Introduction
Worldwide, primary liver cancer (PLC) is the second
leading cause of cancer-associated mortality in poorly developed
countries and the sixth in more developed countries (1). Of the ~782,500 new annual cases of
PLC, China accounts for >50% of the associated incidence
(2). In 2015, PLC was the fourth
most common type of cancer and the third most common cause of
cancer-associated mortality in China (3). The majority of PLC cases occurring
worldwide are cases of hepatocellular carcinoma (HCC) (1). Many risk factors can induce the
development of HCC, including chronic hepatitis B virus or
hepatitis C virus infections, chronic alcoholic cirrhosis and high
doses of aflatoxin B1 (4).
Although a number of studies have identified a few of the molecular
alterations associated with the pathogenesis of HCC, the main
mechanism underlying HCC is still unclear.
Previous studies have demonstrated that epigenetic
alterations are one of the many early events that occur during
tumorigenesis (5,6). DNA methylation is the main epigenetic
feature in regulating gene transcriptional regulation and
preserving genome stability; however, aberrant DNA methylation can
lead to the inactivation of tumor suppressor genes or activation of
oncogenes, which eventually induces the development of many types
of cancer (7,8). A number of studies have also reported
alterations in one or several genes at one time; the abnormal
methylation of genes, including Ras association domain family
member 1 (9), p16, postmeiotic
segregation increased 2, MutL homolog 1, MutS homolog 2 (10), Adenomatosis polyposis coli
(11) and glutathione
S-transferase Pi 1 (12,13), has also been associated with HCC.
Shen et al (14) used
Illumina Infinium HumanMethylation 27K arrays to analyze 27,578 CpG
sites covering 14,495 genes in paired HCC tumor and adjacent
non-tumor tissues. The Illumina Infinium HumanMethylation 450K
BeadChip represents a significant improvement in the detection of
CpG sites (482,421 CpG and 3,091 non-CpG sites), covers 99% of
RefSeq genes with multiple sites in annotated promoters (1,500 or
200 bp upstream of the transcription start site), 5′-untranslated
regions (UTRs), first exons, gene body, and 3′-UTRs (15). Previously, aberrant DNA
hypermethylation of CpG islands was reported to induce the
inactivation of tumor suppressor genes (16), which was thought to contribute to
tumorigenesis (17). Recently,
previous studies have revealed that cancer-associated aberrant DNA
methylation not only occurs within CpG islands but may also be
detected within CpG shores or CpG shelves (18–20).
To the best of our knowledge, no research analyzing
the genome-wide DNA methylation status within a HCC cell line using
the Illumina Infinium HumanMethylation 450K BeadChip has been
conducted. Therefore, in the present study, the Illumina 450K
Methylation BeadChip was employed to screen promoter DNA
methylation and the expression profiles of methylated genes in a
human hepatocellular carcinoma cell line (Huh7 cells) and in a
human normal liver cell line (L02 cells). The results may aid the
characterization of differentially methylated CpG sites, regions
and genes associated with the pathogenesis of HCC, thereby
improving our current understanding of the methylation mechanisms
underlying the development and progression of HCC.
Materials and methods
Cell culture
The human HCC cell line, Huh7 and the human normal
liver cell line, L02, were purchased from the Cell Bank of Type
Culture Collection of Chinese Academy of Sciences (Shanghai,
China). Huh7 cells were maintained in Dulbecco's modified Eagle's
medium (DMEM; Invitrogen; Thermo Fisher Scientific, Inc., Waltham,
MA, USA), while L02 cells were maintained in RPMI-1640 (Invitrogen;
Thermo Fisher Scientific, Inc.). The cell lines were supplemented
with 100 U/ml penicillin and 100 g/ml streptomycin in the presence
of 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc.) and incubated in a humidified atmosphere containing 5%
CO2 at 37°C.
DNA preparation and Illumina Infinium
HumanMethylation 450K BeadChip assay
DNA was extracted from the two cell lines (Huh7 and
L02) using a QIAamp DNA Micro kit (Qiagen GmbH, Hilden, Germany)
according to the manufacturer's protocol. Bisulfite modification of
1 µg DNA was conducted using an EZ DNA Methylation kit (Zymo
Research Corp., Irvine, CA, USA) according to the manufacturer's
protocol. The Illumina Infinium HumanMethylation 450K BeadChip
assay was performed according to Illumina's standard protocol
(Illumina, Inc., San Diego, CA, USA). Experiments with Huh7 and L02
cells were performed in triplicate to avoid false positive and
false negative results.
Statistical analysis
The methylation data were processed with the
Methylation Module of GenomeStudio software (Methylation v1.9;
Illumina, Inc.). The methylation levels of the CpG sites were
calculated as β-values: β=intensity of the methylated allele
(M)/[intensity of the unmethylated allele (U) + M + 100] (15). A t-test, in addition to analysis of
variance with Bonferroni correction for multiple comparisons was
used to compare differentially methylated CpG sites between Huh7
and L02 cells. The differentially methylated CpG sites were defined
as sites with Adjusted P-values of ≤0.05 and |β-Difference|≥0.2.
Methylation measures with a detection P>0.05 and CpG coverage
<95% were excluded (14). For
the selection of candidate CpG sites that had significant
differences between Huh7 and L02 cell methylation levels, the
following additional filtering criteria were applied: i) Adjusted
P≤0.05, which corresponds to a raw P-value of
≤1.06×10−7; ii) for significantly hypermethylated CpG
sites, the |β-Difference| in the methylation levels between Huh7
and L02 cells was >20%, and the mean methylation level for L02
was <25%; and iii) for significantly hypomethylated CpG sites,
the methylation level |β-Difference| between L02 and Huh7 cells was
>20%, and the mean methylation level for Huh7 cells was <25%
(14).
Functional annotation of
differentially methylated genes
The genes for which the CpG sites corresponded with
differential methylation levels were determined using Kyoto
Encyclopedia of Genes and Genomes (KEGG) Pathway (www.genome.jp/kegg/) and Gene Ontology (GO; www.geneontology.org/) databases to analyze the
pathway and enrichment information of these genes.
Results
Global DNA methylation in Huh7 and LO2
cells
Following a t-test with Bonferroni correction for
multiple comparisons, 102,254 differentially methylated CpG sites
(covering 26,511 genes) were detected between Huh7 and L02 cells.
