Introduction
Cytochrome P450 enzymes (P450s) constitute a
multi-gene family of constitutive and inducible heme-containing key
oxidative enzymes (1,2). These enzymes not only have roles in
endogenous functions but also participate in the metabolism of a
wide variety of carcinogens and anti-cancer drugs. Thus, cytochrome
P450s are considered to play important roles in tumor biology.
Specifically, cytochrome P450 family 4 (CYP4) enzymes typically
function as microsomal omega (ω)-hydroxylases that metabolize fatty
acids, eicosanoids, and vitamin D and are important for chemical
defense (3). Six CYP4 subfamilies
exist in mammals: CYP4A, CYP4B, CYP4F, CYP4 V, CYP4X, and CYP4Z
(4). The CYP4A, CYP4B, and CYP4F
subfamilies have been shown to metabolize fatty acids of different
chain lengths, whereas the fatty acid chain length specificity of
CYP4AV, CYP4X, and CYP4Z is currently unknown (5). Subfamilies with large functional
divergence likely perform different functions. Depending on the
type of CYP4 gene, gene expression levels differ significantly
among tissues, and their functions are unique (6). Therefore, CYP4 gene expression data
may be utilized to obtain a better understanding of putative gene
functions (7). Several studies
have revealed marked mRNA upregulation of genes encoding CYP4
enzymes in some cancers, such as thyroid, breast, colon, and
ovarian cancers (2,8). In addition, increased CYP4 gene
expression has important clinical implications in various
carcinomas (9). For example, CYP4
enzymes are expressed and play various metabolic roles in the
liver. As expression of CYP4 family genes is increased in other
cancers, CYP4 gene expression is also expected to be elevated in
hepatocellular carcinoma (HCC); moreover, its elevation may be
associated with prognosis, as in other carcinomas. Therefore,
altered levels of CYP4 gene expression might be related to
hepatocarcinogenesis.
In the present study, we investigated CYP4 mRNA
expression levels and related clinical outcomes in HCC using The
Cancer Genome Atlas (TCGA) cohort. In addition, we analyzed the
underlying mechanism of the clinical outcomes in these patients
using gene set enrichment analysis (GSEA) as well as Database for
Annotation, Visualization and Integrated Discovery (DAVID) and
Cytoscape hierarchical analyses. Differential CYP4F2 and CYP4F3
protein expression between matched pairs of HCC and non-tumor
tissue samples was evaluated. Our data may provide a useful
strategy for identifying therapeutic targets in HCC.
Materials and methods
CYP4 mRNA expression analysis
Patients and data
mRNA sequence profiling
(illuminahiseq_rnaseqv2-RSEM_genes_normalized) and clinical data of
HCC patients were obtained from FIREHOSE (gdac.broadinstitute.org/). The methods of biospecimen
procurement, RNA isolation and RNA sequencing have been previously
described by The Cancer Genome Atlas Research Network (10). The TCGA RNA-seq data were
cross-referenced with the clinical information of the patients.
Patients with missing clinical/expression values were excluded from
further analyses. In total, 377 samples were included in this
study. The clinical characteristics of the patients in the cohort
are presented in Table I. The
mRNA-seq data were normalized using the Rank Normalize module in
GenePattern (broadinstitute.org/cancer/software/genepattern).
 | Table I.Clinicopathologic information of the
HCC patients. |
Table I.
Clinicopathologic information of the
HCC patients.
| Feature | Total n (%) |
|---|
| No. | 377 (100.0) |
| Sex | 377 (100.0) |
|
Female | 122 |
| Male | 255 |
| Age, years | 377 (100.0) |
| ≤60 | 180 |
|
>60 | 196 |
| NA | 1 |
| TNM stage | 377 (100.0) |
| Stage
I | 175 |
| Stage
II | 87 |
| Stage
III | 86 |
| Stage
IV | 5 |
| NA | 24 |
| Histological
grade | 377 (100.0) |
| Grade
1 | 55 |
| Grade
2 | 180 |
| Grade
3 | 124 |
| Grade
4 | 13 |
| NA | 5 |
| Vital status | 377 (100.0) |
|
Alive | 245 |
| Dead | 132 |
| Child-Pugh
classification | 377 (100.0) |
| A | 223 |
| B | 21 |
| C | 1 |
| NA | 132 |
| Fibrosis Ishak
score | 377 (100.0) |
| 0-no
fibrosis | 76 |
|
1,2-portal fibrosis | 31 |
|
3,4-fibrous septa | 30 |
|
5-nodular formation and
incomplete cirrhosis | 9 |
|
6-established cirrhosis | 72 |
| NA | 159 |
|
Thrombocytopeniaa | 377 (100.0) |
|
Yes | 76 |
| No | 234 |
| NA | 67 |
| Albumin level,
g/dl | 377 (100.0) |
|
>3.5 | 217 |
|
≤3.5 | 86 |
| NA | 74 |
| AFP (ng/ml) | 377 (100.0) |
|
≤20 | 152 |
|
>20 | 132 |
| NA | 93 |
| History of
hepatocellular carcinoma risk |
|
|
Hepatitis B | 105 |
|
Hepatitis C | 51 |
|
Hepatitis B + C | 7 |
| Alcohol
consumption | 118 |
|
Non-alcoholic fatty liver
disease | 18 |
GSEA
GSEA was performed to determine the biological
significance of Kyoto Encyclopedia of Genes and Genomes (KEGG)
canonical pathways. Enrichment analysis was performed for 20,502
genes. Phenotype labels, which were defined as CYP4 high 10% or low
10% according to CYP4 gene mRNA expression, were determined.
