Introduction
Atherosclerosis is the most frequent pathological
process resulting in cardiovascular diseases, a disease of medium-
and large-sized arteries characterized by the development of
atherosclerotic plaques consisting of foam cells, leukocytes,
endothelial cells (ECs), inflamed smooth muscle cells (SMCs),
built-up modified lipids, calcified regions and necrotic cores
(1). These characteristics of
atherosclerotic plaques show that atherosclerosis is a complex
disorder, and several compositions of the immune, metabolic and
vascular systems are involved in this process. It is well
demonstrated that diabetes mellitus is an independent risk factor
of cardiovascular disease and atherosclerosis (2). Several studies have shown that
patients with diabetes have higher risks of cardiovascular disease
and atherosclerosis, compared with normal subjects (3).
Endothelial dysfunction (ED) has been identified as
being associated with the clinical course and pathogenesis of all
identified cardiovascular disorders, occurring in response to
cardiovascular risk factors, followed by the occurrence of
atherosclerosis (4,5). ED is active in the process of lesion
formation, facilitating the late and early mechanisms of
atherosclerosis, and resulting in the promotion of EC permeability,
vascular SMC proliferation and migration, platelet activation and
oxidized-low density lipoprotein (ox-LDL), and the upregulation of
leukocyte adherence, cytokine secretion, chemokine and adhesion
molecules (5). For all these
factors, ED is one of the main mechanisms in atherosclerotic
disorders (6). The access of
insulin to the tissue can be decreased by compromised endothelial
functions in metabolic syndrome, and reduces the sensitivity of
insulin independently from the direct effects of the muscle cells
(7). The systemic knockout of
adipose triglyceride lipase (ATGL), a critical enzyme in
triglyceride lipolysis, leads to a murine phenotype featuring
severe heart failure and progredient cardiac steatosis (8). It has been shown that global ATGL
deficiency results in marked vascular endothelial dysfunction,
which is partially due to the downregulated vascular expression of
endothelial nitric oxide synthase (eNOS), possibly through
activation/upregulation of the 26S proteasome (8).
Long non-coding RNAs (lncRNAs) are a major family
that emerges from pervasive transcription, which broadly refer to
RNAs >200 nucleotides in length with no marked ability to code.
Current evidence has annotated and supported the identification of
>14,000 lncRNA gene units in the human genome (9). Of the genome, 1% (gencode v20)
comprises the exonic region of human lncRNAs, which contain
approximately the same quantity of DNA as protein-coding exons
(10). Equivalent, substantial
quantities of lncRNA genes are estimated to present in other
mammalian genomes (11). Certain
lncRNAs have been shown to have a functional role in the
pathogenesis of the occurrence of atherosclerosis; for example,
ANRIL is closely associated with the severity of atherosclerosis
(12). The H19 lncRNA is also
prevalent in the neointima following damage and in human
atherosclerotic lesions, but is rarely produced in normal arteries
(13). Their effects in
atherosclerosis, particularly in foam cells, have not been
reported, although HULC is important in cell lipid afflux. Hu et
al identified two lncRNAs in foam cells, namely
lncRNA-DYNLRB2-2 and lncRNA-RP5-833A20.1, which modulated
inflammation and foam cell cholesterol efflux (14).
It has been shown that SRA functions as a regulator
of ATGL indirectly by suppressing the expression of PPARγ, a
transcription factor of ATGL, and that ATGL deficiency may cause
the dysregulation of inflammatory cytokines, including tumor
necrosis factor (TNF)-α, and interleukin (IL)-6 (15,16).
The downregulation of ATGL is responsible for the dysfunction of
endothelial cells, which is one of the major causes of
atherosclerosis (8). In addition,
resveratrol (RSV) has been reported to suppress the inflammatory
reaction caused by the deficiency of ATGL (16). In the present study, the effects of
SRA and ATGL on endothelial cells were examined, and the effects of
alterations of SRA and ATGL on inflammatory cytokines and
endothelial cell dysfunction were investigated.
Materials and methods
Cell culture
HUVECs were obtained from Lonza (Basel,
Switzerland), and radioimmunoprecipitation assay (RPMI)-1640 medium
(Gibco; Thermo Fisher Scientific, Inc. Waltham, MA, USA) containing
10% fetal bovine serum (FBS; Sijiqing Biological Engineering
Materials Co. Ltd., Hangzhou, China) and 1% streptomycin-penicillin
was utilized to maintain the cells under an atmosphere of 5%
CO2/95% air at 37°C.
Vector construction
The pGL3-SRA human SRA expression vector was
generated by inserting the full sequence of SRA into the pGL3-Basic
vector (Promega Corporation, Madison, WI, USA) using BamHI
and HindIII endonucleases, and the full sequence of SRA was
amplified by PCR using DNA polymerase (Promega Corporation) and the
following primer sequences: Forward, 5′-CTCCTGAAGTGGGAAACGAAG-3′;
and reverse, 5′-GAGGTTGGCTTCCATGTCTAA-3′). The thermocycling
conditions used for PCR were as follows: 95°C for 3 min; followed
by 30 cycles of 94°C for 40 sec, 56°C for 35 sec and a final
extension at 72°C for 60 sec. All experiments were performed three
times.
Transfection
The cells were seeded into 96-well plates without
antibiotics at a final density of 2×104 per well. When
the HUVECs had grown to 80% confluence, Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) was utilized to
perform transfection. Three independent tests were run.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) was utilized to extract total RNA from HUVECs
according to the manufacturer's protocol. Subsequently, TaqMan
One-Step RT-PCR master mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.) was used to perform RT-qPCR analysis with a 25 µl
reaction mixture [2 µl cDNA template (2 µl), standard Taq reaction
buffer (2.5 µl), 10 mM dNTPs (0.5 µl), 10 µM forward primer (0.5
µl; 5′-CTCCTGAAGTGGGAAACGAAG-3′), 10 µM reverse primer (0.5 µl;
5′-GAGGTTGGCTTCCATGTCTAA-3′) and Taq DNA polymerase (1.25 µl), and
the remaining volume consisted of nuclease-free water]. The RT-qPCR
cycling conditions were as follows: Initial polymerase activation
at 95°C for 10 min, followed by 40 cycles for 15 sec at 95°C and 60
sec at 60°C. For the lncRNA SRA assay, a TaqMan RNA reverse
transcription kit (Applied Biosystems, Thermo Fisher Scientific,
Inc.) was used to reverse transcribe 1 µg of purified RNA to
single-stranded cDNA, the temperature protocol of which was
performed at 16°C for 30 min, followed by 42°C for 30 min, 85°C for
5 min. Following this, qPCR was performed in 48-well plates with a
Step One Plus sequence detection system (Applied Biosystems, Thermo
Fisher Scientific, Inc.), and the thermocycling conditions used
were as follows: 2 min at 50°C and 10 min at 95°C; followed by 30
cycles of 15 sec at 95°C and 60 sec at 60°C. U6 served as an
internal control. The comparative quantification (Cq) method
(17) was utilized to calculate
the relative lncRNA expression of SRA, and mRNA expression of ATGL
and PPARγ. Three independent reactions were repeated.