Within these CpG sites, 62,702 (61.3%) sites were hypermethylated
(covering 12,665 genes), and 39,552 (38.7%) sites were
hypomethylated (covering 13,846 genes). The results suggested that
aberrant DNA methylation may a very common event in Huh7 cells, and
alterations in hypermethylation were more frequently observed than
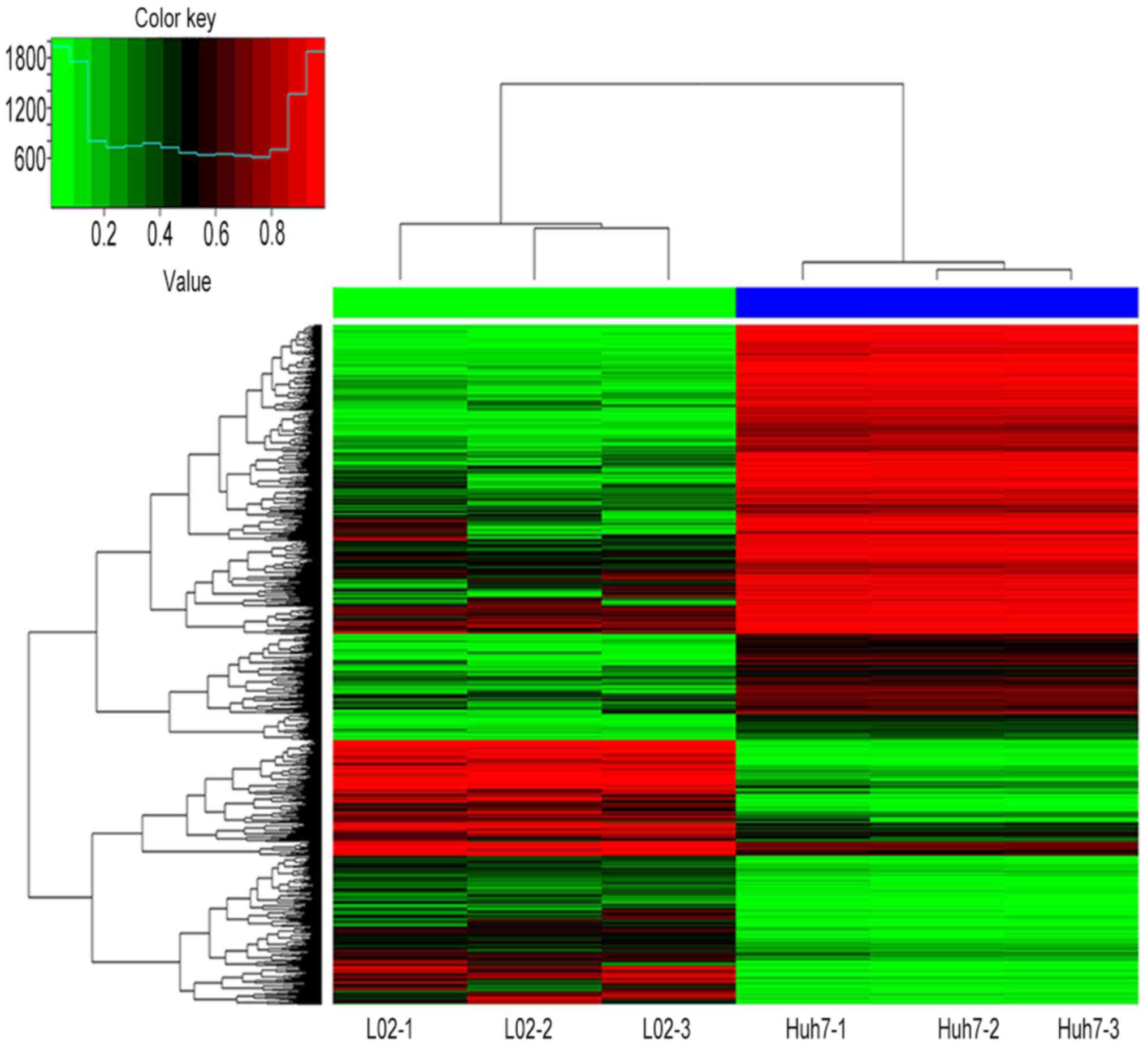
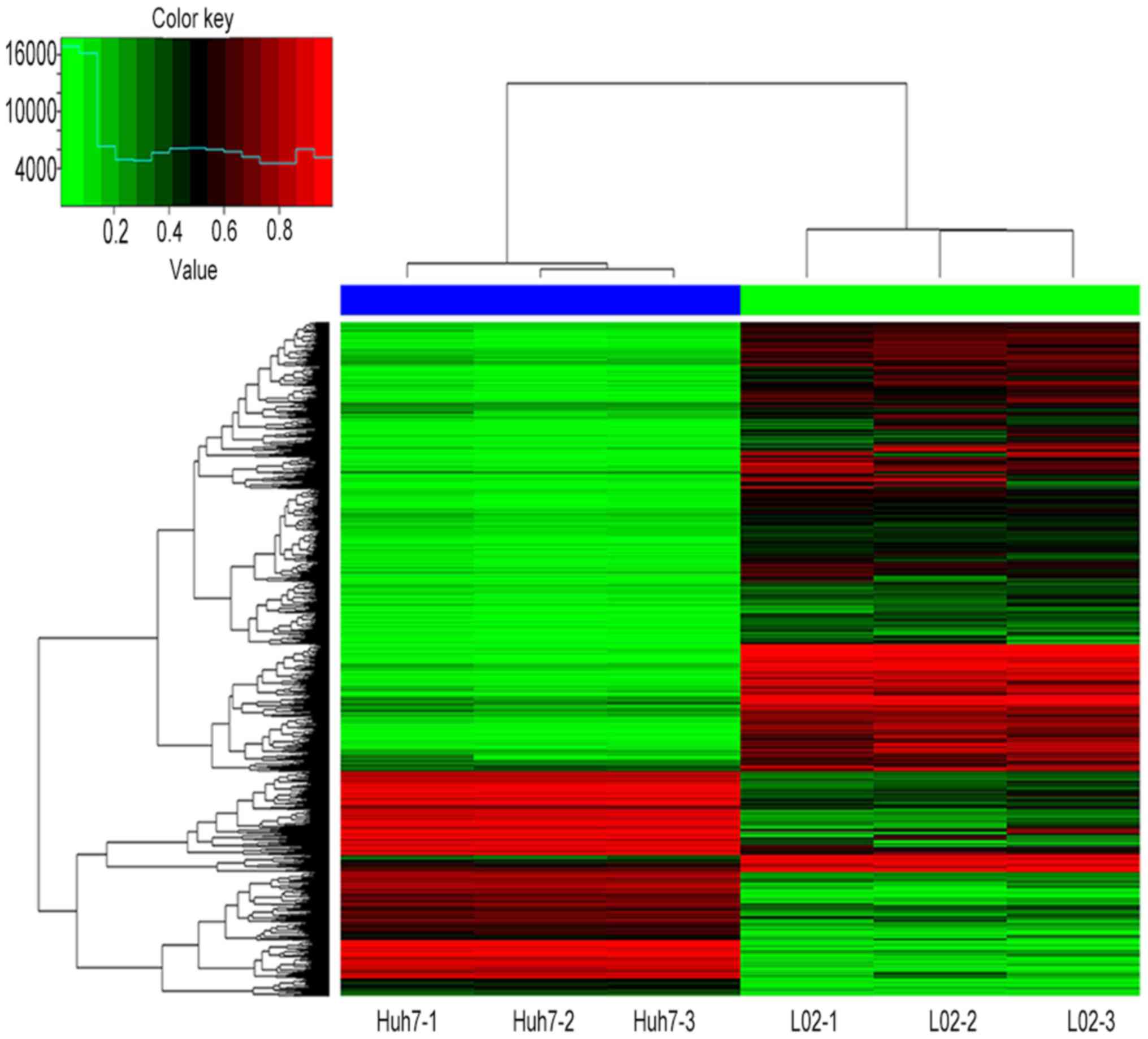
hypomethylation. Figs. 1 and
2 present hierarchical cluster
analysis of the differentially methylated CpG sites and genes that
distinguish Huh7 from L02 cells.
