Introduction
Thyroid hormones (triiodothyronine and thyroxine)
are major regulators of diverse metabolic pathways via interactions
with thyroid hormone nuclear receptors in various tissues (1–3).
Maintaining thyroid hormone levels is essential for development and
metabolism. Hypothyroidism, one of several thyroid diseases, is a
pathological state where thyroid hormone levels are decreased
systemically or locally in one or more tissues. The prevalence of
hypothyroidism is sizeable and ranges between 3–18% in the adult
population, with women, elderly persons and populations with iodine
deficiency or excess being more often affected (4,5).
Generally, hypothyroidism is diagnosed either in the subclinical or
clinical form. Congenital hypothyroidism, if not treated, may lead
to severe and irreversible mental retardation (6).
Hypothyroidism is associated with various symptoms,
including cold sensitivity, fatigue and lethargy, cognitive
dysfunction and delayed growth; these symptoms can be accompanied
by distinct signs of tachycardia, weight loss and attention
deficit-hyperactivity disorder (7,8). Two
of the main characteristics of hypothyroidism are the marked
impairment of lipid metabolism and dyslipidemia (9,10).
Furthermore, hypercholesterolemia induces increased concentrations
of total and low-density lipoprotein cholesterol (11), which will affect normal metabolism.
At present, the genetic mechanism implicated in the pathogenesis of
hypothyroidism remains poorly understood. It has been reported that
mutations in the NK2 homeobox 1 and forkhead box E1 (FOXE1) genes
cause hypothyroidism (12,13). In addition, a mutation in
immunoglobulin superfamily member 1 has been revealed to be
associated with central hypothyroidism (X-linked syndrome)
(14). Loss-of-function mutations
in thyroglobulin, paired box 8, thyroid-stimulating hormone
receptor, FOXE1, NK2 homeobox (NKX2)-1 and NKX2-5 genes are also
associated with inherited congenital hypothyroidism (15,16).
Therefore, identifying candidate gene mutations may be helpful in
understanding the pathology of hypothyroidism.
Whole exome sequencing (WES) is a useful tool for
exploring the genetic mechanism of different diseases (17–19).
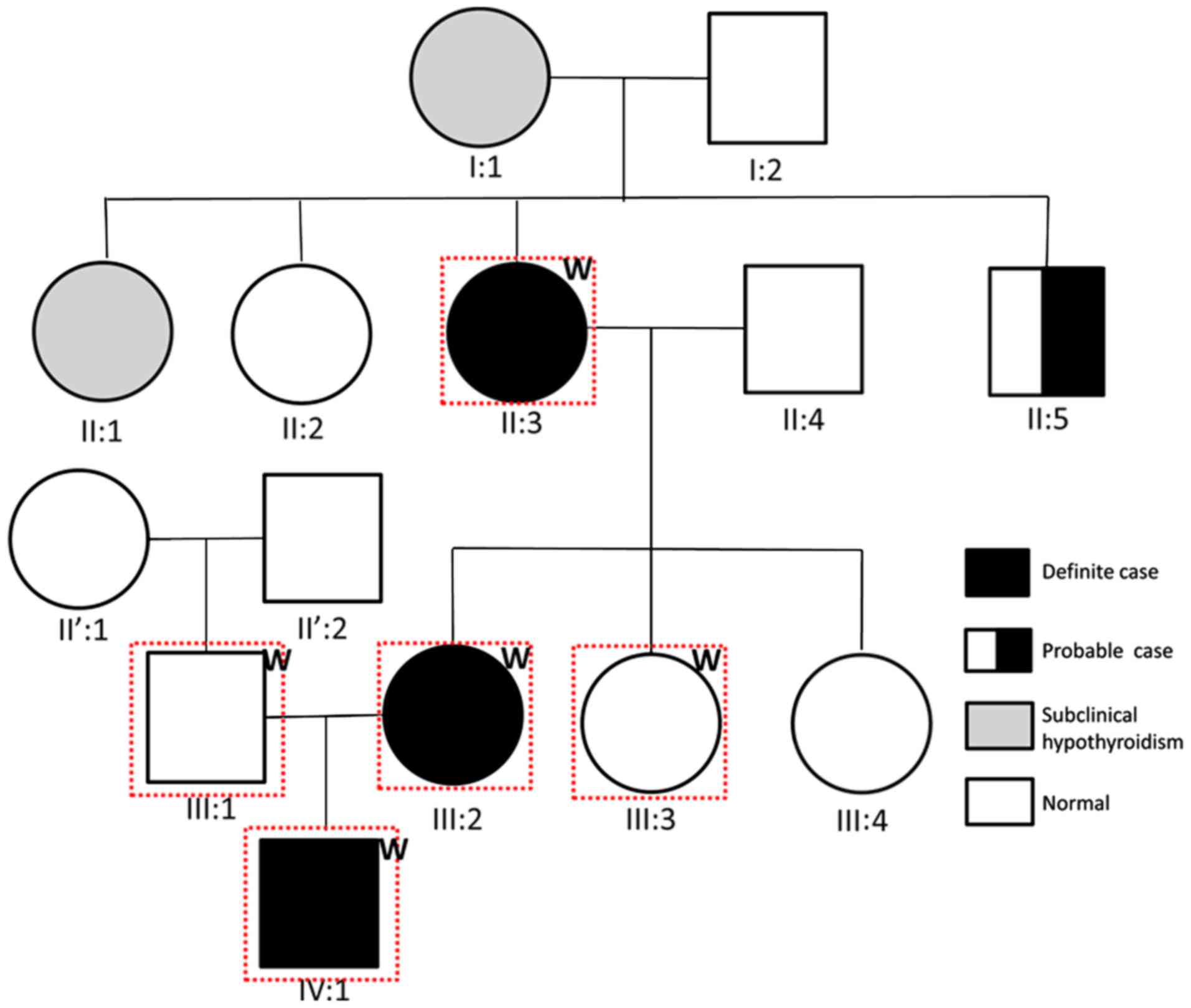
The present study investigated a Chinese hypothyroidism pedigree,
which included an affected proband, mother and maternal
grandmother; other relatives were unaffected. The results indicated
that the c.G7192T (p.A2398S) mutation in fatty acid synthase (FASN)
and the c.C1883G (p.T628R) mutation in apolipoprotein B receptor
(APOBR) may be the most likely causes of the disease. These results
on the inheritance of mutant genes may provide novel information
regarding the pathological mechanism underlying hypothyroidism.