Luciferase assay
PCR was performed to amplify the promoter region of
the ATGL human gene from the genomic DNA, and the full region of
the ATGL promoter was treated with endonuclease digestion sites
overhangs. The PCR-amplified fragments were then ligated to the
pGL3-Basic vector (Promega Corporation) to generate the
pGL3-ATGL-1LUC reporter construct. The expression vector of the
human SRA pGL3-SRA expression vector was obtained by inserting full
sequence of SRA into the pGL3-Basic vector (Promega Corporation).
HUVECs were seeded into 96-well plates at a final density of
2×104 per well. When the HUVECs had grown to 80%
confluence, Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) was used for co-transfection with the
pGL3-SRA SRA expression vector or luciferase reporter plasmids and
the pRL-TK-Renilla vector for luciferase assays. The
Dual-Luciferase reporter system (Promega Corporation) was used to
detect the luciferase activity of Renilla and firefly
luciferase at 48 h post-transfection according to the
manufacturer's protocol. All experiments were performed three
times.
Western blot analysis
RIPA lysis buffer (Roche Diagnostics, Basel,
Switzerland) was utilized to lyse HUVECs in accordance with the
manufacturer's protocol. A Pierce bicinchoninic acid assay protein
assay kit (Thermo Fisher Scientific, Inc.) was performed to
quantify total protein. A 10% (w/v) polyacrylamide gel was utilized
to separate protein extracts with SDS-PAGE, which were then
electro-transferred onto a BioTrace NT membrane (Pall Life
Sciences, Port Washington, NY, USA). PBS containing 0.1% Tween-20
and 5% fat-free milk was utilized to block the membrane for 60 min.
Primary anti-ATGL antibody at a dilution of 1:5,000 (cat. no.
3370-1; Epitomics, Burlingame CA, USA), anti-PPARγ antibody at a
dilution of 1:5,000 (cat. no. RN075PW; Medical & Biological
Laboratories Co., Ltd., Tokyo, Japan), and anti-β-actin antibody at
a dilution of 1:10,000 (cat. no. 4970; Cell Signaling Technology,
Inc., Danvers, MA, USA) were utilized to maintain membrane
overnight at 4°C. Subsequently, secondary horseradish peroxidase
(HRP)-labeled antibody at a dilution of 1:15,000 (cat. no. AP187P;
EMD Millipore, Billerica, MA, USA) was utilized to treat the
membrane at room temperature for 2 h. Electrochemiluminescence
blotting detection reagent (Thermo Fisher Scientific, Inc.) was
utilized to detect the bands of the target proteins. A Chemioscope
Mini system (ChemiQ 4800; Bioshine, Shanghai, China) was utilized
to quantify and capture images of the protein bands of ATGL and
PPARγ. Three independent tests were performed.
ELISA
Carbonate-bicarbonate buffer (50 mM) with mouse
polyclonal IgG against ATGL (1:5,000; cat. no. 2138; Cell Signaling
Technology, Inc.)/PPARγ (1:5,000; cat. no. sc-398394; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) was utilized to treat cells
(2×103 /well) in a 24-well plate, followed by incubation
for 12 h at 4°C, and PBS with 4% bovine serum albumin (BSA;
Invitrogen; Thermo Fisher Scientific, Inc.) was then utilized to
treat the plates for 1 h. PBS with 0.1% Tween-20 was utilized to
wash the plates twice for 3–5 min. A 50 µl volume of purified
intercellular adhesion molecule-1 (ICAM-1), tumor necrosis factor
(TNF)-α, IL-6 and monocyte chemotactic protein-1 (MCP-1) antigen as
well as clinical samples were distributed into 96-wells, and
incubated for 2 h at 37°C. This was followed by washing and culture
for 60 min at 37°C in 100 µl PBS containing l% BSA and rabbit
monoclonal IgG against ICAM-1 (cat. no. 4915; Cell Signaling
Technology, Inc.), TNF-α (cat. no. 6945; Cell Signaling Technology,
Inc.), IL-6 (cat. no. 12153) and MCP-1 (cat. no. 2027; both Cell
Signaling Technology, Inc.) at dilutions of 1:1,500. Subsequently,
secondary antibodies conjugated to HRP (1:3,000; cat. no. 7074;
Cell Signaling Technology, Inc.) and 1% BSA (LI-COR Biosciences,
Lincoln, NE, USA) were utilized to maintain the plate for 24 h at
37°C. A microplate reader was utilized to determine the immunoplate
HRP activity with the use of O-phenylenediamine in accordance with
the absorbance at 492 nm. H2O2 served as an
internal control. Three independent tests were performed.