In addition, the results revealed that there were
41,178 (40.3%) CpG sites located in CpG island regions, 21,150
(20.7%) CpG sites were within CpG shores, 9,030 (8.8%) CpG sites
were in shelves, and 30,896 (30.2%) CpG sites were in open sea
(Table I). Furthermore, the
results also demonstrated that within different regions, the
distribution of hypo- or hypermethylated CpG sites differed: In CpG
island regions, 19,462 (47.3%) CpG sites were hypermethylated and
21,716 (52.7%) CpG sites were hypomethylated. In CpG shore regions,
13,729 (64.9%) CpG sites were hypermethylated and 7,421 (35.1%) CpG
sites were hypomethylated. In shelf regions, 7,340 (81.3%) CpG
sites were hypermethylated and 1,690 (18.7%) CpG sites were
hypomethylated.
 | Table I.Distribution of all of the
differentially methylated CpG sites. |
Table I.
Distribution of all of the
differentially methylated CpG sites.
| Type | All methylated CpG
sites, n (%) | Hypermethylated CpG
sites, n (%) | Hypomethylation CpG
sites, n (%) |
|---|
| CpG island | 41,178 (40.3) | 19,462 (47.3) | 21,716 (52.7) |
| CpG shores | 21,150 (20.7) | 13,729 (64.9) | 7,421 (35.1) |
| CpG shelves | 9,030 (8.8) | 7,340 (81.3) | 1,690 (18.7) |
| Open sea | 30,896 (30.2) | 22,171 (71.8) | 8,725 (28.2) |
| Total | 102,254 | 62,702 | 39,552 |
Frequency distribution of differentially methylated
CpG sites between Huh7 and L02 cells. In the results of the present
study, 35,937 (57.3%) hypermethylated CpG sites and 15,587 (39.4%)
hypomethylated CpG sites were observed to have an
|β-Difference|≥50%. A total of 18,529 (29.5%) of hypermethylated
CpG sites and 14,177 (35.9%) hypomethylated CpG sites had an
|β-Difference|≥30% but <50%. A total of 8,236 (13.1%)
hypermethylated CpG sites and 9,788 (24.7%) of hypomethylated CpG
sites had an |β-Difference|<30% but ≥20% (Table II). Collectively, these results
revealed that DNA aberrant hypermethylation in Huh7 cells was more
frequent than in L02 cells, which may serve a potential role in
genomic instability.
| Table II.Frequency distribution of all of the
differentially methylated CpG sites in Huh7 and L02 cells by
methylation status. |
Table II.
Frequency distribution of all of the
differentially methylated CpG sites in Huh7 and L02 cells by
methylation status.
| |β-difference|,
% | Hypermethylated CpG
sites, n (%) | Cumulative, % | Hypomethylated CpG
sites, n (%) | Cumulative, % | Total CpG sites, n
(%) | Cumulative, % |
|---|
| ≥60 | 25,585 (40.8) | 40.8 | 10,796 (27.3) | 27.3 | 36,381 (35.6) | 35.6 |
| 50≤x<60 | 10,352 (16.5) | 57.3 | 4,791 (12.1) | 39.4 | 15,134 (14.8) | 50.4 |
| 40≤x<50 | 9,681 (15.4) | 72.7 | 6,159 (15.6) | 54.0 | 15,840 (15.5) | 65.9 |
| 30≤x<40 | 8,848 (14.1) | 86.8 | 8,018 (20.3) | 75.3 | 16,866 (16.5) | 82.4 |
| 20≤x<30 | 8,236 (13.1) | 100.0 | 9,788 (24.7) | 100.0 | 18,024 (17.6) | 100.0 |
| Total | 62,702 | – | 39,552 | – | 102,254 | – |
Significant differentially methylated
CpG sites and genes
To reduce the potential impact of an extreme β value
on methylation differences, the present study applied stringent
criteria to select potentially biologically important CpG sites
(14). A total of 5,285
significantly hypermethylated CpG sites (covering 3,222 genes) and
2,659 significantly hypomethylated CpG sites (covering 2,204 genes)
were observed. For the significantly hypermethylated CpG sites,
there were 1,544 sites in CpG islands, 1,137 sites in CpG shores,
655 sites within CpG shelves and 1,949 sites in open sea regions.
By contrast, for the significantly hypomethylated CpG sites, there
were 1,201 sites in CpG islands, 632 sites in CpG shores, 133 sites
in CpG shelves and 693 sites in open sea regions (Table III). The top 20 differentially
hypermethylated and hypomethylated sites and genes are presented in
the Tables IV and V.
| Table III.Distribution of genomic regions for
significant differentially methylated CpG sites in Huh7 cells when
compared with L02 cells. |
Table III.
Distribution of genomic regions for
significant differentially methylated CpG sites in Huh7 cells when
compared with L02 cells.
| Type | Hypermethylated CpG
sites, n | Hypomethylated CpG
sites, n |
|---|
| CpG island | 1,544 | 1,201 |
| CpG shores | 1,137 |
632 |
| CpG shelves |
655 |
133 |
| Open sea | 1,949 |
693 |
| Total | 5,285 | 2,659 |
| Table IV.Top 20 significant hypermethylated
CpG sites and genes within differentially methylated regions in
Huh7 cells when compared with L02 cells. |
Table IV.
Top 20 significant hypermethylated
CpG sites and genes within differentially methylated regions in
Huh7 cells when compared with L02 cells.
| CpG sites | Adjust P-value | |β-difference| | Mean Huh | Mean L02 | Hypermethylated
genes |
|---|
| cg11058366 |
2.09×10−6 | 0.924 | 0.934 | 0.010 | ERBB4 |
| cg13245152 |
1.69×10−6 | 0.863 | 0.876 | 0.013 | PAX6 |
| cg25758545 |
1.69×10−6 | 0.863 | 0.876 | 0.013 | SALL4 |
| cg14950829 |
1.69×10−6 | 0.925 | 0.940 | 0.015 | PCDH8 |
| cg09260089 |
1.69×10−6 | 0.952 | 0.968 | 0.016 | NKX6-2 |
| cg11459773 |
1.69×10−6 | 0.876 | 0.891 | 0.015 | BCL3 |
| cg12989574 |
1.69×10−6 | 0.965 | 0.983 | 0.018 | GPC6 |
| cg03129384 |
1.69×10−6 | 0.956 | 0.974 | 0.018 | FAM196A; DOCK1 |
| cg03396151 |
1.69×10−6 | 0.929 | 0.947 | 0.018 | MEIS2 |
| cg04556126 |
1.69×10−6 | 0.920 | 0.938 | 0.018 | ZIC4 |
| cg20317123 |
1.69×10−6 | 0.947 | 0.966 | 0.019 | TCF21 |
| cg21062760 |
1.69×10−6 | 0.876 | 0.894 | 0.018 | ZBTB32 |
| cg09454560 |
1.99×10−6 | 0.624 | 0.636 | 0.013 | LRFN2 |
| cg12090740 |
1.69×10−6 | 0.892 | 0.911 | 0.020 | BCL2 |
| cg24249411 |
1.69×10−6 | 0.887 | 0.907 | 0.020 | BDNF |
| cg00057722 |
1.69×10−6 | 0.929 | 0.950 | 0.021 | – |
| cg08640046 |
1.69×10−6 | 0.810 | 0.828 | 0.018 | – |
| cg03283124 |
2.03×10−6 | 0.898 | 0.920 | 0.021 | PCDH9 |
| cg13087076 |
1.69×10−6 | 0.890 | 0.912 | 0.021 | DYDC2 |
| cg25453154 |
2.03×10−6 | 0.820 | 0.839 | 0.020 | ZCCHC24 |
| Table V.Top 20 significant hypomethylated CpG
sites and genes within differentially methylated regions in Huh7
cells when compared with L02 cells. |
Table V.