P<0.05 indicated statistical significance. After performing
GSEA, Enrichment Map Visualization was performed using Cytoscape
(v3.5.1) to show the networks between the GSEA results. cBioPortal
(www.cbioportal.org/) was also employed
to analyze gene alterations and networks of CYP4 genes in HCC.
Survival analysis
Cutoff Finder (molpath.charite.de/cutoff) was utilized to determinate
cutoff values for HCC mRNA expression. The CYP4 gene mRNA-seq data
were uploaded from tab-separated files in which the rows
represented patients and the columns represented variables
(molpath.charite.de/cutoff/load.jsp). In Cutoff Finder,
the cutoff value determination for survival significance was based
on the fit of a mixture model that is conveniently applicable to
molecular variables with bimodal-shaped distributions and is
optimized based on the hypothesis that the variables are
distributed according to a mixture of two Gaussian distributions.
The cumulative event (death) rate was calculated using the
Kaplan-Meier method, and the time to the first event was considered
the outcome variable. The cutoff point optimization, briefly
defined as the point with the most significant split, was
determined by the significance of correlation with the survival
variable. Additionally, the hazard ratio (HR) was calculated
(11). The difference in overall
survival between the poor and better survival groups, which were
defined by the computed cutoff point for CYP4 expression, was
depicted using Kaplan-Meier curves with calculated P-values
(log-rank test, P<0.05).
Statistical analysis
Statistical analyses were performed using Prism v5.0
software (GraphPad Software, Inc., La Jolla, CA, USA) and SPSS v24
(IBM Corp., Armonk, NY, USA). Distributions between two groups were
compared using t-tests (or the Kolmogorov-Smirnov test if the
expected frequency within any cell was <5) for continuous
variables and the χ2 test (or Fisher's exact test if the
expected frequency within any cell was <5) for categorical
variables. Distributions of the characteristics among three or more
groups were compared using analysis of variance. P<0.05 was
considered to indicate a statistically significant difference.
Assessment of CYP4F2 protein expression
in matched pairs of HCC and non-tumor tissue samples
Tissue samples
We retrospectively screened 113 cases of HCC between
1999 and 2014 at Chungnam National University Hospital in Daejeon,
South Korea. All formalin-fixed paraffin-embedded (FFPE) tissue
samples were isolated from HCC patients who underwent segmentectomy
or lobectomy. The two most representative viable tumor areas and
one non-neoplastic area were selected and marked on hematoxylin and
eosin (H&E)-stained slides. Tissue microarrays (TMAs) were
created by punching tissue columns (3.0 mm in diameter) from the
original paraffin blocks and inserting the columns into new
recipient paraffin blocks (each containing 30 holes to receive the
tissue columns). All clinical data were obtained from the National
Biobank of Korea at Chungnam National University Hospital. The use
of FFPE tissue for immunohistochemical analysis waived the
prerequisite for informed consent for a retrospective comparison
study using these tissues. In addition, all experimental procedures
in this study were performed in accordance with relevant guidelines
and regulations approved by the Institutional Review Board of
Chungnam National University Hospital. The present study was
approved by the Institutional Review Board of Chungnam National
University Hospital (CNUH 2018-02-017).
Immunohistochemical staining
analysis
Immunohistochemical staining of the tissue sections
from the TMA paraffin blocks was performed as previously described
(12). A primary rabbit polyclonal
antibody against human CYP4F2 (ab111741, diluted 1:100; Abcam,
Cambridge, UK) was used; the reactions were incubated at room
temperature for 1 h. A modified Allred et al (13) method was applied to evaluate both
the intensity of the immunohistochemical staining and the
proportion of stained neoplastic or non-neoplastic hepatocytes on
each slide. The proportion scores ranged from 0 to 5 (0, 0; 1,
>0 to 1/100; 2, >1/100 to 1/10; 3, >1/10 to 1/3; 4,
>1/3 to 2/3; and 5, >2/3 to 1), and the intensity scores
ranged from 0 to 3 (0, negative; 1, weak; 2, moderate; and 3,
strong). To generate the total immunohistochemical score, the
intensity score and proportional score were multiplied for each
specimen (range, 0–15). The results were examined separately and
scored by KHK and IOS, who were blinded to the patient details.
Discrepancies in scores were discussed to obtain a consensus.