Statistical analysis
All data are presented as the mean ± standard error
of mean of three independent experiments. SPSS 23.0 statistical
software (IBM SPSS, Armonk, NY, USA) was utilized to perform all
statistical analysis. Student's t-test (two-tailed) was utilized to
analyze differences between two groups, and the Wilcoxon signed
rank test was utilized to compare relative expression levels of
lncRNA SRA ATGL and PPARγ among groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
SRA suppresses the transactivation of
ATGL via PPARγ
SRA has been shown to inhibit the transcription
activity of the ATGL promoter, and it was well established that
PPARγ serves as a transcription factor of ATGL. Therefore, the
present study aimed to examine whether SRA repressed the
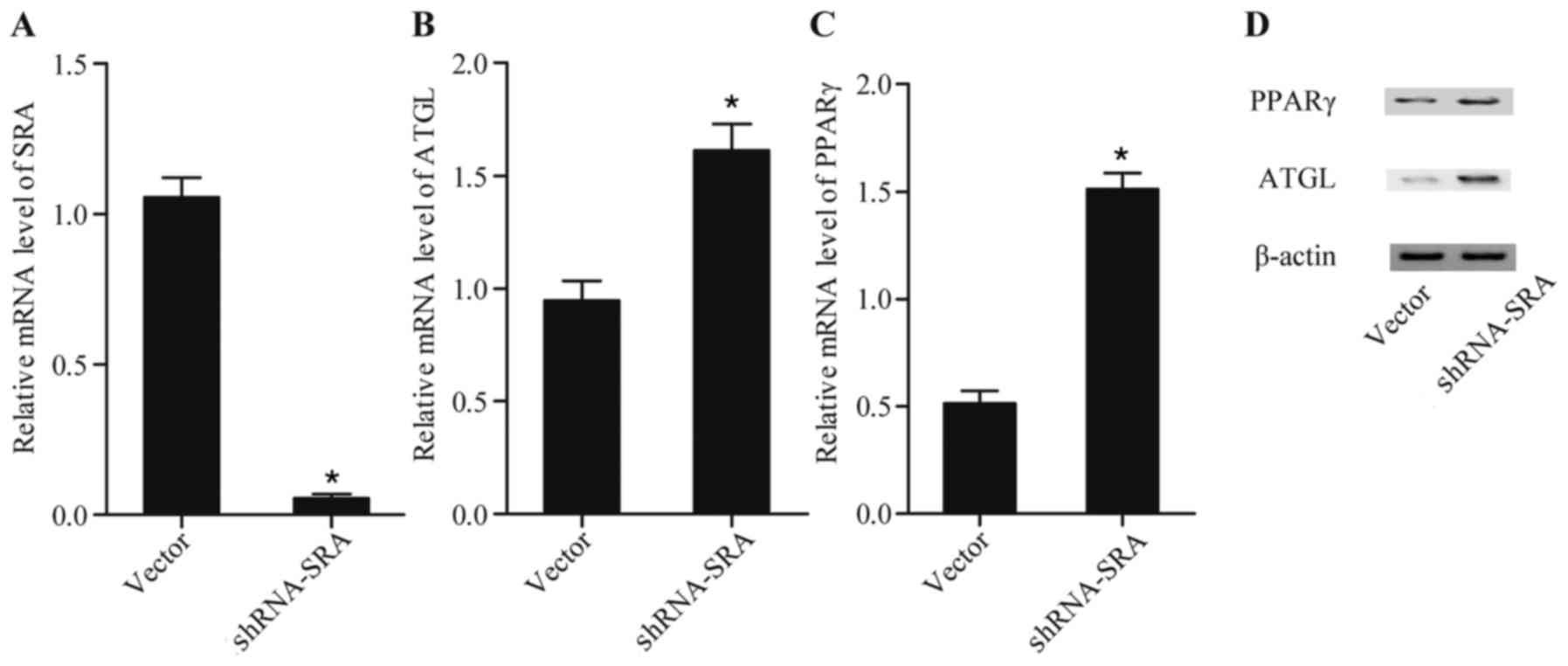
transactivation of ATGL through PPARγ. As shown in Fig. 1, knockdown of the expression of SRA
(Fig. 1A) using shRNA against SRA
substantially reduced the mRNA levels of expression ATLG (Fig. 1B) and PPARγ (Fig. 1C), and the protein (Fig. 1D) levels of the two. This suggested
that lncRNA-SRA suppressed the production of ATGL via inhibiting
the expression of PPARγ.
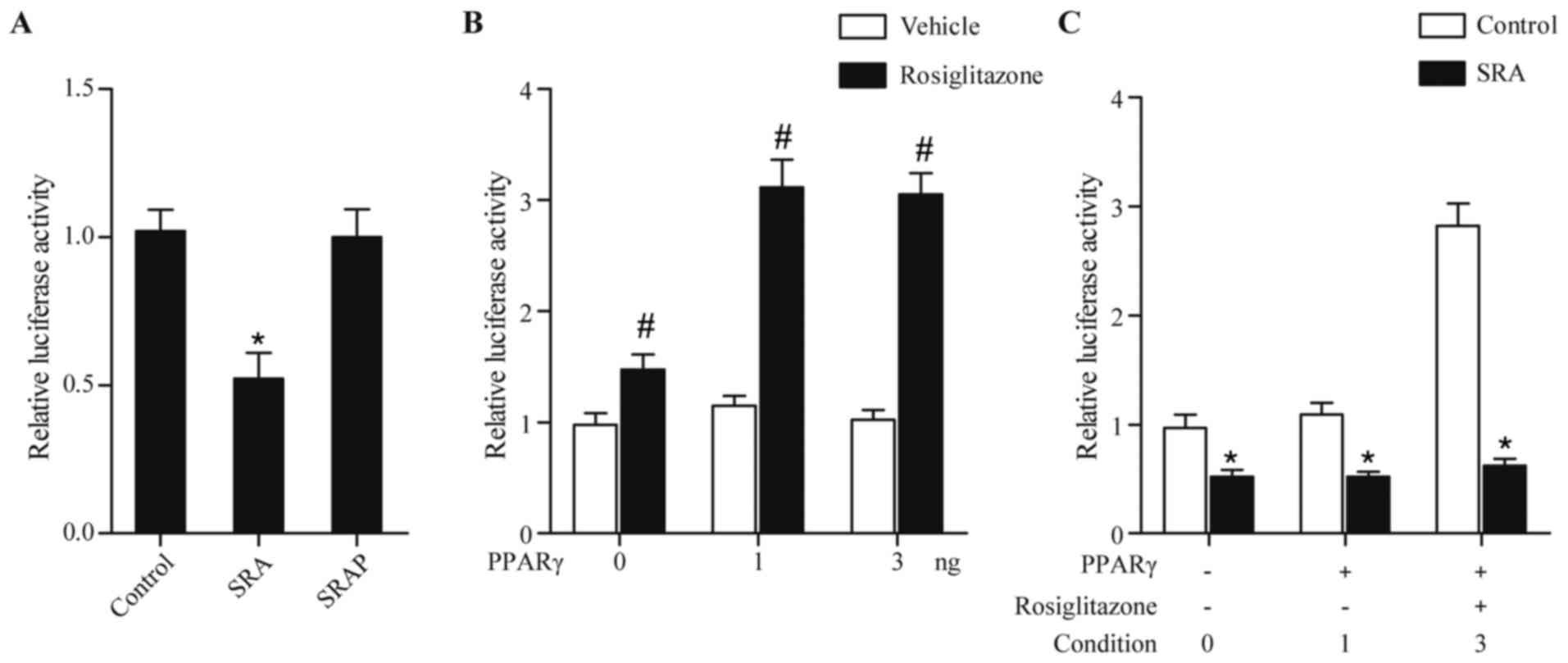
SRA represses the transcription
ability of the ATGL promoter via modulating PPARγ
The endothelial cells were transfected with vectors
encompassing SRA or SRAP. As shown in Fig. 2A, transfection of cells with a
vector coding SRA RNA but not SRAP protein reduced the luciferase
activity driven by the ATGL promoter, compared with control. PPARγ
is also considered to be a transcription factor of ATGL. Therefore,
it was crucial to clarify the effect of PPARγ on the expression of
ATGL. As shown in Fig. 2B, without
rosiglitazone, a PPARγ ligand, the upregulated expression of PPARγ
had no effect on the luciferase activity driven by the ATGL
promoter (Fig. 2B). However, in
the presence of rosiglitazone, PPARγ increased the luciferase
activity of the ATGL promoter, compared with that in the vehicle
control (Fig. 2B). As shown in
Fig. 2C, the promoting effect of
PPARγ and rosiglitazone on the activity of ATGL promoter was
eliminated by the overexpression of SRA (condition 3 in Fig. 2C), suggesting that SRA repressed
the luciferase activity driven by ATGL via suppressing PPARγ.