Top 20 significant hypomethylated CpG
sites and genes within differentially methylated regions in Huh7
cells when compared with L02 cells.
| CpG sites | Adjust P-value | |β-difference| | Mean Huh | Mean L02 | Hypomethylated
gene |
|---|
| cg10739344 |
1.70×10−6 | 0.894 | 0.019 | 0.913 | WDR76 |
| cg00618865 |
1.69×10−6 | 0.945 | 0.023 | 0.967 | PLXND1 |
| cg16267343 |
1.69×10−6 | 0.898 | 0.024 | 0.922 | NPR3 |
| cg00138041 |
1.69×10−6 | 0.943 | 0.029 | 0.972 | PRDM8 |
| cg01529365 |
1.69×10−6 | 0.930 | 0.031 | 0.961 | – |
| cg09564253 |
1.69×10−6 | 0.902 | 0.031 | 0.933 | LASP1 |
| cg08176368 |
1.69×10−6 | 0.926 | 0.033 | 0.959 | MMP9 |
| cg08812555 |
2.05×10−6 | 0.784 | 0.029 | 0.813 | DKK1 |
| cg25612391 |
1.77×10−6 | 0.741 | 0.028 | 0.769 | SLC25A42 |
| cg22417879 |
1.70×10−6 | 0.908 | 0.035 | 0.943 | SDCBP2 |
| cg15019790 |
1.69×10−6 | 0.924 | 0.036 | 0.960 | SIX2 |
| cg07407787 |
1.69×10−6 | 0.922 | 0.036 | 0.958 | ARSG; SLC16A6 |
| cg08361684 |
1.69×10−6 | 0.911 | 0.038 | 0.949 | FJX1 |
| cg16195157 |
1.69×10−6 | 0.900 | 0.038 | 0.938 | DNAJB1 |
| cg15842502 |
1.69×10−6 | 0.913 | 0.040 | 0.953 | RB1 |
| cg13848566 |
1.95×10−6 | 0.934 | 0.041 | 0.975 | GAS1 |
| cg27454412 |
1.81×10−6 | 0.781 | 0.034 | 0.815 | C7orf50 |
| cg02152578 |
1.69×10−6 | 0.931 | 0.041 | 0.972 | AHCYL1 |
| cg13355248 |
1.69×10−6 | 0.792 | 0.038 | 0.829 | NPTX1 |
| cg16443866 |
1.69×10−6 | 0.878 | 0.042 | 0.920 | STC2 |
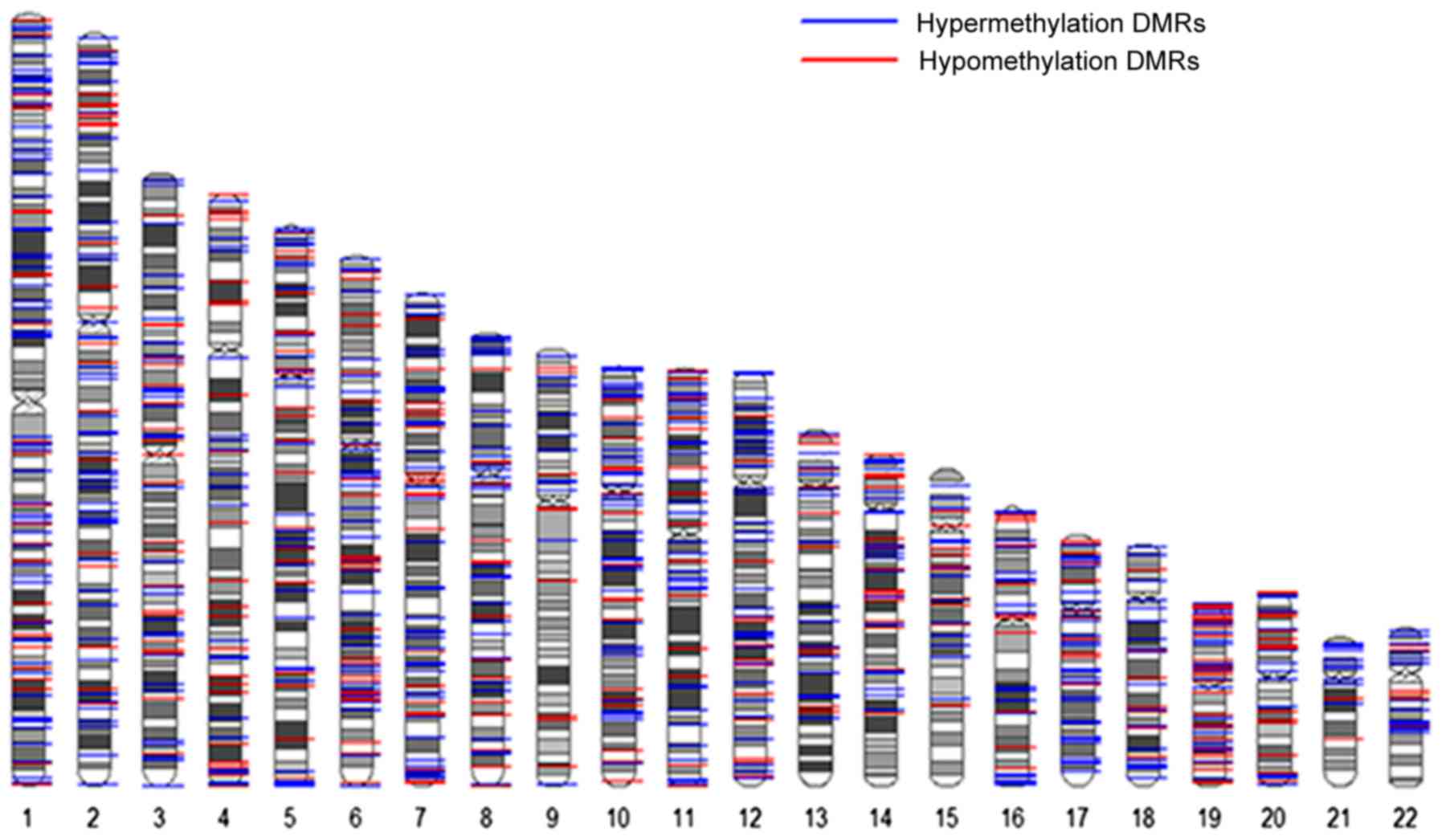
Significant differentially methylated
regions (DMRs)
The results of the present study revealed that 390
significantly hypermethylated CpG sites (covering 287 genes) and
208 significantly hypomethylated CpG sites (covering 203 genes)
were in DMRs. For the significantly hypermethylated CpG sites, 64
sites were in cancer-specific (c)-DMRs, 125 sites were in
reprogramming-specific (r)-DMRs and 201 sites were in DMRs; for the
significantly hypomethylated CpG sites, 30 were located within
cDMRs, 74 were located within rDMRs and 104 were located within
DMRs (Table VI; Fig. 3).
| Table VI.Significant differentially methylated
regions. |
Table VI.