Materials and methods
Subjects and family members
The present study examined a three-generation
hypothyroidism pedigree residing in China. The proband (IV:1) was a
3.9-year-old boy who was diagnosed with hypothyroidism.
Furthermore, his mother (III:2) and maternal grandmother (II:3)
also had hypothyroidism, and all of them shared common
characteristics (Fig. 1). In
addition, family members I:1 and II:1 were diagnosed with
subclinical hypothyroidism, and II:5 was a probable case. The
father, maternal aunt and paternal grandparents of the proband were
unaffected. Therefore, it was hypothesized that the etiology of
hypothyroidism in this family had an autosomal dominant mode of
inheritance. Therefore, the proband, along with his parents,
maternal aunt and maternal grandmother were enrolled for whole
exome sequencing (Fig. 1). Written
informed consent was obtained from all individuals enrolled in this
study. In addition, the present study was approved by the ethical
approval committee of Jinan Central Hospital Affiliated to Shandong
University (2016-053-01; Jinan, China).
Exome sequencing and variant
calling
Venous blood was obtained from the affected
individuals (IV:1, III:2 and II:3) and two unaffected individuals
(III:1 and III3) in the hypothyroidism pedigree. The collected
blood was stored in EDTA, followed by DNA extraction using a kit
(Tiangen Biotech Co., Ltd., Beijing, China). The Agilent SureSelect
Human All Exon 50Mb Exon kit (Agilent Technologies, Inc., Santa
Clara, CA, USA) was used to perform exome target enrichment of
quantified genomic DNA. The amplification process was performed
under the following conditions: 15 min at 95°C followed by 40
cycles of 10 sec at 95°C, 30 sec at 55°C, 32 sec at 72°C, and 15
sec at 95°C, 60 sec at 60°C, 15 sec extension at 95°C. The Illumina
HiSeq 4000 Sequencer (Illumina, San Diego, CA, USA) was utilized
for WES. Sequencing reads that had paired-ends, were 200-bp long
and had a mean coverage of 100× were generated for each sample.
After filtering out adapter sequences, and contaminated and low
quality reads, the clean paired reads were then mapped to the
reference human genome sequence, hg19 (20), using the Burrows-Wheeler alignment
tool, generating the sequence alignment/map file. Picard software
program (version 1.07) was used to mark and remove polymerase chain
reaction (PCR) duplicate reads. MuTect (version 1.1.4) and Genome
Analysis Toolkit software (version 3.1) (21,22)
were used to identify single nucleotide variants (SNVs), deletions
and insertions throughout the genome.
Variant filtering
To identify potential candidate genes, the variants
were annotated in a systematic manner. Variant information was
annotated using various genetic variation databases by the program
ANNOVAR (23). From the reported
variant frequencies, variants with a minor allele frequency
>0.01 in the 1000 Genomes Project (https://www.nature.com/articles/nature15393) were
excluded. Based on the variant location within genes, higher
priority was given to the variants in the coding region and
variants that altered the coding sequence (nonsynonymous variants)
were identified. The deleteriousness of identified variants was
then predicted by bioinformatics analysis [e.g. Oncotator v1.5.3.0,
sift (http://sift.jcvi.org/) and polyphen
(http://genetics.bwh.harvard.edu/pph2/)], and the
harmful nonsynonymous variants were obtained. The eligible
nonsynonymous variants were identified using the following
filtering parameter criteria in the Genome Analysis Toolkit
software (v3.1) (https://software.broadinstitute.org/gatk/):
QualByDepth>2.0, FisherStrand<60.0, StrandOddsRatio<4.0,
RMSMappingQuality (MQ)>40.0, MQRankSumTest>-12.5,
ReadPosRankSumTest>-8.0. In addition, minor allele frequency
<0.05 was also included.
In silico analysis
The multiple sequence alignment of FASN and APOBR in
different species was performed using an online tool (https://www.ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi?LINK_LOC=BlastHomeLink).
In addition, the Conserved Domain Search Service (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi)
was used to identify the conserved protein domains.
Sanger sequencing
After the systematic filtering, two candidate
variants (FASN c.G7192T, p.A2398S and APOBR c.C1883G, p.T628R) were
confirmed. Genomic DNA was prepared from the proband, ten family
members and twenty additional, unrelated, affected individuals.
Primer-Premier 5.0 (Premier Biosoft International, Palo Alto, CA,
USA) was used to design oligonucleotide primer sets for the
variants. PCR analyses and Sanger sequencing were then performed.
The amplification process was: 15 min at 95°C followed by 40 cycles
of 10 sec at 95°C, 30 sec at 55°C, 32 sec at 72°C, and 15 sec at
95°C, 60 sec at 60°C, 15 sec extension at 95°C. The PCR products
were used for Sanger sequencing.
