
| SNHG11 | 1 |
| SLC37A4 | 1 |
| DGCR5 | 1 |
| SLC38A3 | 1 |
| LINC00176 | 1 |
| SNHG3 | 1 |
| POLDIP2 | 1 |
In silico validation in the other
independent AS microarray data
With the attempt to verify the significant
sub-pathways identified above, the other AS data from the publicly
available microarray dataset E-GEOD-25101 was used.
Following reweighting, a total of 28 lncRNAs, 123
mRNAs and 141 co-expressed interactions were extracted. The 123
mRNAs were entered into the reference pathways to identify the seed
pathways. There were 11 seed pathways that differed between
subjects with AS and normal subjects, based on the FDR <0.05.
These pathways included the PI3K-Akt signaling pathway, focal
adhesion, pathways in cancer, pancreatic cancer, cell cycle,
influenza A, insulin signaling pathway, p53 signaling pathway,
glioma, small cell lung cancer and prostate cancer. Significantly,
it was identified that there were three common pathways between the
top five pathways in the E-GEOD-41038 and the seed pathways in the
E-GEOD-25101, including the PI3K-Akt signaling pathway, pathways in
cancer and pancreatic cancer (Table
III).
| Table III.Common seed pathways in the top five
seed pathways of E-GEOD-41038 and the top five seed pathways in
E-GEOD-25101. |
Table III.
Common seed pathways in the top five
seed pathways of E-GEOD-41038 and the top five seed pathways in
E-GEOD-25101.
| Top five seed
pathways in E-GEOD-41038 | Top five seed
pathways in E-GEOD-25101 | Common seed
pathways |
|---|
| Pathways in
cancer | PI3K-Akt signaling
pathway | Pathways in
cancer |
| Hepatitis B | Focal adhesion | Pancreatic
cancer |
| Prostate
cancer | Pathways in
cancer | PI3K-Akt signaling
pathway |
| Pancreatic
cancer | Pancreatic
cancer |
|
| PI3K-Akt signaling
pathway | Cell cycle |
|
Following obtaining the seed pathways, the LCRP was
established, which included lncRNA nodes and lncRNA-mRNA edges.
Within the LCRP network, a total of 21 significant lncRNAs
competitively regulating sub-pathways involved in 11 complete
pathways were identified. In further analysis, the top three
sub-pathways that were competitively regulated by lncRNAs in the
comparison between the AS and normal groups were investigated. The
first most significant sub-pathway was path: 04115_1, which was a
subregion of the p53 signaling pathway. The second significant
sub-pathway was path: 05222_1, an important subregion in small cell
lung cancer. The third sub-pathway, path: 05214_1, was involved in
glioma. Notably, the top three sub-pathways in the E-GEOD-41038 and
E-GEOD-25101 were identified as cancer-associated pathways. Based
on the degree distribution, three hub lncRNAs were screened out,
including ZNF761, DCP1A and C14orf169. Notably, it was observed
that the hub lncRNA C14orf169 was the most common in the
E-GEOD-41038 and E-GEOD-25101 (data not shown). These findings
demonstrated that the contents of the present study are
reliable.
Discussion
Previously, a number of studies have implied that
disruption of cellular pathways competitively mediated by lncRNAs
may lead to the onset of disorders (28–30).
Therefore, understanding this regulation mechanism may offer novel
opportunities for detecting key signatures for disease and for
developing novel target therapies. However, the research regarding
lncRNA functions involved in SpA/AS is in its infancy. Furthermore,
more attention to crucial sub-pathways instead of entire pathways
may be more applicable to reveal the roles of lncRNAs in a given
disease (13). Additionally, this
subregion strategy integrating lncRNA-mRNA data and pathway
topologies has a number of advantages. Firstly, lncRNA as a type of
novel regulatory layer is covered in the pathway analysis.