Significant differentially methylated
regions.
| DMRs | Significantly
hypermethylated sites (n) | Significantly
hypermethylated sites (n) |
|---|
| cDMRs | 64 | 30 |
| Island | 16 | 4 |
| Shores | 36 | 18 |
| Shelves | 5 | 3 |
| Open sea | 7 | 5 |
| rDMRs | 125 | 74 |
| Island | 38 | 13 |
| Shores | 70 | 50 |
| Shelves | 7 | 3 |
| Open sea | 10 | 8 |
| DMRs | 201 | 104 |
| Island | 179 | 70 |
| Shores | 13 | 21 |
| Shelves | 3 | 0 |
| Open sea | 6 | 13 |
| Total | 390 | 208 |
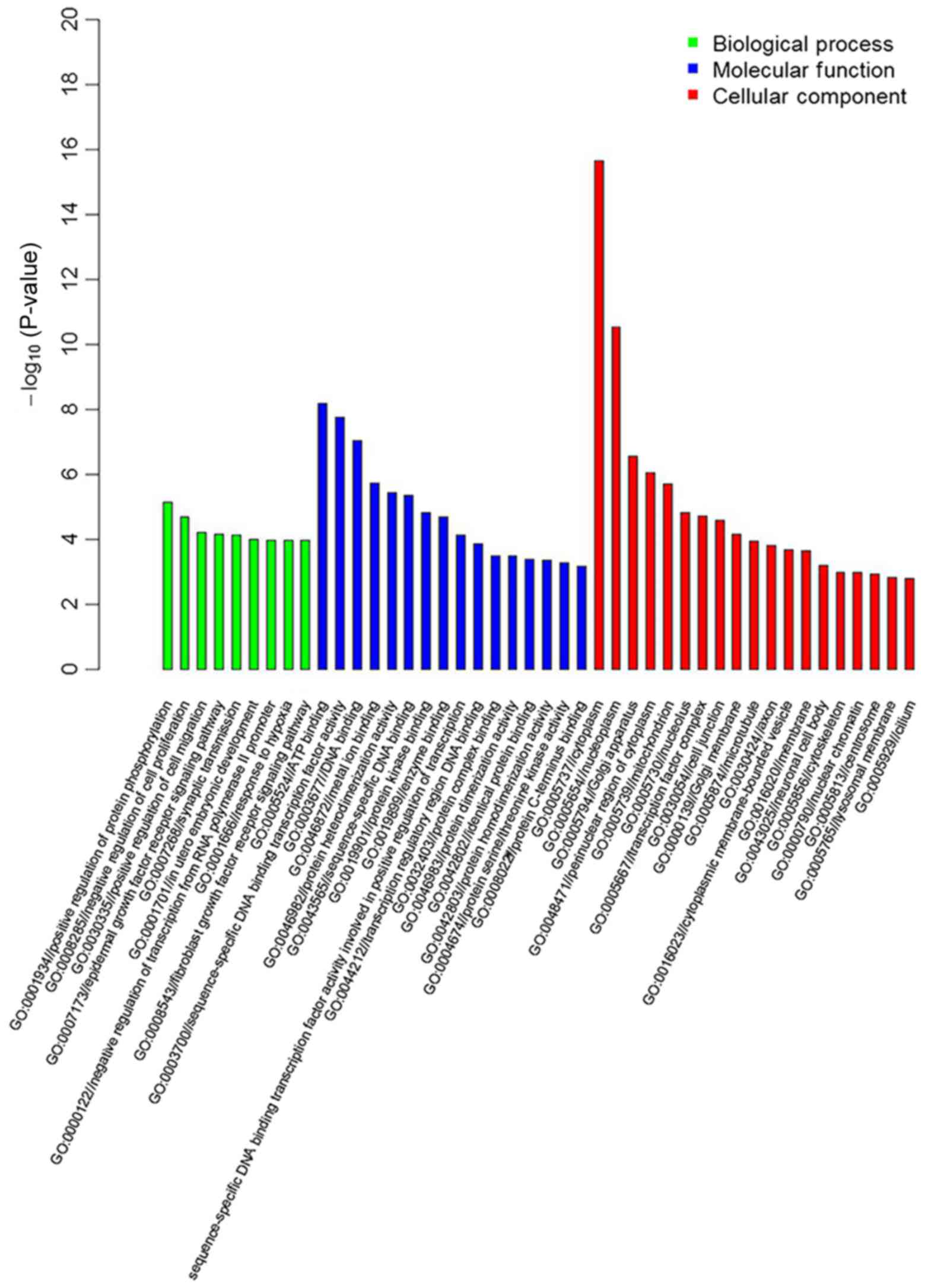
GO enrichment and KEGG pathway
analysis
GO enrichment and the KEGG Pathway database were
employed to analyze information regarding the differentially
methylated genes. The results of GO enrichment revealed that there
were 2,107 differentially methylated genes associated with
‘biological process’, and the most enriched groups included
negative regulators of cell proliferation, negative regulators of
transcription such as RNA polymerase II promoter, and synaptic
transmission. A total of 13,351 differentially methylated genes
were associated with ‘molecular function’, and the most enriched
groups included protein binding, DNA binding and metal ion binding.
A total of 18,041 differentially methylated genes were associated
with ‘cellular component’, and the most enriched groups included
the nucleus, cytoplasm and cytosol (Fig. 4). The top 20 significant
differentially methylated genes obtained from GO enrichment
analysis are listed in Table
VII.
| Table VII.Top 20 significant differentially
methylated genes in Gene Ontology enrichment. |
Table VII.
Top 20 significant differentially
methylated genes in Gene Ontology enrichment.