Results
Genetic and in silico analysis
Genomic DNA samples from IV:1, III:1, III:2, III:3
and II:3 were analyzed. A total of 50 nonsynonymous variants were
identified (Table I). Based on the
hypothyroidism in the pedigree and subsequent Sanger sequencing
results (Table II), two SNVs, the
c.G7192T mutation (a substitution from C to A) in FASN (Chr17:
80037439) and the c.C1883G mutation (a substitution from C to G) in
APOBR (Chr16: 28508245), were likely to be associated with the
disease. In pedigree individuals, the results of II:3, II:5, III:2
and IV:1 showed the c.G7192T mutation in FASN (Chr17: 80037439) and
the c.C1883G mutation in APOBR.
 | Table I.A total of 50 nonsynonymous variants
in the pedigree. |
Table I.
A total of 50 nonsynonymous variants
in the pedigree.
| Chr | Start | End | Ref | Alt | Gene.refGene | DbSNP138 |
|---|
| 1 | 16057042 | 16057042 | G | A | PLEKHM2 | . |
| 1 | 23238971 | 23238971 | G | A | EPHB2 | . |
| 2 | 167760021 | 167760021 | A | G | XIRP2 | . |
| 2 | 169801131 | 169801131 | G | A | ABCB11 | rs118109635 |
| 2 | 172409920 | 172409920 | T | C | CYBRD1 | rs16859487 |
| 2 | 175618404 | 175618404 | C | T | CHRNA1 | . |
| 2 | 179300979 | 179300979 | A | T | PRKRA | rs77419724 |
| 2 | 179597657 | 179597657 | T | G | TTN | . |
| 2 | 179632619 | 179632619 | C | T | TTN | rs141258018 |
| 2 | 204304489 | 204304489 | G | T | RAPH1 | rs191393494 |
| 2 | 234869499 | 234869499 | G | C | TRPM8 | . |
| 2 | 238732983 | 238732983 | G | T | RBM44 | . |
| 3 | 13670739 | 13670739 | G | A | FBLN2 | rs201340643 |
| 3 | 38087142 | 38087142 | G | A | DLEC1 | rs117463277 |
| 3 | 49701035 | 49701035 | G | A | BSN | rs141950704 |
| 3 | 56597830 | 56597830 | G | A | CCDC66 | rs146224729 |
| 4 | 74363387 | 74363387 | A | T | AFM | rs2276444 |
| 6 | 90578678 | 90578678 | A | C | CASP8AP2 | . |
| 6 | 168294583 | 168294583 | G | A | MLLT4 | rs150936076 |
| 6 | 170886768 | 170886768 | C | G | PDCD2 | rs140493653 |
| 9 | 140323749 | 140323749 | C | T | NOXA1 | rs201388549 |
| 10 | 86132217 | 86132217 | G | T | CCSER2 | . |
| 11 | 3249828 | 3249828 | C | T | MRGPRE | rs200334859 |
| 11 | 3681054 | 3681054 | G | A | ART1 | rs2280133 |
| 11 | 3744621 | 3744621 | C | T | NUP98 | rs148384795 |
| 11 | 4411560 | 4411560 | A | C | TRIM21 | . |
| 11 | 44069816 | 44069816 | G | A | ACCSL | rs182257970 |
| 11 | 58605757 | 58605757 | C | T | GLYATL2 | . |
| 11 | 62381857 | 62381857 | G | A | ROM1 | . |
| 11 | 68748268 | 68748268 | A | G | MRGPRD | rs74390416 |
| 11 | 73067275 | 73067275 | C | T | ARHGEF17 | . |
| 11 | 77838424 | 77838424 | T | C | ALG8 | rs138293432 |
| 11 | 93754643 | 93754643 | T | A | HEPHL1 | rs192979315 |
| 13 | 96242562 | 96242562 | T | G | DZIP1 | . |
| 15 | 65490592 | 65490592 | C | T | CILP | rs148582730 |
| 16 | 28508245 | 28508245 | C | G | APOBR | rs13306186 |
| 16 | 28948654 | 28948654 | T | C | CD19 | . |
| 17 | 4076694 | 4076694 | A | G | ANKFY1 | . |
| 17 | 4856390 | 4856390 | G | C | ENO3 | rs143945974 |
| 17 | 10312678 | 10312678 | A | T | MYH8 | rs151091483 |
| 17 | 19246867 | 19246867 | T | C | B9D1 | rs7221577 |
| 17 | 79167744 | 79167744 | C | T | CEP131 | rs138784674 |
| 17 | 80037439 | 80037439 | C | A | FASN | rs200842352 |
| 18 | 3094191 | 3094191 | C | T | MYOM1 | rs149588924 |
| 20 | 76700 | 76700 | G | A | DEFB125 | rs116934569 |
| 20 | 60575227 | 60575227 | C | A | TAF4 | . |
| 20 | 60892518 | 60892518 | C | T | LAMA5 | rs200632605 |
| 20 | 61167658 | 61167658 | C | G | MIR1-1HG | rs145416632 |
| 21 | 34951831 | 34951831 | C | A | DONSON | rs190773441 |
| 21 | 40568847 | 40568847 | T | C | BRWD1 | rs73357824 |
| Table II.Sanger sequencing validation
results. |
Table II.