Secondly, the joint effect of lncRNAs, pathway topologies, in
addition to lncRNA competitively regulated genes was
comprehensively measured. The sub-pathway method may detect more
meaningful pathways. SpA, including AS and non-radiographic SpA, is
connected with a significant burden of disease and typically
affects patients with AS of working age. Therefore, it is urgently
required to identify molecular targets to prevent SpA/AS
development, and further improve the prognosis of patients with
SpA/AS. In the present study, in order to reveal the
etiopathogenesis of SpA/AS, gene expression data E-GEOD-41038 were
investigated to identify significant sub-pathways, which may be
involved in SpA/AS progression by combining lncRNA-mRNA expression
data with pathway topologies using the sub-pathway strategy. A
total of 35 significant lncRNAs competitively regulating
sub-pathways were involved in 56 complete pathways. The first was
the most significant sub-pathway path: 04010_1, which is a
subregion of the MAPK signaling pathway. The second significant
sub-pathway was path: 04062-1, an important subregion in the
chemokine signaling pathway.
The MAPK signaling pathway is known to mediate
stress responses and is activated by the proinflammatory cytokines
interleukin-1 or tumor necrosis factor-α (31). There are no reports, to the best of
the author's knowledge, demonstrating the direct association
between the MAPK pathway and SpA/AS. The MAPK signaling pathway has
been demonstrated to be highly associated with the functioning of
the immune response (32).
Furthermore, Chen et al (33) demonstrated that one MAPK pathway
serves a key function in the induction of the proinflammatory
response, which is involved in SpA. Furthermore, inflammation is
suggested to be associated with novel bone formation, which is
highly associated with the development of SpA and AS (34,35).
Bone formation requires differentiation of osteoblasts (36). Notably, the MAPK pathway is
implicated in the regulation of osteoblast differentiation
(37,38). Inactivation of the pro-osteogenic
MAPK pathway has been reported to inhibit osteoblast
differentiation (39). Therefore,
it may be inferred that MAPK may serve crucial roles in SpA/AS,
partially by regulating the resolution of inflammation and the
subsequent new bone formation.
The second sub-pathway was the chemokine signaling
pathway in the present analysis. Chemokines are crucial mediators
in the inflammatory response, and in parallel, members of the
chemokine system serve important roles in AS occurrence and
progression (40,41). In addition, Chen et al
(33) reported that SpA is
associated with certain proinflammatory pathways, for example, the
chemokine signaling pathway. Furthermore, Duftner et al
(42) reported that type 1 and
type 2 chemokines and lymphocytic expression of chemokine receptors
serve important roles in AS. Yang et al (43) additionally demonstrated that the
chemokine receptor CCR4 is increased in AS. Accordingly, it is
speculated that the chemokine signaling pathway may contribute to
the progression of SpA/AS, thereby suggesting that this created
sub-pathway method is a good approach for biomarker prediction.
C14orf169 was one of the hub lncRNAs in the present
study for E-GEOD-41038. In the in silico validation using
E-GEOD-25101, C14orf169 was additionally identified as the hub node
in the LCRP. The alias of C14orf169 is NO66. NO66 proteins are
believed to exhibit enzymatic activity, which regulates gene
expression and chromatin remodeling (44). Chromatin remodeling is crucial for
controlling Osterix function, which is an osteoblast-specific
transcription factor required for osteoblast differentiation and
bone formation (45). In
accordance with the aforementioned study, a different previous
study strongly supported the physiological role of NO66 during
osteoblast differentiation (46).
C14orf169 may account, at least partially, for the progression of
SpA/AS, by regulating bone formation and differentiation.
The present study was the first, to the best of the
authors' knowledge, to conduct an analysis on SpA/AS based on a
sub-pathway strategy by systematically integrating pathway
information with lncRNA-mRNA data. This may be considered the
primary strength of the present study. Overall, a number of
significant sub-pathways were successfully identified based on this
computational method. However, numerous limitations must be taken
into consideration in the present study. To begin with, the sample
data were recruited from the open access database. The SpA/AS
samples used for microarray analysis were not obtained by the
present study. Although a number of key sub-pathways and lncRNAs of
interest were identified in the present study, it must be
considered as an exploratory study at the present time. In
addition, the present study only used a bioinformatics approach to
select significant sub-pathways to reveal the etiopathogenic
process of SpA/AS; however, the association between sub-pathways
and SpA/AS has not been validated by experiments. This was the
principal limitation. As a result, further independent confirmation
studies are required to prove the significance of the present
initial findings. Although these drawbacks existed, it was
confirmed that the predicted sub-pathways offer researchers
valuable resources for providing guidance for focusing research
efforts to elucidate disease mechanisms, and detect potential
biomarkers for early diagnosis and therapy of SpA/AS. Furthermore,
this strategy may be useful for the study of other diseases.