Repression of inflammatory-related
cytokine levels by SRA
It is well established that inflammation is
important in the development and progression of atherosclerosis,
therefore ELISA and western-blot analysis were performed in the
present study to investigate whether SRA was associated with
atherosclerosis via affecting the production of inflammatory
cytokines. As shown in Fig. 3,
higher protein levels of TNF-α, IL-6, MCP-1 and ICAM-1 (Fig. 3A and B) were observed in the HUVECs
with loss of the expression of SRA, compared with control
group.
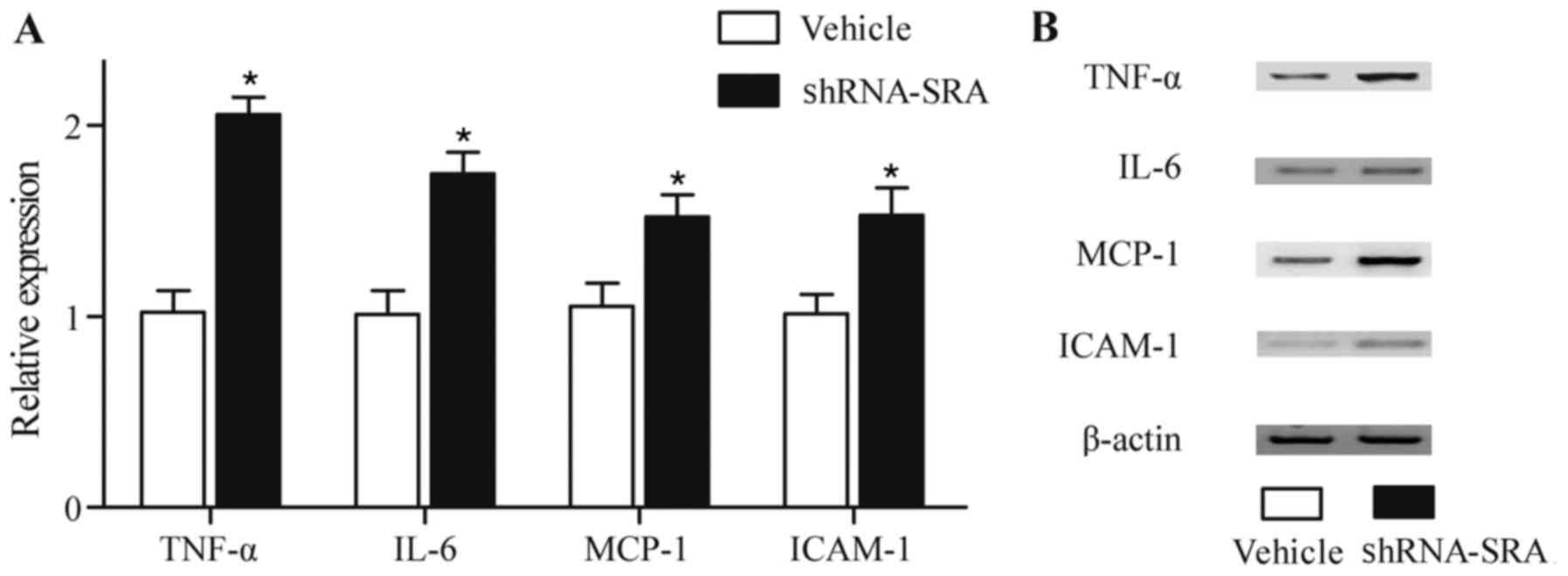 | Figure 3.Repression of inflammatory-related
cytokines by SRA. (A) Higher protein levels of TNF-α, IL-6, MCP-1
and ICAM-1 were detected in HUVECs following knockdown of the
expression of SRA, detected using ELISA. (B) Higher protein levels
of TNF-α, IL-6, MCP-1 and ICAM-1 were detected in HUVECs following
knockdown of the expression of SRA, detected using western blot
analysis. *P<0.01 vs. vehicle control group. SRA, steroid
receptor RNA activator; TNF-α, tumor necrosis factor-α; IL-6,
interleukin-6; MCP-1, monocyte chemotactic protein-1; ICAM-1,
intercellular adhesion molecule-1; shRNA, short hairpin RNA. |