Sanger sequencing validation
results.
| Individuals | CHRNA1 | ABCB11 | APOBR | FASN | TRPM8 |
|---|
| Affected sporadic
individuals |
| 1 | GG | CC | CC | CC | GG |
| 2 | GG | CC | CC | CC | GG |
| 3 | GG | CC | CC | CC | GG |
| 4 | GG | CC | CC | CC | GG |
| 5 | GG | CC | CC | CC | GG |
| 6 | GG | CC | CC | CC | GG |
| 7 | GG | CC | CC | CC | GG |
| 8 | GG | CC | CC | CC | GG |
| 9 | GG | CC | CC | CC | GG |
| 10 | GG | CC | CC | CC | GG |
| 11 | GG | CC | CC | CC | GG |
| 12 | GG | CC | CC | CC | GG |
| 13 | GG | CC | CC | CC | GG |
| 14 | GG | CC | CC | CC | GG |
| 15 | GG | CC | CC | CC | GG |
| 16 | GG | CC | CC | CC | GG |
| 17 | GG | CC | CC | CC | GG |
| 18 | GG | CC | CC | CC | GG |
| 19 | GG | CC | CC | CC | GG |
| 20 | GG | CC | CC | CC | GG |
| Pedigree
individuals |
|
III-2 | GG | CC | CG | AC | GG |
|
II-1 | GA | CT | CC | CC | GC |
|
II-2 | GG | CC | CC | CC | GG |
|
II-1 | GG | CC | CC | CC | GG |
|
III-3 | GG | CC | CC | CC | GG |
|
III-1 | GA | CT | CC | CC | GC |
|
II-5 | GA | CT | CG | AC | GC |
|
II-4 | GG | CC | CC | CC | GG |
|
II-3 | GA | CT | CG | AC | GC |
|
I-1 | GG | CC | CC | CC | GG |
|
IV-1 | GA | CT | CG | AC | GC |
In silico analysis
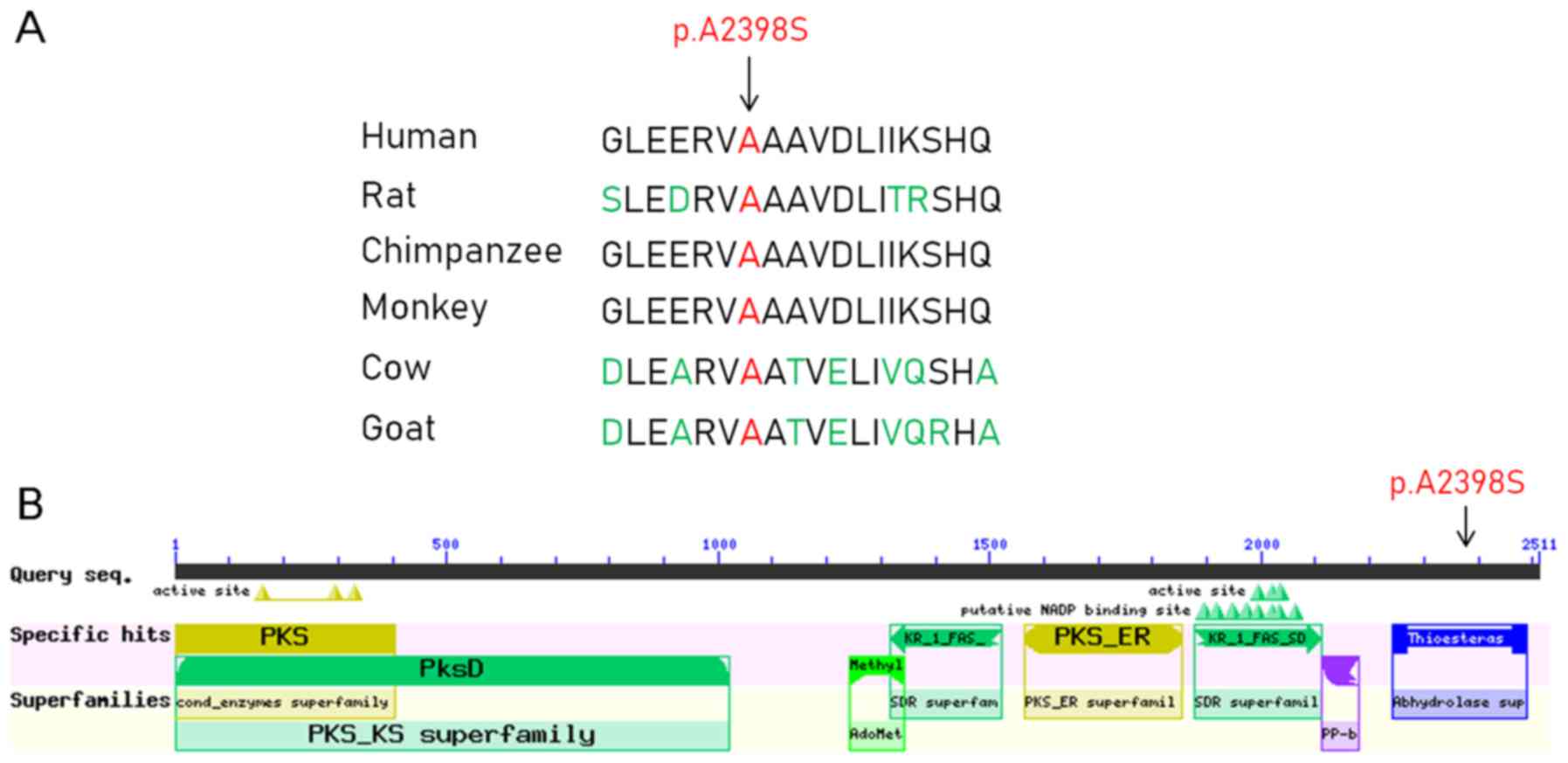
The c.G7192T mutation in FASN resulted in a protein
alteration of p.A2398S. In addition, the c.C1883G mutation in APOBR
resulted in a protein alteration of p.T628R. Furthermore, multiple
sequence alignment and conserved protein domain analyses indicated
that the amino acid at position 2,398 in the FASN protein sequence
was located in the α/β-hydrolase fold, and it was highly conserved
across several species, including humans, rats, chimpanzees,
monkeys, cows and goats (Fig. 2).