In conclusion, sub-pathways, including the MAPK
signaling pathway and chemokine signaling pathway, may be potential
biomarkers for SpA/AS therapy. The identified sub-pathways and
lncRNAs may provide valuable diagnostic and therapeutic targets for
SpA/AS.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MD and TJG performed the experiments, analyzed the
data and drafted the manuscript. CYW and BHC conceptualized the
study design and critically revised the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Garg N, van den Bosch F and Deodhar A: The
concept of spondyloarthritis: Where are we now? Best Pract Res Clin
Rheumatol. 28:663–672. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Park R, Kim TH and Ji JD: Gene expression
profile in patients with axial spondyloarthritis: Meta-analysis of
publicly accessible microarray datasets. J Rheum Dis. 23:363–372.
2016. View Article : Google Scholar
|
|
3
|
Guo YY, Yang LL, Cui HD, Zhao S and Zhang
N: Coexisting ankylosing spondylitis and rheumatoid arthritis: A
case report with literature review. Chin Med J (Engl).
124:3430–3432. 2011.PubMed/NCBI
|
|
4
|
Bauer JW, Bilgic H and Baechler EC:
Gene-expression profiling in rheumatic disease: Tools and
therapeutic potential. Nat Rev Rheumatol. 5:257–265. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Häupl T, Stuhlmüller B, Grützkau A,
Radbruch A and Burmester GR: Does gene expression analysis inform
us in rheumatoid arthritis? Ann Rheum Dis. 69:37–42. 2010.
View Article : Google Scholar
|
|
6
|
Duan R, Leo P, Bradbury L, Brown MA and
Thomas G: Gene expression profiling reveals a downregulation in
immune-associated genes in patients with AS. Ann Rheum Dis.
69:17242010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sharma SM, Choi D, Planck SR, Harrington
CA, Austin CR, Lewis JA, Diebel TN, Martin TM, Smith JR and
Rosenbaum JT: Insights in to the pathogenesis of axial
spondyloarthropathy based on gene expression profiles. Arthritis
Res Ther. 11:R1682009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Assassi S, Reveille JD, Arnett FC, Weisman
MH, Ward MM, Agarwal SK, Gourh P, Bhula J, Sharif R, Sampat K, et
al: Whole-blood gene expression profiling in ankylosing spondylitis
shows upregulation of toll-like receptor 4 and 5. J Rheumatol.
38:87–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Y and Agarwal P: A pathway-based view
of human diseases and disease relationships. PLoS One. 4:e43462009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ulitsky I, Krishnamurthy A, Karp RM and
Shamir R: DEGAS: De novo discovery of dysregulated pathways in
human diseases. PLoS One. 5:e133672010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li X, Chai W, Zhang G, Ni M, Chen J, Dong
J, Zhou Y, Hao L, Bai Y and Wang Y: Down-regulation of
lncRNA-AK001085 and its influences on the diagnosis of ankylosing
spondylitis. Med Sci Monit. 23:11–16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sui W, Li H, He H, Xue W, Zhao X and Dai
Y: Microarray analysis of long non-coding RNA expression in
ankylosing spondylitis. https://doi.org/10.15761/IMM.1000172simple10.15761/IMM.1000172
|
|
13
|
Lin S, Li T, Zhu D, Ma C, Wang Y, He L,
Zhu C and Xing Q: The association between GAD1 gene polymorphisms
and cerebral palsy in Chinese infants. Tsitol Genet. 47:22–27.