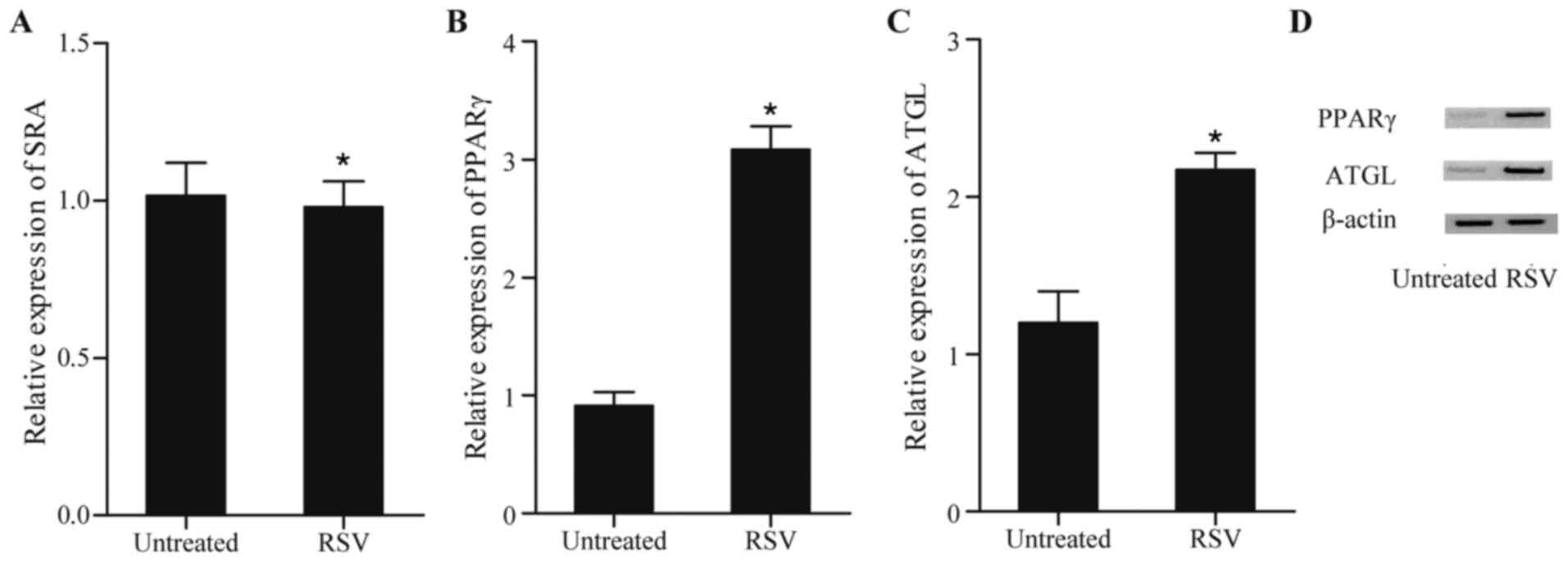
RSV alters the expression of SRA and
downstream of SRA
RSV is known as an inhibitor of inflammation and is
widely used as a drug to treat diseases caused by chronic
inflammation, including atherosclerosis (18). To examine whether RSV was involved
in inflammation through SRA and downstream, RT-qPCR analysis was
performed to examine the levels of SRA, PPARγ and ATGL in cells
treated with RSV. As shown in Fig.
4A, no significant difference in the levels of SRA were found
between the cells treated with RSV and the untreated cells. By
contrast, RSV markedly enhanced the expression of PPARγ (Fig. 4B) and ATGL (Fig. 4C) at the mRNA levels and protein
(Fig. 4D) levels, compared with
the levels in the untreated cells, confirming that RSV was involved
in inflammation through promoting the expression of PPARγ and
ATGL.
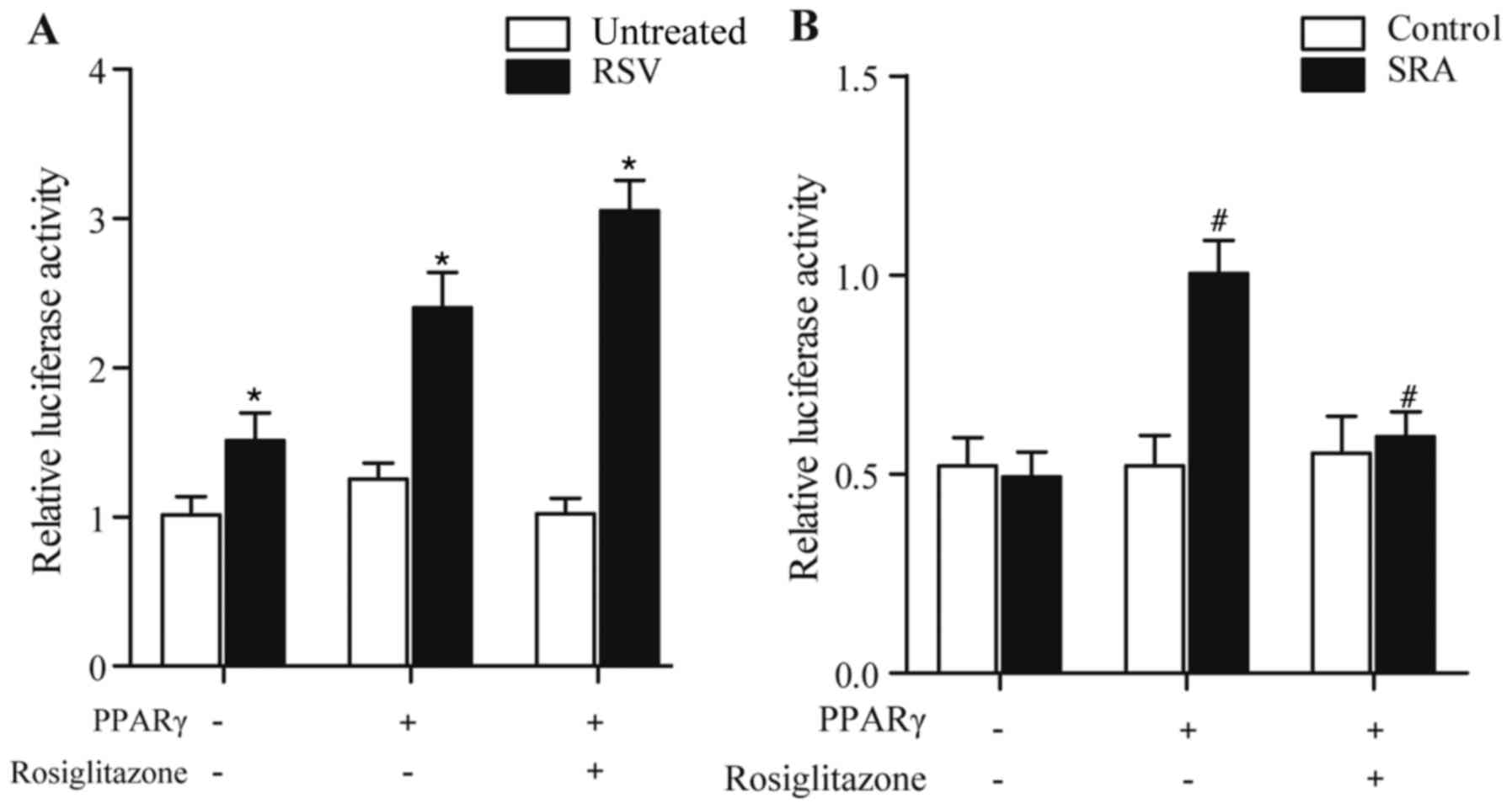
RSV represses the luciferase activity
driven by ATGL
As it was found that RSV enhanced the expression of
ATGL, luciferase constructs were generated to analyze this further,
following which the HUVECs were treated with different doses of RSV
and rosiglitazone. As shown in Fig.