Furthermore, the amino acid at position 628 in the APOBR protein
sequence was located in the Na+/Ca2+
exchanger superfamily, and it was highly conserved across several
species, including humans, monkeys, camels, chimpanzees, pigs and
rats (Fig. 3).
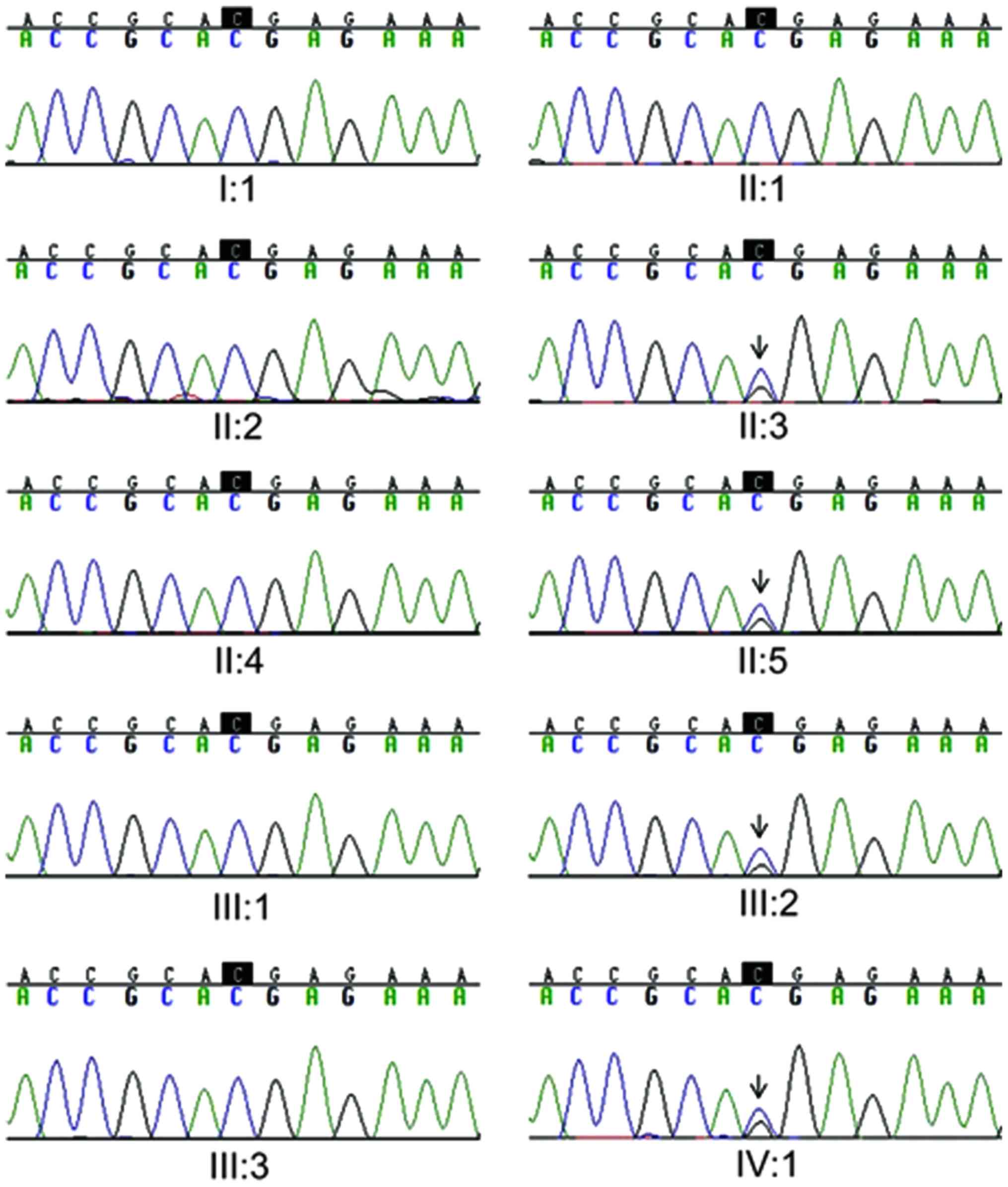
Sanger sequencing of the candidate
causative variants
In order to further confirm the c.G7192T and
c.C1883G mutations in hypothyroidism, Sanger sequencing was
performed on the 11 family members and on 20 additional, unrelated
individuals with primary hypothyroidism. The clinical information
of the 20 additional, unrelated individuals with primary
hypothyroidism is presented in Table
III. The results demonstrated that the c.G7192T mutation in
FASN and the c.C1883G mutation in APOBR were fully co-segregated
with established hypothyroidism phenotypes in the family recruited
for WES (Figs. 4 and 5). Furthermore, the FASN c.G7192T and
APOBR c.C1883G mutations were detected in the patient with probable
hypothyroidism (II:5). It is worth mentioning that the FASN
c.G7192T and APOBR c.C1883G mutations were not observed in
individuals with subclinical hypothyroidism (I:1, II:1). In
addition, the mutations were not detected in the unrelated,
affected individuals with hypothyroidism (date not shown), which
supported the hypothesis of genetic heterogeneity of hypothyroidism
in this family.
| Table III.Clinical information of 20
additional, unrelated individuals with primary hypothyroidism. |
Table III.