2013.PubMed/NCBI
|
|
14
|
Thomas GP, Ran D, Pettit AR, Helen W,
Simranpreet K, Malcolm S and Brown MA: Expression profiling in
spondyloarthropathy synovial biopsies highlights changes in
expression of inflammatory genes in conjunction with tissue
remodelling genes. BMC Musculoskel Disord. 14:3542013. View Article : Google Scholar
|
|
15
|
Pimentelsantos FM, Ligeiro D, Matos M,
Mourão AF, Costa J, Santos H, Barcelos A, Godinho F, Pinto P, Cruz
M, et al: Whole blood transcriptional profiling in ankylosing
spondylitis identifies novel candidate genes that might contribute
to the inflammatory and tissue-destructive disease aspects.
Arthritis Res Ther. 13:R572011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42(Database Issue): D92–D97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hsu SD, Lin FM, Wu WY, Liang C, Huang WC,
Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al: Mirtarbase: A
database curates experimentally validated microrna-target
interactions. Nucleic Acids Res. 39(Database Issue): D163–D169.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xiao F, Zuo Z, Cai G, Kang S, Gao X and Li
T: Mirecords: An integrated resource for microrna-target
interactions. Nucleic Acids Res. 37(Database Issue): D105–D110.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vergoulis T, Vlachos IS, Alexiou P,
Georgakilas G, Maragkakis M, Reczko M, Gerangelos S, Koziris N,
Dalamagas T and Hatzigeorgiou AG: TarBase 6.0: Capturing the
exponential growth of miRNA targets with experimental support.
Nucleic Acids Res. 40(Database Issue): D222–D229. 2011.PubMed/NCBI
|
|
21
|
Jiang Q, Wang Y, Hao Y, Juan L, Teng M,
Zhang X, Li M, Wang G and Liu Y: miR2Disease: A manually curated
database for microRNA deregulation in human disease. Nucleic Acids
Res. 37(Database Issue): D98–D104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi X, Xu Y, Zhang C, Feng L, Sun Z, Han
J, Su F, Zhang Y, Li C and Li X: Subpathway-LNCE: Identify
dysfunctional subpathways competitively regulated by lncRNAs
through integrating lncRNA-mRNA expression profile and pathway
topologies. Oncotarget. 7:69857–69870. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nahler G: Pearson correlation coefficient.
Dictionary of Pharmaceutical Medicine. 1322010.
|
|
24
|
Best DJ and Roberts DE: Algorithm AS 89:
The upper tail probabilities of Spearman's Rho. J Royal Stat Soc
Series C (Appl Stat). 24:377–379. 1975.
|
|
25
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J Royal Stat Soc Series B (Methodological).
57:289–300. 1995.
|
|
26
|
Li C, Han J, Yao Q, Zou C, Xu Y, Zhang C,
Shang D, Zhou L, Zou C, Sun Z, et al: Subpathway-GM: Identification
of metabolic subpathways via joint power of interesting genes and
metabolites and their topologies within pathways. Nucleic Acids
Res. 41:e1012013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mar JC, Matigian NA, Quackenbush J and
Wells CA: Attract: A method for identifying core pathways that
define cellular phenotypes. PLoS One. 6:e254452011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu AL, Fan MP and Liu DQ: Dysfunctional
subpathways of osteoarthritis identified through combining
lncRNA-mRNA expression profile with pathway topologies. Int J Clin
Exp Med. 11:1260–1269. 2018.
|
|
29
|
Chen X, Dong H, Liu S, Yu L, Yan D, Yao X,
Sun W, Han D and Gao G: Long noncoding RNA MHENCR promotes melanoma
progression via regulating miR-425/489-mediated PI3K-Akt pathway.
Am J Transl Res. 9:90–102. 2017.PubMed/NCBI
|
|
30
|
Wang Y, He L, Du Y, Zhu P, Huang G, Luo J,
Yan X, Ye B, Li C, Xia P, et al: The long noncoding RNA lncTCF7
promotes self-renewal of human liver cancer stem cells through
activation of Wnt signaling. Cell Stem Cell. 16:413–425. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Freshney NW, Rawlinson L, Guesdon F, Jones
E, Cowley S, Hsuan J and Saklatvala J: Interleukin-1 activates a
novel protein kinase cascade that results in the phosphorylation of
hsp27. Cell. 78:1039–1049. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee MS and Kim YJ: Signaling pathways
downstream of pattern-recognition receptors and their cross talk.