5A, treatment with RSV elevated the luciferase activity driven
by ATGL in a concentration-dependent manner, compared with that in
the HUVECs not treated with RSV. The effect of RSV on the
luciferase activity driven by ATGL was inhibited by treatment with
rosiglitazone (Fig. 5B),
demonstrating that RSV attenuated the luciferase activity driven by
ATGL via PPARγ.
RSV eliminates the effect on
inflammatory-related cytokines induced by SRA
ELISA and western blot analysis were performed to
investigate whether RSV can eliminate the effect of SRA on the
production of inflammatory-related cytokines. As shown in Fig. 6A and B, treatment with RSV
eliminated the effect of treatment with SRA shRNA, suggesting that
RSV eliminated the effect on the production of TNF-α, IL-6, MCP-1
and ICAM-1 induced by SRA.
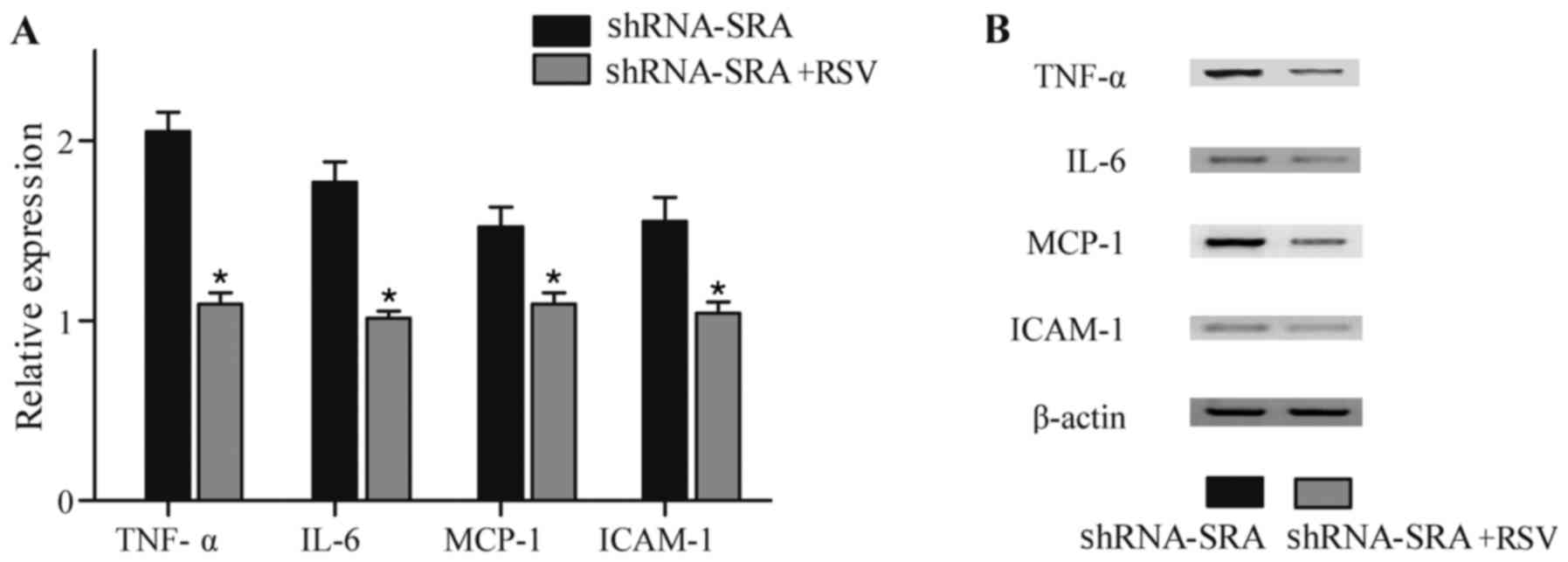 | Figure 6.RSV eliminates the effect of SRA on
inflammatory-related cytokines. (A) Based on the results of the
ELISA, the knockdown of SRA significantly increased the production
of TNF-α, IL-6, MCP-1 and ICAM-1, whereas RSV eliminated the
effects of treatment with SRA shRNA. (B) Base on results of the
western blot analysis, knockdown of SRA significantly upregulated
the production of TNF-α, IL-6, MCP-1 and ICAM-1, whereas RSV
eradicated the effect of SRA shRNA. *P<0.01 vs. shRNA-SRA group.
SRA, steroid receptor RNA activator; TNF-α, tumor necrosis
factor-α; IL-6, interleukin-6; MCP-1, monocyte chemotactic
protein-1; ICAM-1, intercellular adhesion molecule-1; shRNA, short
hairpin RNA; RSV, resveratrol. |
Discussion
Several lncRNAs have been reported to be involved in
cardiovascular disorders. SRA, a gene encoding an lncRNA and
initially identified as a steroid receptor coactivator in steroid
hormone target tissues, is situated in a 600-kb region of linkage
disequilibrium associated with cardiomyopathy. Depletion of the
SRA homolog in zebrafish leads to serious myocardial
dysfunction (19). It has also
been suggested that SRA enhances insulin-stimulated glucose uptake
and adipocyte differentiation in adipocytes in vitro via
multiple mechanisms, including suppressing the production of
adipocyte-related inflammatory genes or enhancing the expression of
insulin receptors, and coactivating the transcriptional activity of
PPARγ (20). Previously, to
evaluate the function of SRA in vivo, global Sra1
gene-knockout mice were established; these mice exhibited decreased
inflammation, improved insulin sensitivity and reduced hepatic
triacylglycerol contents (21).
Another clinical manifestation of atherosclerosis is erectile
dysfunction (ED), particularly in type II diabetes mellitus
(22,23). One key mechanism of ED in diabetes
mellitus is a change in the signaling pathways, which results in
the activation of eNOS in the endothelium. This process has been
widely investigated and SRA has been found to be a regulator of
this signaling pathway (24–27).