Clinical information of 20
additional, unrelated individuals with primary hypothyroidism.
| Case | Sex | Age (years) | TSH (mU/l) | FT4 (pmol/l) |
|---|
| 1 | Male | 75 | 27.7 | 7.31 |
| 2 | Male | 49 | 4.85 | 16.2 |
| 3 | Male | 47 | 72 | 0.88 |
| 4 | Male | 67 | 9.15 | 10.7 |
| 5 | Male | 47 | 1.71 | 15.3 |
| 6 | Female | 64 | 3.07 | 15.7 |
| 7 | Female | 31 | 6.21 | 12.3 |
| 8 | Female | 21 | 5.56 | 11.5 |
| 9 | Female | 29 | 1.59 | 10.3 |
| 10 | Female | 25 | 5.52 | 13.2 |
| 11 | Female | 27 | 4.56 | 12.7 |
| 12 | Female | 31 | 3.97 | 16 |
| 13 | Female | 32 | 2.57 | 10 |
| 14 | Female | 68 | 37 | 7.08 |
| 15 | Female | 54 | 5.08 | 11.5 |
| 16 | Female | 30 | 57.5 | 7.16 |
| 17 | Female | 36 | 4.3 | 12.2 |
| 18 | Female | 29 | 0.969 | 11.4 |
| 19 | Female | 35 | 9.74 | 12.4 |
| 20 | Female | 27 | 4.26 | 11.5 |
Discussion
Hypothyroidism is the most common endocrine disorder
caused by thyroid hormone deficiency, which can induce metabolic
dysfunction (24). At present, the
genetic mechanisms underlying hypothyroidism pathogenesis remain
poorly understood. It has been reported that the c.1184_1187dup4
mutation in the thyroid peroxidase gene, c.40A>G and c.94G>A
mutations in the thyroid-stimulating hormone β gene, p.G488R,
p.A649E, p.R885Q, p.I1080T and p.A1206T mutations in the dual
oxidase 2 gene, and the p.Y138X mutation in the dual oxidase
maturation factor 2 gene are associated with congenital
hypothyroidism (25–27). WES technology is an effective
method for identifying potential causative genes in disease
phenotypes. In the present study, WES was performed to identify
potential causative genes in the affected individual, his parents,
maternal aunt and maternal grandmother in a hypothyroidism
pedigree. Two mutations (c.G7192TT and c.C1883G) in the FASN and
APOBR genes, respectively, were revealed to be associated with
hypothyroidism. In addition, Sanger sequencing validated these FASN
and APOBR mutations in the proband and his mother. Furthermore, the
mutations were fully co-segregated with established hypothyroidism
phenotypes in the family.
Fatty acid synthesis is a process that entails
producing de novo fatty acids from carbohydrate- and amino
acid-derived carbon sources. The liver serves a key role in fatty
acid modulation. The key enzymes in the lipogenic and lipolytic
pathways are regulated by thyroid hormones in the liver and adipose
tissues (28,29). Disrupted thyroid hormone levels
alter hepatic fatty acid composition (30). In addition, it has been reported
that subclinical and clinical hypothyroidism are associated with
hepatic steatosis (31). Numerous
studies have indicated that hypothyroidism is a risk factor for
nonalcoholic fatty liver disease and results in metabolic syndrome
(32–34). In addition, the effects of
hypothyroidism to enhance lipogenesis is amplified in the presence
of physiological concentrations of fatty acids (35). FASN is an enzyme involved in fatty
acid biosynthesis (36). In
mammals, cells manufacture de novo fatty acids using
different pathways, which synthesize fatty acids from acetyl and
malonyl esters of CoA that are catalyzed by dimerized FASN
(37). Furthermore, fatty acid
synthesis is controlled by FASN (38), and the downregulation of FASN
protein levels causes markedly decreased regulation of de
novo lipogenesis (30).
Notably, FASN is downregulated in the livers of patients with
hypothyroidism (39). In addition,
a microarray analysis revealed that FASN, one of the hepatic target
genes, is preferentially activated by triiodothyronine during
transition from the hypothyroid state to the euthyroid state
(40). The c.G7192T mutation is
located in the conserved α/β-hydrolase fold of FASN and was highly
conversed among several diverse species, including humans, rats,
chimpanzees, monkeys, cows and goats. Therefore, this FASN mutation
suggested that FASN may have roles in hypothyroidism.
APOBR is an apolipoprotein E-independent receptor
that binds APOB48, allowing cells to uptake postprandial
triglyceride-rich lipoproteins, for which APOBR has a high
affinity; APOBR functions as a nutritional receptor that provides
dietary fatty acids and lipid-soluble vitamins to cells (41). The APOBR protein is distributed in
the moieties of plasma triglyceride-rich lipoproteins. It has been
reported that APOBR is an important molecule in lipid metabolism,
and that it primarily serves roles in the sterol transport and
metabolism pathways (42).
Mutations in the APOBR gene (c.934-960/del and A419P) have been
identified as novel hyperlipidemia-associated variants, which may
be involved in regulating plasma total cholesterol levels in
patients with hypercholesterolemia (43). APOBR is also involved in
atherogenesis, which is a lipid metabolism disorder (44). Therefore, it may be hypothesized
that the APOBR pathway is a novel therapeutic target, particularly
in hypertriglyceridemic patients, such as those with
hypothyroidism. In the present study, a mutation (c.C1883G,
p.T628R) in the APOBR gene was detected in patients with
hypothyroidism. The mutation is located in the conserved
Na+/Ca2+ exchanger domain of APOBR and was
highly conversed among different species, including humans,
monkeys, camels, chimpanzees, pigs and rats. Therefore, this APOBR
mutation may be considered another candidate molecule in the
pathogenesis of hypothyroidism.
In conclusion, the findings reported here
demonstrated that patients with mutations in FASN (c.G7192T,
p.A2398S) and APOBR (c.C1883G, p.T628R) may be predisposed to
hypothyroidism. These mutations may disrupt the regulation of fatty
acid biosynthesis and lipid metabolism. These findings may reveal
the high degree of genetic heterogeneity in hypothyroidism
phenotypes. Future work will be performed to improve understanding
of this disorder. In addition, there is a limitation of the present
study; the identified SNVs, FASN (c.G7192T, p.A2398S) and APOBR
(c.C1883G, p.T628R), require validation in animal models of
hypothyroidism.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
All data generate or analyzed during the present
study are included in this published article.