Annu Rev Biochem. 76:447–480. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen Z, Cheng K, Walton Z, Wang Y, Ebi H,
Shimamura T, Liu Y, Tupper T, Ouyang J, Li J, et al: A murine lung
cancer co-clinical trial identifies genetic modifiers of
therapeutic response. Nature. 483:613–617. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lories RJ, Luyten FP and de Vlam K:
Progress in spondylarthritis. Mechanisms of new bone formation in
spondyloarthritis. Arthritis Res Ther. 11:2212009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pedersen SJ, Chiowchanwisawakit P, Lambert
RG, Østergaard M and Maksymowych WP: Resolution of inflammation
following treatment of ankylosing spondylitis is associated with
new bone formation. J Rheumatol. 38:1349–1354. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee EJ, Lee EJ, Chung YH, Song DH, Hong S,
Lee CK, Yoo B, Kim TH, Park YS, Kim SH, et al: High level of
interleukin-32 gamma in the joint of ankylosing spondylitis is
associated with osteoblast differentiation. Arthritis Res Ther.
17:3502015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ge C, Xiao G, Jiang D and Franceschi RT:
Critical role of the extracellular signal-regulated kinase-MAPK
pathway in osteoblast differentiation and skeletal development. J
Cell Biol. 176:709–718. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hu Y, Chan E, Wang SX and Li B: Activation
of p38 mitogen-activated protein kinase is required for osteoblast
differentiation. Endocrinology. 144:2068–2074. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Redlich K and Smolen JS: Inflammatory bone
loss: Pathogenesis and therapeutic intervention. Nat Rev Drug
Discov. 11:234–250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang J, Zhao Q, Wang G, Yang C, Xu Y, Li Y
and Yang P: Circulating levels of Th1 and Th2 chemokines in
patients with ankylosing spondylitis. Cytokine. 81:10–14. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tao K, Tang X, Wang B, Li RJ, Zhang BQ,
Lin JH and Li H: Distinct expression of chemokine-like factor 1 in
synovium of osteoarthritis, rheumatoid arthritis and ankylosing
spondylitis. J Huazhong Univ Sci Technology Med Sci. 36:70–76.
2016. View Article : Google Scholar
|
|
42
|
Duftner C, Dejaco C, Kullich W, Klauser A,
Goldberger C, Falkenbach A and Schirmer M: Preferential type 1
chemokine receptors and cytokine production of CD28- T cells in
ankylosing spondylitis. Ann Rheum Dis. 65:647–653. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang PT, Kasai H, Zhao LJ, Xiao WG, Tanabe
F and Ito M: Increased CCR4 expression on circulating CD4(+) T
cells in ankylosing spondylitis, rheumatoid arthritis and systemic
lupus erythematosus. Clin Exp Immunol. 138:342–347. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Eilbracht J, Reichenzeller M, Hergt M,
Schnölzer M, Heid H, Stöhr M, Franke WW and Schmidt-Zachmann MS:
NO66, a highly conserved dual location protein in the nucleolus and
in a special type of synchronously replicating chromatin. Mol Biol
Cell. 15:1816–1832. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nakashima K, Zhou X, Kunkel G, Zhang Z,
Deng JM, Behringer RR and de Crombrugghe B: The novel zinc
finger-containing transcription factor osterix is required for
osteoblast differentiation and bone formation. Cell. 108:17–29.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sinha KM, Yasuda H, Coombes MM, Dent SY
and De Crombrugghe B: Regulation of the osteoblast-specific
transcription factor Osterix by NO66, a Jumonji family histone
demethylase. EMBO J. 29:68–79. 2010. View Article : Google Scholar : PubMed/NCBI
|