The present study examined whether SRA affected the transactivation
of ATGL through PPARγ using RT-qPCR and western blot analyses, and
it was found that SRA inhibited the expression of ATGL through
inhibiting the production of PPARγ. A luciferase assay was
performed to evaluate the effect of SRA on its transcription
ability, and it was found that the luciferase activity of the ATGL
promoter was downregulated following cell transfection with a
vector coding SRA RNA.
The dysfunction of endothelial cells is considered
to be important in the development of atherosclerosis, and the
dysregulated production of inflammatory cytokines may be a sign of
the dysfunction of endothelial cells (28,29).
In the present study, ELISA and western blot analysis were
performed to investigate whether SRA affected the production of
inflammatory cytokines, and it was revealed that SRF increased the
expression of TNF-α, IL-6, MCP-1 and ICAM-1.
ATGL was first identified as the main TG hydrolase
in heart and adipose tissue. Reports indicate it also has a primary
role in other tissues, including the liver (30). ATGL affects the downstream
partitioning of its fatty acid (FA) products. ATGL functions as a
critical modulator of PPAR in addition to its effects on FA
trafficking. It has been demonstrated that mice or primary
hepatocytes, which are exposed to ATGL shRNA have decreased
production of PPARα in addition to its downstream target genes
(30). These findings are in
accordance with microarray analysis of tissues in whole-body
ATGL-knockout mice and previous results revealing that ATGL
modulates brown adipose tissue, small intestine, and PPAR in the
heart (31,32).
FAs are considered the main physiological ligands to
stimulate PPAR, although a number of metabolites can bind to PPAR
(33). It has been shown that they
are not sufficient to eliminate the chronic inflammatory changes in
adipose tissue, which develop in the context of diet-induced
obesity, although the suppression of ATGL-mediated adipocyte
lipolysis decreases the adipose tissue inflammatory reaction to
acute lipolysis (34). Of note,
regardless of a comparable reduction in adipocyte lipolysis, global
ATGL-knockout mice exhibit elevated, not lower, susceptibility to
hepatic inflammation, likely due to the effect of hepatic ATGL
being necessary for hepatic lipid homeostasis and for stimulation
of the anti-inflammatory effects of PPARα (35). Compared with wild-type bone
marrow-transplanted LDL receptor (Ldlr)−/− mice following feeding
with an atherogenic Western-type diet for nine weeks, grafting of
Atgl−/− bone marrow into γ-irradiated Ldlr−/− mice led to
substantially reduced formation of atherosclerotic lesions.
However, compromised clearance of apoptotic macrophages in advanced
atherosclerotic lesions causes the formation of a necrotic core and
secondary necrosis. A similar effect was found without the presence
of ATGL, and TG built-up in macrophages with elevations in typical
markers of apoptosis, including the externalization of
phosphatidylserine in poly-(ADP-ribose) polymerase cleavage and
plasma membrane, and caspase-3 (36). In the present study, it was found
that RSV upregulated the luciferase activity of ATGL in a
concentration-dependent manner, whereas the luciferase activity of
ATGL in the cells treated with RSV and rosiglitazone was comparable
with that in the untreated cell. The present study also
investigated whether RSV can eliminate effect of SRA on the
production of inflammatory-related cytokines using ELISA and
western blot analysis, and it as revealed that RSV eliminated the
promoting effect of SRA on the production of TNF-α, IL-6, MCP-1 and
ICAM-1.
In conclusion, the results of the present study
indicated that lncRNA SRA promoted atherosclerosis in through
repressing the expression of ATGL. The upregulated SRA suppressed
the expression of ATGL, which induced endothelial dysfunction, and
endothelial dysfunction has been established to be involved in the
pathogenesis and clinical course of atherosclerosis. Therefore,
lncRNA SRA may be used as a novel biomarker of atherosclerosis in
patients with diabetes mellitus.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SY performed the experiments, collected, analyzed
and visualized the data, collected the literature and prepared the
manuscript. JS designed the study, performed the experiments,
analyzed the data, prepared the manuscript and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ross R: Atherosclerosis-an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Orchard TJ, Stevens LK, Forrest KY and
Fuller JH: Cardiovascular disease in insulin dependent diabetes
mellitus: Similar rates but different risk factors in the US
compared with Europe. Int J Epidemiol. 27:976–983. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Himmelmann A, Hansson L, Svensson A,
Harmsen P, Holmgren C and Svanborg A: Predictors of stroke in the
elderly. Acta Med Scand. 224:439–443. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Suwaidi JA, Hamasaki S, Higano ST,
Nishimura RA, Holmes DR Jr and Lerman A: Long-term follow-up of
patients with mild coronary artery disease and endothelial
dysfunction. Circulation. 101:948–954. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Butany JW, Verma S, Leask RL, Mohsen B and
Asa SL: Genetic abnormalities of the endothelium. Microsc Res Tech.
60:30–37. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Esper RJ, Nordaby RA, Vilariño JO,
Paragano A, Cacharrón JL and Machado RA: Endothelial dysfunction: A
comprehensive appraisal. Cardiovasc Diabetol. 5:42006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kolka CM and Bergman RN: The endothelium
in diabetes: Its role in insulin access and diabetic complications.
Rev Endocr Metab Disord. 14:13–19. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schrammel A, Mussbacher M, Wölkart G,
Stessel H, Pail K, Winkler S, Schweiger M, Haemmerle G, Al Zoughbi
W, Höfler G, et al: Endothelial dysfunction in adipose triglyceride
lipase deficiency. Biochim Biophys Acta. 1841:906–917. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Derrien T, Johnson R, Bussotti G, Tanzer
A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG,
et al: The GENCODE v7 catalog of human long noncoding RNAs:
Analysis of their gene structure, evolution, and expression. Genome
Res. 22:1775–1789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Harrow J, Frankish A, Gonzalez JM,
Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa
A, Searle S, et al: GENCODE: The reference human genome annotation
for The ENCODE Project. Genome Res. 22:1760–1774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Young RS, Marques AC, Tibbit C, Haerty W,
Bassett AR, Liu JL and Ponting CP: Identification and properties of
1,119 candidate lincRNA loci in the Drosophila melanogaster genome.