| GO enrichment | Top 20
significantly hypermethylated genes in GO enrichment | Top 20
significantly hypomethylated genes in GO enrichment |
|---|
| Biological
process |
|
Positive regulation of protein
phosphorylation | ERBB4 | – |
|
Negative regulation of cell
proliferation | ERBB4 | – |
|
Epidermal growth factor
receptor signaling | ERBB4 | – |
| pathway |
|
Synaptic transmission | PCDH8 | – |
|
Negative regulation of
transcription from RNA | SALL4; NKX6-2; | – |
| polymerase II
promoter | MEIS2; TCF21;
ZBTB32; |
|
|
Fibroblast growth factor
receptor signaling pathway | ERBB4 | – |
| Molecular
function |
| Protein
binding | ERBB4; PAX6; BCL3;
DOCK1; ZBTB32; BCL2 | PLXND1; LASP1;
MMP9; DKK1; DNAJB1; RB1; GAS1; AHCYL1 |
| ATP
binding | ERBB4 | – |
|
Sequence-specific DNA binding
transcription factor activity | PAX6; NKX6-2;
BCL3 | SIX2; RB1 |
| DNA
binding | PAX6; SALL4; BCL3;
ZIC4; ZBTB32 | PRDM8; RB1 |
| Metal
ion binding | SALL4; ZIC4;
ZBTB3 | PRDM8; ARSG;
NPTX1 |
| Protein
heterodimerization activity | BCL2 | NPR3; SDCBP2 |
|
Sequence-specific DNA
binding | BCL2 | SIX2 |
| Protein
kinase binding | PAX6 | – |
|
Transcription regulatory
region DNA binding | ERBB4; TCF21 | – |
| Protein
complex binding | – | SIX2 |
| Protein
dimerization activity | TCF21 | – |
|
Identical protein binding | BCL2 | MMP9; RB1 |
| Protein
homodimerization activity | ERBB4; BCL2 | SDCBP2 |
| Protein
C-terminus binding | – | SDCBP2 |
| Cellular
component |
|
Nucleus | ERBB4; PAX6; SALL4;
NKX6-2; BCL3; DOCK1; MEIS2; ZIC4; TCF21; ZBTB32; BCL2 | PRDM8; SIX2;
RB1 |
|
Cytosol | ERBB4 | – |
|
Cytoplasm | ERBB4; PAX6; SALL4;
BCL3; DOCK1; BCL2; BDNF | SDCBP2; DNAJB1 |
|
Nucleoplasm | ERBB4; ZBTB32 | RB1 |
| Golgi
apparatus | – | STC2 |
|
Perinuclear region of
cytoplasm | BCL3; BDNF | NPTX1 |
|
Mitochondrion | ERBB4; BCL2 | SLC25A42 |
|
Nucleolus | ERBB4; PAX6 | DNAJB1; RB1 |
|
Transcription factor
complex | LRFN2 | – |
| Cell
junction | PCDH | – |
|
Membrane | ERBB4; DOCK1;
BCL2 | SLC16A6 |
| Nuclear
chromatin | PAX6 | – |
KEGG Pathway-based analyses revealed that 43
signaling pathways involved 5,195 differentially methylated genes,
and these genes were significantly enriched in specific pathways,
including the cancer, metabolic, mitogen-activated protein kinase
(MAPK), calcium, Wnt, hepatitis C, Erb-B2 receptor tyrosine kinase
(ErbB), transforming growth factor (TGF)-β, vascular endothelial
growth factor (VEGF), p53 and Notch signaling pathways (Table VIII).
| Table VIII.Kyoto encyclopedia of genes and
genomes pathway analysis of differentially methylated genes. |
Table VIII.
Kyoto encyclopedia of genes and
genomes pathway analysis of differentially methylated genes.
| Pathway | Number of
differentially methylated genes (n) |
|---|
| Pathways in
cancer | 309 |
| Focal adhesion | 186 |
| MAPK signaling
pathway | 251 |
| Wnt signaling
pathway | 143 |
| Axon guidance | 120 |
| TGF-β signaling
pathway | 81 |
| Basal cell
carcinoma | 55 |
| Regulation of actin
cytoskeleton | 191 |
| Colorectal
cancer | 61 |
| Adherens
junction | 71 |
| Chronic myeloid
leukemia | 71 |
| ECM-receptor
interaction | 81 |
| Endocytosis | 186 |
| Pyrimidine
metabolism | 97 |
| Non-small cell lung
cancer | 57 |
| Hedgehog signaling
pathway | 54 |
| Neurotrophin
signaling pathway | 116 |
| Glioma | 63 |
| Endometrial
cancer | 52 |
| VEGF signaling
pathway | 71 |
| ErbB signaling
pathway | 80 |
| Small cell lung
cancer | 80 |
| Lysosome | 110 |
| Metabolic
pathways | 964 |
| Calcium signaling
pathway | 159 |
| Purine
metabolism | 150 |
| Ubiquitin mediated
proteolysis | 128 |
| Insulin signaling
pathway | 128 |
| Notch signaling
pathway | 45 |
| Protein processing
in endoplasmic reticulum | 156 |
| RNA polymerase | 32 |
| Hepatitis C | 116 |
| Renal cell
carcinoma | 61 |
| Aminoacyl-tRNA
biosynthesis | 42 |
| B cell receptor
signaling pathway | 69 |
| Thyroid cancer | 29 |
| Melanoma | 65 |
| Oocyte meiosis | 103 |
| Adipocytokine
signaling pathway | 64 |
| Melanogenesis | 94 |
| Vascular smooth
muscle contraction | 115 |
| Selenocompound
metabolism | 26 |
| p53 signaling
pathway | 63 |
Discussion
DNA methylation is the main epigenetic modification
and regulator of gene expression in humans. Aberrant changes in
genomic methylation patterns have been observed in many cancer cell
lines; these are regarded as the major type of molecular aberration
in malignancies (7,8). Previous studies have evaluated
aberrant DNA methylation in HCC by analyzing tumor tissues and
adjacent non-tumor tissues (14,21–23).
Their main aims were to identify novel potential biomarkers for the
diagnosis of HCC or to study the associations between methylation
and cirrhosis-associated HCC; the study design and enrolment
criteria differed when selecting various patients for study. In
addition, variations in ethnicity and the effects of the cutting
edge of adjacent non-tumor tissues may lead to differing results
observed across the different studies. Although previous studies
have indicated a few novel DNA methylation markers that are
associated with HCC, specific DNA methylation patterns associated
with the progression of HCC and alterations in methylation between
HCC and normal liver cells have yet to be identified. In the
present study, Illumina Infinium HumanMethylation 450K BeadChip was
used to identify global DNA methylation profiles in Huh7 and L02
cells.
In the present study, a total of 102,254
differentially methylated CpG sites were detected across the
whole-genome of Huh7 and L02 cells; more hypermethylated CpG sites
(62,702; 61.3%) were observed than hypomethylated (39,552; 38.7%)
CpG sites. The results indicated that within Huh7 cells, aberrant
DNA methylation was a very common event and that hypermethylation
of CpG sites occurred more frequently than hypomethylation. In
addition, stringent criteria were employed to select the
significantly differentially methylated CpG sites, genes and DMRs.
Finally, 5,285 (66.5%) significantly hypermethylated and 2,659
(33.5%) hypomethylated CpG sites were identified. It has been
reported that, in many types of diseases including cancers,
aberrant DNA methylation is a common event, particularly aberrant
DNA methylation of CpG islands or within promoter regions, which
are associated with tumor suppressor gene inactivation or oncogene
activation (16). The results of
the present study indicated that within a CpG island, a greater
number of significantly hypermethylated CpG sites (1,544) were
observed than significantly hypomethylated CpG sites (1,201). This
result is consistent with previous HCC genome-wide methylation
studies (14,24–27).
Yates et al (18) and
Dudziec et al (19)
demonstrated that aberrant DNA methylation occurs in CpG islands,
but can also be detected in the regions adjacent to CpG islands,
CpG shores and CpG shelves (15),
and may lead to tumorigenesis (20,28,29).
The present study also demonstrated these points; significantly
hypermethylated CpG sites in the CpG shores regions were more
abundant than significantly hypomethylated CpG sites (1,137 to
632). In addition, in the CpG shelf regions, the significantly
hypermethylated CpG sites were more frequent than significantly
hypomethylated CpG sites (655 to 133).
DMRs are stretches of DNA in the genome. Varied DNA
methylation patterns are seen between different organisms, and
adjacent sites or a group of sites in proximity to each other tend
to have different methylation patterns between different diseases
(30). DMRs are associated with
many diseases including several types of cancer (31). There are also many types of DMRs:
Tissue-specific DMRs, cDMRs, rDMRs, imprinting-specific DMRs and
aging-specific DMRs (20). In the
present study, there were 390 differentially hypermethylated CpG
sites located within DMRs, 233 (59.7%) were in island regions, 119
(30.5%) were in shore regions, and 15 (3.8%) were in shelf regions.