Authors' contributions
JS, LS, WC, XY and YL analyzed and interpreted the
data. JS was a major contributor in writing the manuscript. QJ
designed the project. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
individuals enrolled in this study. In addition, the present study
was approved by the ethical approval committee of Jinan Central
Hospital Affiliated to Shandong University (2016-053-01).
Patient consent for publication
Informed written consent was obtained from all
subjects.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
APOBR
|
apolipoprotein B receptor
|
|
FASN
|
fatty acid synthase
|
|
SNVs
|
single nucleotide variants
|
|
WES
|
whole exome sequencing
|
References
|
1
|
Yen PM: Physiological and molecular basis
of thyroid hormone action. Physiol Rev. 81:1097–1142. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang J and Lazar MA: The mechanism of
action of thyroid hormones. Annu Rev Physiol. 62:439–466. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Song Y, Yao X and Ying H: Thyroid hormone
action in metabolic regulation. Protein Cell. 2:358–368. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Virgini V, Baumgartner C, Bischoff T,
Haller DM, Frey P, Rosemann T, Collet TH, Sykiotis G, Pitteloud N
and Rodondi N: Comment les médecins de famille prennent-ils en
charge l'hypthyroïdie infraclinique? Rev Med Suisse. 10:526–529.
2014.(In French). PubMed/NCBI
|
|
5
|
Baumgartner C, Blum MR and Rodondi N:
Subclinical hypothyroidism: Summary of evidence in 2014. Swiss Med
Wkly. 144:w140582014.PubMed/NCBI
|
|
6
|
Fuhrer D, Brix K and Biebermann H:
Understanding the healthy thyroid state in 2015. Eur Thyroid J. 4
Suppl 1:S1–S8. 2015. View Article : Google Scholar
|
|
7
|
Yu H, Yang Y, Zhang M, Lu H, Zhang J, Wang
H and Cianflone K: Thyroid status influence on adiponectin,
acylation stimulating protein (ASP) and complement C3 in
hyperthyroid and hypothyroid subjects. Nutr Metab(Lond). 3:132006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miller LD, McPhie P, Suzuki H, Kato Y, Liu
ET and Cheng SY: Multi-tissue gene-expression analysis in a mouse
model of thyroid hormone resistance. Genome Biol. 5:R312004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang N, Yao Z, Miao L, Liu J, Gao X, Xu Y
and Wang G: Homocysteine diminishes apolipoprotein A-I function and
expression in patients with hypothyroidism: A cross-sectional
study. Lipids Health Dis. 15:1232016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roberts CG and Ladenson PW:
Hypothyroidism. Lancet. 363:793–803. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y, Yu X, Zhao QZ, Zheng S, Qing WJ,
Miao CD and Sanjay J: Thyroid dysfunction, either hyper or
hypothyroidism, promotes gallstone formation by different
mechanisms. J Zhejiang Univ Sci B. 17:515–525. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Veneziano L, Parkinson MH, Mantuano E,
Frontali M, Bhatia KP and Giunti P: A novel de novo mutation of the
TITF1/NKX2-1 gene causing ataxia, benign hereditary chorea,
hypothyroidism and a pituitary mass in a UK family and review of
the literature. Cerebellum. 13:588–595. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mendieta-Zeron H, Jimenez-Rosales A,
Perez-Amado CJ and Jimenez-Morales S: FOXE1 mutation screening in a
case with cleft lip, hypothyroidism, and thyroid carcinoma: A new
syndrome? Case Rep Genet. 2017:63905452017.PubMed/NCBI
|
|
14
|
Sun Y, Bak B, Schoenmakers N, van
Trotsenburg AS, Oostdijk W, Voshol P, Cambridge E, White JK, le
Tissier P, Gharavy SN, et al: Loss-of-function mutations in IGSF1
cause an X-linked syndrome of central hypothyroidism and testicular
enlargement. Nat Genet. 44:1375–1381. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Watanabe Y, Sharwood E, Goodwin B, Creech
MK, Hassan HY, Netea MG, Jaeger M, Dumitrescu A, Refetoff S, Huynh
T and Weiss RE: A novel mutation in the TG gene (G2322S) causing
congenital hypothyroidism in a sudanese family: A case report. BMC
Med Genet. 19:692018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vincenzi M, Camilot M, Ferrarini E,
Teofoli F, Venturi G, Gaudino R, Cavarzere P, De Marco G, Agretti
P, Dimida A, et al: Identification of a novel pax8 gene sequence
variant in four members of the same family: From congenital
hypothyroidism with thyroid hypoplasia to mild subclinical
hypothyroidism. BMC Endocr Disord. 14:692014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bamshad MJ, Ng SB, Bigham AW, Tabor HK,
Emond MJ, Nickerson DA and Shendure J: Exome sequencing as a tool
for mendelian disease gene discovery. Nat Rev Genet. 12:745–755.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ng SB, Turner EH, Robertson PD, Flygare
SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler
EE, et al: Targeted capture and massively parallel sequencing of 12
human exomes. Nature. 461:272–276. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang JL, Yang X, Xia K, Hu ZM, Weng L, Jin
X, Jiang H, Zhang P, Shen L, Guo JF, et al: TGM6 identified as a
novel causative gene of spinocerebellar ataxias using exome
sequencing. Brain. 133:3510–3518. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A mapreduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cibulskis K, Lawrence MS, Carter SL,
Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES
and Getz G: Sensitive detection of somatic point mutations in
impure and heterogeneous cancer samples. Nat Biotechnol.