Genome Biol Evol. 4:427–442. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Holdt LM, Beutner F, Scholz M, Gielen S,
Gäbel G, Bergert H, Schuler G, Thiery J and Teupser D: ANRIL
expression is associated with atherosclerosis risk at chromosome
9p21. Arterioscler Thromb Vasc Biol. 30:620–627. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han DK, Khaing ZZ, Pollock RA,
Haudenschild CC and Liau G: H19, a marker of developmental
transition, is reexpressed in human atherosclerotic plaques and is
regulated by the insulin family of growth factors in cultured
rabbit smooth muscle cells. J Clin Invest. 97:1276–1285. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu YW, Yang JY, Ma X, Chen ZP, Hu YR, Zhao
JY, Li SF, Qiu YR, Lu JB, Wang YC, et al: A
lincRNA-DYNLRB2-2/GPR119/GLP-1R/ABCA1-dependent signal transduction
pathway is essential for the regulation of cholesterol homeostasis.
J Lipid Res. 55:681–697. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen G, Yu D, Nian X, Liu J, Koenig RJ, Xu
B and Sheng L: LncRNA SRA promotes hepatic steatosis through
repressing the expression of adipose triglyceride lipase (ATGL).
Sci Rep. 6:355312016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barbato Lettieri D, Tatulli G, Aquilano K
and Ciriolo MR: Inhibition of age-related cytokines production by
ATGL: A mechanism linked to the anti-inflammatory effect of
resveratrol. Mediators Inflamm. 2014:9176982014.PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feng Q, Su Z, Song S, Χu H, Zhang B, Yi L,
Tian M and Wang H: Histone deacetylase inhibitors suppress RSV
infection and alleviate virus-induced airway inflammation. Int J
Mol Med. 38:812–822. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Friedrichs F, Zugck C, Rauch GJ, Ivandic
B, Weichenhan D, Müller-Bardorff M, Meder B, El Mokhtari NE,
Regitz-Zagrosek V, Hetzer R, et al: HBEGF, SRA1, and IK: Three
cosegregating genes as determinants of cardiomyopathy. Genome Res.
19:395–403. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu B, Gerin I, Miao H, Vu-Phan D, Johnson
CN, Xu R, Chen XW, Cawthorn WP, MacDougald OA and Koenig RJ:
Multiple roles for the non-coding RNA SRA in regulation of
adipogenesis and insulin sensitivity. PLoS One. 5:e141992010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu S, Sheng L, Miao H, Saunders TL,
MacDougald OA, Koenig RJ and Xu B: SRA gene knockout protects
against diet-induced obesity and improves glucose tolerance. J Biol
Chem. 289:13000–13009. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Creager MA, Lüscher TF, Cosentino F and
Beckman JA: Diabetes and vascular disease: Pathophysiology,
clinical consequences, and medical therapy: Part I. Circulation.
108:1527–1532. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Benjamin EJ, Larson MG, Keyes MJ, Mitchell
GF, Vasan RS, Keaney JF Jr, Lehman BT, Fan S, Osypiuk E and Vita
JA: Clinical correlates and heritability of flow-mediated dilation
in the community: The Framingham Heart Study. Circulation.
109:613–619. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tabit CE, Chung WB, Hamburg NM and Vita
JA: Endothelial dysfunction in diabetes mellitus: Molecular
mechanisms and clinical implications. Rev Endocr Metab Disord.
11:61–74. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wheatcroft SB, Williams IL, Shah AM and
Kearney MT: Pathophysiological implications of insulin resistance
on vascular endothelial function. Diabet Med. 20:255–268. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Festa A, Hanley AJ, Tracy RP, D'Agostino R
Jr and Haffner SM: Inflammation in the prediabetic state is related
to increased insulin resistance rather than decreased insulin
secretion. Circulation. 108:1822–1830. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shoelson SE, Lee J and Yuan M:
Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity-
and diet-induced insulin resistance. Int J Obes Relat Metab Disord.
27 Suppl 3:S49–S52. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sourris KC, Lyons JG, de Courten MP,
Dougherty SL, Henstridge DC, Cooper ME, Hage M, Dart A, Kingwell
BA, Forbes JM and de Courten B: c-Jun NH2-terminal kinase activity
in subcutaneous adipose tissue but not nuclear factor-kappaB
activity in peripheral blood mononuclear cells is an independent
determinant of insulin resistance in healthy individuals. Diabetes.
58:1259–1265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Georgiou HM, Lappas M, Georgiou GM, Marita
A, Bryant VJ, Hiscock R, Permezel M, Khalil Z and Rice GE:
Screening for biomarkers predictive of gestational diabetes
mellitus. Acta Diabetol. 45:157–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ong KT, Mashek MT, Bu SY, Greenberg AS and
Mashek DG: Adipose triglyceride lipase is a major hepatic lipase
that regulates triacylglycerol turnover and fatty acid signaling
and partitioning. Hepatology. 53:116–126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ahmadian M, Abbott MJ, Tang T, Hudak CS,
Kim Y, Bruss M, Hellerstein MK, Lee HY, Samuel VT, Shulman GI, et
al: Desnutrin/ATGL is regulated by AMPK and is required for a brown
adipose phenotype. Cell Metab. 13:739–748. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Haemmerle G, Lass A, Zimmermann R,
Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C,
Eder S, et al: Defective lipolysis and altered energy metabolism in
mice lacking adipose triglyceride lipase. Science. 312:734–737.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chakravarthy MV, Lodhi IJ, Yin L, Malapaka
RR, Xu HE, Turk J and Semenkovich CF: Identification of a
physiologically relevant endogenous ligand for PPARalpha in liver.
Cell. 138:476–488. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schoiswohl G, Stefanovic-Racic M, Menke
MN, Wills RC, Surlow BA, Basantani MK, Sitnick MT, Cai L, Yazbeck
CF, Stolz DB, et al: Impact of reduced ATGL-mediated adipocyte
lipolysis on obesity-associated insulin resistance and inflammation
in male mice. Endocrinology. 156:3610–3624. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jha P, Claudel T, Baghdasaryan A, Mueller
M, Halilbasic E, Das SK, Lass A, Zimmermann R, Zechner R, Hoefler G
and Trauner M: Role of adipose triglyceride lipase (PNPLA2) in
protection from hepatic inflammation in mouse models of
steatohepatitis and endotoxemia. Hepatology. 59:858–869. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lammers B, Chandak PG, Aflaki E, Van
Puijvelde GH, Radovic B, Hildebrand RB, Meurs I, Out R, Kuiper J,
Van Berkel TJ, et al: Macrophage adipose triglyceride lipase
deficiency attenuates atherosclerotic lesion development in
low-density lipoprotein receptor knockout mice. Arterioscler Thromb
Vasc Biol. 31:67–73. 2011. View Article : Google Scholar : PubMed/NCBI
|