In addition, there were 208 differentially hypomethylated CpG sites
located within DMRs, 87 (41.8%) were in CpG island regions, 89
(42.8%) were in shore regions and 6 (2.9%) were in shelf regions.
These results indicated that within HCC cells, aberrant DNA
methylation may occur within CpG shore regions, which can also
cause DNA transcriptional silencing and inactivation of gene
function. Hepatocarcinogenesis was also associated with genomic
instability and inactivation of gene function; the results of the
present study concerning DMRs suggests that aberrant methylation
within these sites may be an important epigenetic mechanism
associated with hepatocarcinogenesis. These results may provide
more information regarding the associations between HCC and
aberrant DNA methylation.
Furthermore, the present study listed the top 20
significantly hyper- and hypo-methylated CpG sites, and genes in
DMRs within Huh7 cells compared with L02 cells. The top 20
significantly hypermethylated genes, which were high-ranking with
notable differences in the absolute value of β-difference, included
the following: ERBB4, paired box 6, splat like transcription factor
4, protocadherin (PCDH)-8, NK2 homeobox 6, B-cell lymphoma (BCL)-3,
glypican 6, family with sequence similarity 196 member A, dedicator
of cytokinesis 1, Meis homeobox 2, Zic family member 4,
transcription factor 21, zinc finger and BTB domain containing 32,
leucine rich repeat and fibronectin type III domain containing 2,
BCL2, PCDH9, DPY30 domain-containing protein 2, zinc finger
CCHC-type containing 24, brain-derived neurotrophic factor,
cg00057722 and cg08640046. The functional role of these genes in
HCC requires further study. The top 20 significant differentially
hyper- and hypo-methylated genes from GO enrichment were also
listed. These genes, which were located within DMRs, were mainly
associated with ‘cell differentiation development’, ‘transcription
factor activity’, ‘sequence-specific DNA binding’, ‘cellular
development process’ and ‘cell junction’.
Additionally, through GO enrichment analysis, the
present study revealed that aberrant DNA methylation in HCC was
associated with cell differentiation and proliferation, and through
KEGG pathway analysis, 43 signaling pathways associated with HCC
were identified, including pathways in cancer, MAPK signaling, Wnt
signaling, VEGF signaling and p53 signaling pathways. Previous
studies have demonstrated that aberrant DNA hypermethylation can
downregulate the expression of cell cycle inhibitors,
p16INK4A, p53 and factors involved in TGF-β/mothers
against decapentaplegic signaling (32,33).
Thus far, researchers have revealed that the inactivation of Wnt
pathway-associated antagonists is linked to the aberrant DNA
hypermethylation of some genes (34,35).
Activation of the ERB receptor and MAPK signaling pathways, as well
as the regulation of epigenetic proteins that were previously
demonstrated to promote cancer growth and metastasis, have been
reported to be possible candidate targets for anticancer treatment
in multiple types of cancer, including HCC (32,36).
In addition, HCC cells may escape or become tolerant
to chemotherapy via various mechanisms, therefore, identifying
novel drugs is very important for the future therapy of HCC. The
application of inhibitors of DNA methylated drugs in the treatment
of cancer has gradually attracted the attention of researchers
(37), including 5-azacytidine
(5-aza-C), decitabine (5-aza-2′-deoxycytidine, 5-aza-dC),
1-β-D-arabinofuranosyl-5-azacytosine, dihydro-5-azacytosine
(38), SGI-110 (previously known
as S110), a dinucleotide of 5-aza-2′-deoxycytidine and
deoxyguanosine, containing 5-azaCdR moiety, which has been revealed
to be very effective in inhibiting DNA methylation, though its
stability and cytotoxicity are comparable to that of decitabine
(39), and a non-nucleoside DNA
methyltransferase inhibitor, SGI-1027 (40,41).
To the best of our knowledge, there have been only a few studies
investigating the effects of demethylation agents on HCC in
vitro.
In conclusion, the present study detected
genome-wide DNA methylation patterns occurring in Huh7 cells, and
identified numerous differentially hypo- and hypermethylated CpG
sites, genes, DMRs and signaling pathways associated with HCC.
Additionally, the diversity in methylation within Huh7 cells was
also observed. The results of the present study may provide
important information regarding the molecular mechanisms underlying
methylation in Huh7 cells, which may be useful in future research
into the underlying mechanisms associated with HCC. In addition,
HCC cells may escape or develop tolerance to chemotherapy via
various mechanisms, therefore, identifying novel drugs is very
important for future therapies of HCC. The application of
inhibitors of DNA methylation for the treatment of cancer has
gradually attracted more attention within the field (37), and there have been a few studies
investigating the effects of demethylation agents in HCC in
vitro. The results of the present study may provide a useful
basis for future research into effective HCC therapies.
Acknowledgements
Not applicable.
Funding
The present study was supported by The Science and
Technology project of Shenyang (grant no. F13-212-9-00).
Availability of data and materials
The analyzed datasets generated during this study
are available from the corresponding author on reasonable
request.
Authors' contributions
JZ conceived and designed the study. NS, CZ, YS, BZ
and BC performed the experiments. NS and AJ analyzed the data. NS
wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PLC
|
primary liver cancer
|
|
HCC
|
hepatocellular carcinoma
|
|
DMR
|
differentially methylated regions
|
|
GO
|
Gene Ontology
|
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zuo TT, Zheng RS, Zhang SW, Zeng HM and
Chen WQ: Incidence and mortality of liver cancer in China in 2011.
Chin J Cancer. 34:562015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tanaka M, Katayama F, Kato H, Tanaka H,
Wang J, Qiao YL and Inoue M: Hepatitis B and C virus infection and
hepatocellular carcinoma in China: A review of epidemiology and
control measures. J Epidemiol. 21:401–416. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Panayiotidis MI: Cancer epigenetics as
biomarkers of clinical significance. Cancer Lett. 342:168–169.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Udali S, Guarini P, Moruzzi S, Ruzzenente
A, Tammen SA, Guglielmi A, Conci S, Pattini P, Olivieri O,
Corrocher R, et al: Global DNA methylation and hydroxymethylation
differ in hepatocellular carcinoma and cholangiocarcinoma and
relate to survival rate. Hepatology. 62:496–504. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jones PA and Takai D: The role of DNA
methylation in mammalian epigenetics. Science. 293:1068–1070. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jain S, Xie L, Boldbaatar B, Lin SY,
Hamilton JP, Meltzer SJ, Chen SH, Hu CT, Block TM, Song W and Su
YH: Differential methylation of the promoter and first exon of the
RASSF1A gene in hepatocarcinogenesis. Hepatol Res. 45:110–123.
2015. View Article : Google Scholar
|
|
10
|
Hinrichsen I, Kemp M, Peveling-Oberhag J,
Passmann S, Plotz G, Zeuzem S and Brieger A: Promoter methylation
of MLH1, PMS2, MSH2 and p16 is a phenomenon of advanced-stage HCCs.