31:213–219. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Iwen KA, Schroder E and Brabant G: Thyroid
hormones and the metabolic syndrome. Eur Thyroid J. 2:83–92. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cangul H, Aydin BK and Bas F: A homozygous
TPO gene duplication (c.1184_1187dup4) causes congenital
hypothyroidism in three siblings born to a consanguineous family. J
Pediatr Genet. 4:194–198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ozhan B, Anlas Boz O, Sarikepe B, Albuz B
and Gunduz Semerci N: Congenital central hypothyroidism caused by a
novel thyroid-stimulating hormone-beta subunit gene mutation in two
siblings. J Clin Res Pediatr Endocrinol. 9:278–282. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park KJ, Park HK, Kim YJ, Lee KR, Park JH,
Park JH, Park HD, Lee SY and Kim JW: DUOX2 mutations are frequently
associated with congenital hypothyroidism in the korean population.
Ann Lab Med. 36:145–153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Diamant S, Gorin E and Shafrir E: Enzyme
activities related to fatty-acid synthesis in liver and adipose
tissue of rats treated with triiodothyronine. Eur J Biochem.
26:553–559. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Oppenheimer JH, Schwartz HL, Lane JT and
Thompson MP: Functional relationship of thyroid hormone-induced
lipogenesis, lipolysis, and thermogenesis in the rat. J Clin
Invest. 87:125–132. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yao X, Hou S, Zhang D, Xia H, Wang YC,
Jiang J, Yin H and Ying H: Regulation of fatty acid composition and
lipid storage by thyroid hormone in mouse liver. Cell Biosci.
4:382014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ludwig U, Holzner D, Denzer C, Greinert A,
Haenle MM, Oeztuerk S, Koenig W, Boehm BO, Mason RA, Kratzer W, et
al: Subclinical and clinical hypothyroidism and non-alcoholic fatty
liver disease: A cross-sectional study of a random population
sample aged 18 to 65 years. BMC Endocr Disord. 15:412015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Amarapurkar D, Kamani P, Patel N, Gupte P,
Kumar P, Agal S, Baijal R, Lala S, Chaudhary D and Deshpande A:
Prevalence of non-alcoholic fatty liver disease: Population based
study. Ann Hepatol. 6:161–163. 2007.PubMed/NCBI
|
|
33
|
Law K and Brunt EM: Nonalcoholic fatty
liver disease. Clin Liver Dis. 14:591–604. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ortiz-Lopez C, Lomonaco R, Orsak B, Finch
J, Chang Z, Kochunov VG, Hardies J and Cusi K: Prevalence of
prediabetes and diabetes and metabolic profile of patients with
nonalcoholic fatty liver disease (NAFLD). Diabetes Care.
35:873–878. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baht HS and Saggerson ED: A
tissue-specific increase in lipogenesis in rat brown adipose tissue
in hypothyroidism. Biochem J. 251:553–557. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rusinek D, Swierniak M, Chmielik E, Kowal
M, Kowalska M, Cyplinska R, Czarniecka A, Piglowski W, Korfanty J,
Chekan M, et al: BRAFV600E-associated gene expression profile:
Early changes in the transcriptome, based on a transgenic mouse
model of papillary thyroid carcinoma. PLoS One. 10:e01436882015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Smith S: The animal fatty acid synthase:
One gene, one polypeptide, seven enzymes. FASEB J. 8:1248–1259.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Suburu J, Shi L, Wu J, Wang S, Samuel M,
Thomas MJ, Kock ND, Yang G, Kridel S and Chen YQ: Fatty acid
synthase is required for mammary gland development and milk
production during lactation. Am J Physiol Endocrinol Metab.
306:E1132–E1143. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Blennemann B, Leahy P, Kim TS and Freake
HC: Tissue-specific regulation of lipogenic mRNAs by thyroid
hormone. Mol Cell Endocrinol. 110:1–8. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yen PM, Feng X, Flamant F, Chen Y, Walker
RL, Weiss RE, Chassande O, Samarut J, Refetoff S and Meltzer PS:
Effects of ligand and thyroid hormone receptor isoforms on hepatic
gene expression profiles of thyroid hormone receptor knockout mice.
EMBO Rep. 4:581–587. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Brown ML, Yui K, Smith JD, LeBoeuf RC,
Weng W, Umeda PK, Li R, Song R, Gianturco SH and Bradley WA: The
murine macrophage apoB-48 receptor gene (Apob-48r): Homology to the
human receptor. J Lipid Res. 43:1181–1191. 2002.PubMed/NCBI
|
|
42
|
Wang K, Edmondson AC, Li M, Gao F, Qasim
AN, Devaney JM, Burnett MS, Waterworth DM, Mooser V, Grant SF, et
al: Pathway-wide association study implicates multiple sterol
transport and metabolism genes in HDL cholesterol regulation. Front
Genet. 2:412011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fujita Y, Ezura Y, Bujo H, Nakajima T,
Takahashi K, Kamimura K, Iino Y, Katayama Y, Saito Y and Emi M:
Association of nucleotide variations in the apolipoprotein B48
receptor gene (APOB48R) with hypercholesterolemia. J Hum Genet.
50:203–209. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Brown ML, Ramprasad MP, Umeda PK, Tanaka
A, Kobayashi Y, Watanabe T, Shimoyamada H, Kuo WL, Li R, Song R, et
al: A macrophage receptor for apolipoprotein B48: Cloning,
expression, and atherosclerosis. Proc Natl Acad Sci USA.
97:7488–7493. 2000. View Article : Google Scholar : PubMed/NCBI
|