PLoS One. 9:e844532014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu B, Nie Y, Liu X, Feng S, Yang Z, Wang
Z, Zheng Q and Luo X: Quantitative analysis of APC promoter
methylation in hepatocellular carcinoma and its prognostic
implications. Oncol Lett. 7:1683–1688. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jain S, Chen S, Chang KC, Lin YJ, Hu CT,
Boldbaatar B, Hamilton JP, Lin SY, Chang TT, Chen SH, et al: Impact
of the location of CpG methylation within the GSTP1 gene on its
specificity as a DNA marker for hepatocellular carcinoma. PLoS One.
7:e357892012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qu Z, Jiang Y, Li H, Yu DC and Ding YT:
Detecting abnormal methylation of tumor suppressor genes GSTP1,
P16, RIZ1, and RASSF1A in hepatocellular carcinoma and its clinical
significance. Oncol Lett. 10:2553–2558. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen J, Wang S, Zhang YJ, Wu HC, Kibriya
MG, Jasmine F, Ahsan H, Wu DP, Siegel AB, Remotti H and Santella
RM: Exploring genome-wide DNA methylation profiles altered in
hepatocellular carcinoma using Infinium HumanMethylation 450
BeadChips. Epigenetics. 8:34–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bibikova M, Barnes B, Tsan C, Ho V,
Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et
al: High density DNA methylation array with single CpG site
resolution. Genomics. 98:288–295. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Esteller M: Epigenetic gene silencing in
cancer: The DNA hypermethylome. Hum Mol Genet. 16:R50–R59. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yates DR, Rehman I, Meuth M, Cross SS,
Hamdy FC and Catto JW: Methylational urinalysis: A prospective
study of bladder cancer patients and age stratified benign
controls. Oncogene. 25:1984–1988. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dudziec E, Miah S, Choudhry HM, Owen HC,
Blizard S, Glover M, Hamdy FC and Catto JW: Hypermethylation of CpG
islands and shores around specific microRNAs and mirtrons is
associated with the phenotype and presence of bladder cancer. Clin
Cancer Res. 17:1287–1296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z,
Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al:
The human colon cancer methylome shows similar hypo- and
hypermethylation at conserved tissue-specific CpG island shores.
Nat Genet. 41:178–186. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nishida N, Kudo M, Nagasaka T, Ikai I and
Goel A: Characteristic patterns of altered DNA methylation predict
emergence of human hepatocellular carcinoma. Hepatology.
56:994–1003. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shen J, Wang S, Zhang YJ, Kappil M, Wu HC,
Kibriya MG, Wang Q, Jasmine F, Ahsan H, Lee PH, et al: Genome-wide
DNA methylation profiles in hepatocellular carcinoma. Hepatology.
55:1799–1808. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Udali S, Guarini P, Ruzzenente A,
Ferrarini A, Guglielmi A, Lotto V, Tononi P, Pattini P, Moruzzi S,
Campagnaro T, et al: DNA methylation and gene expression profiles
show novel regulatory pathways in hepatocellular carcinoma. Clin
Epigenetics. 7:432015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao W, Kondo Y, Shen L, Shimizu Y, Sano T,
Yamao K, Natsume A, Goto Y, Ito M, Murakami H, et al: Variable DNA
methylation patterns associated with progression of disease in
hepatocellular carcinomas. Carcinogenesis. 29:1901–1910. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shin SH, Kim BH, Jang JJ, Suh KS and Kang
GH: Identification of novel methylation markers in hepatocellular
carcinoma using a methylation array. J Korean Med Sci.
25:1152–1159. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ammerpohl O, Pratschke J, Schafmayer C, et
al: Distinct DNA methylation patterns in cirrhotic liver and
hepatocellular carcinoma. Int J Cancer. 130:1319–1328. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kohles N, Nagel D, Jüngst D, Durner J,
Stieber P and Holdenrieder S: Prognostic relevance of oncological
serum biomarkers in liver cancer patients undergoing transarterial
chemoembolization therapy. Tumour Biol. 33:33–40. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Doi A, Park IH, Wen B, Murakami P, Aryee
MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al:
Differential methylation of tissue- and cancer-specific CpG island
shores distinguishes human induced pluripotent stem cells,
embryonic stem cells and fibroblasts. Nat Genet. 41:1350–1353.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ogoshi K, Hashimoto S, Nakatani Y, Qu W,
Oshima K, Tokunaga K, Sugano S, Hattori M, Morishita S and
Matsushima K: Genome-wide profiling of DNA methylation in human
cancer cells. Genomics. 98:280–287. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rakyan VK, Down TA, Balding DJ and Beck S:
Epigenome-wide association studies for common human diseases. Nat
Rev Genet. 12:529–541. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Weber M, Davies JJ, Wittig D, Oakeley EJ,
Haase M, Lam WL and Schübeler D: Chromosome-wide and
promoter-specific analyses identify sites of differential DNA
methylation in normal and transformed human cells. Nat Genet.
37:853–862. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Calvisi DF, Pascale RM and Feo F:
Dissection of signal transduction pathways as a tool for the
development of targeted therapies of hepatocellular carcinoma. Rev
Recent Clin Trials. 2:217–236. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kuo KK, Jian SF, Li YJ, Wan SW, Weng CC,
Fang K, Wu DC and Cheng KH: Epigenetic inactivation of transforming
growth factor-beta1 target gene HEYL, a novel tumor suppressor, is
involved in the P53-induced apoptotic pathway in hepatocellular
carcinoma. Hepatol Res. 45:782–793. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Umer M, Qureshi SA, Hashmi ZY, Raza A,
Ahmad J, Rahman M and Iqbal M: Promoter hypermethylation of Wnt
pathway inhibitors in hepatitis C virus-induced multistep
hepatocarcinogenesis. Virol J. 11:1172014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ding SL, Yang ZW, Wang J, Zhang XL, Chen
XM and Lu FM: Integrative analysis of aberrant Wnt signaling in
hepatitis B virus-related hepatocellular carcinoma. World J
Gastroenterol. 21:6317–6328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stefanska B, Cheishvili D, Suderman M,
Arakelian A, Huang J, Hallett M, Han ZG, Al-Mahtab M, Akbar SM,
Khan WA, et al: Genome-wide study of hypomethylated and induced
genes in patients with liver cancer unravels novel anticancer
targets. Clin Cancer Res. 20:3118–3132. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yan W, Herman JG and Guo M:
Epigenome-based personalized medicine in human cancer. Epigenomics.
8:119–133. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ghoshal K and Bai S: DNA
methyltransferases as targets for cancer therapy. Drugs Today
(Barc). 43:395–422. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yoo CB, Jeong S, Egger G, Liang G,
Phiasivongsa P, Tang C, Redkar S and Jones PA: Delivery of
5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer
Res. 67:6400–6408. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Datta J, Ghoshal K, Denny WA, Gamage SA,
Brooke DG, Phiasivongsa P, Redkar S and Jacob ST: A new class of
quinoline-based DNA hypomethylating agents reactivates tumor
suppressor genes by blocking DNA methyltransferase 1 activity and
inducing its degradation. Cancer Res. 69:4277–4285. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gros C, Fleury L, Nahoum V, Faux C,
Valente S, Labella D, Cantagrel F, Rilova E, Bouhlel MA,
David-Cordonnier MH, et al: New insights on the mechanism of
quinoline-based DNA Methyltransferase inhibitors. J Biol Chem.
290:6293–6302. 2015. View Article : Google Scholar : PubMed/NCBI
|