Statistical analyses
Relationships between CYP4F2 expression and
clinicopathological parameters were evaluated using Pearson's
chi-square test and the Mann-Whitney U test. Differences in CYP4F2
expression levels between paired HCC tissue and non-tumor tissue
sections were assessed using the Wilcoxon signed-rank test. One-way
analysis of variance with a Newman-Keuls post hoc test was
performed to analyze three or more groups. P<0.05 was considered
to indicate a statistically significant difference. SPSS v24
software was used for analyses (IBM Corp.).
Results
CYP4 mRNA expression in HCC
CYP4 mRNA expression levels in HCC were examined,
and the results are shown in Fig.
1. Interestingly, although the mRNA expression levels of
CYP4B1, CYP4F8, CYP4Z2P and CYP4F22 were similar among normal
controls, greatly decreased levels compared to normal controls were
found for other CYP4 family genes, including CYP4A11, CYP4A22,
CYP4F2, CYP4X1 and CYP4V2. The gene alteration in CYP4 did not
reveal a significant difference. In addition, mRNA expression
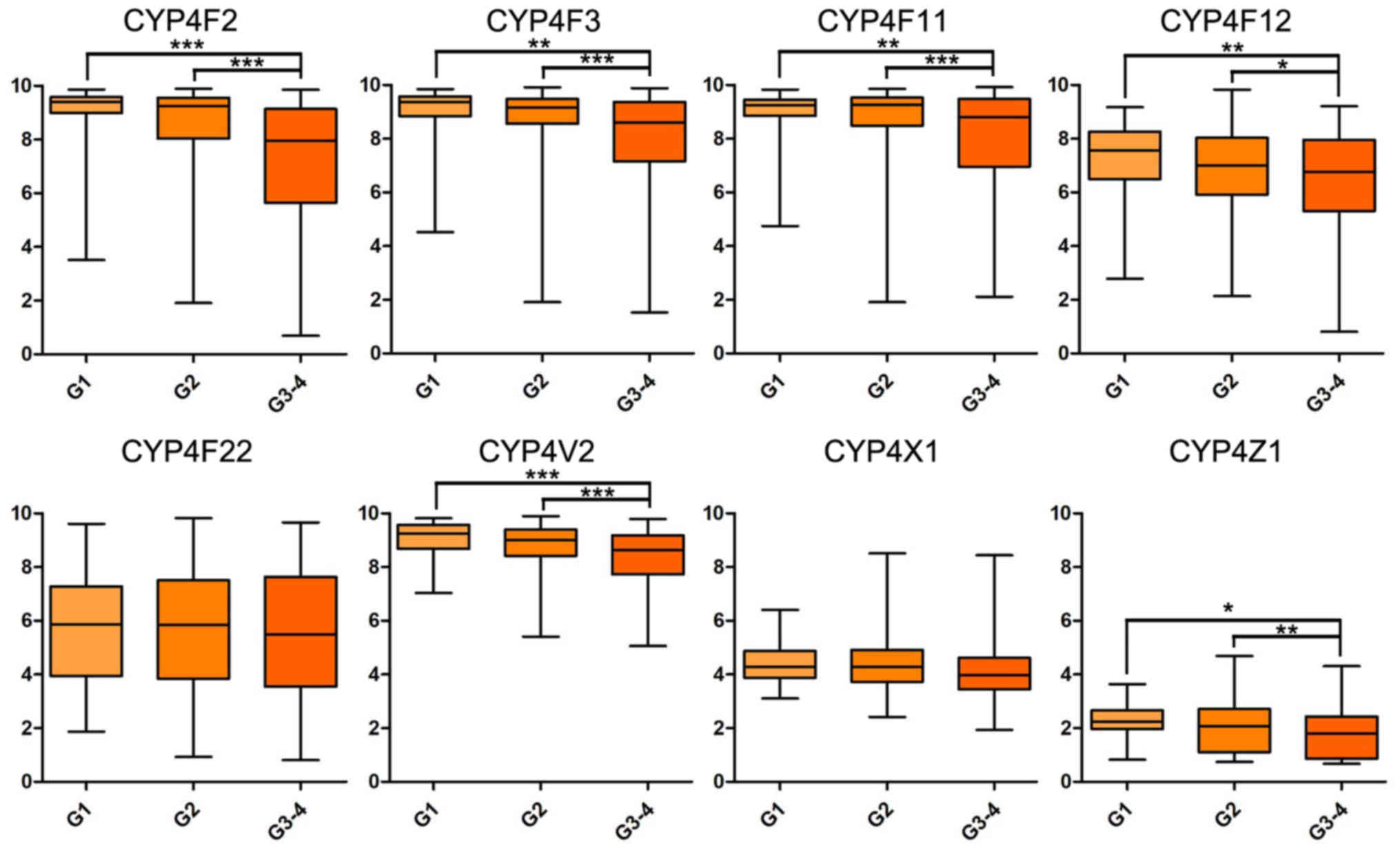
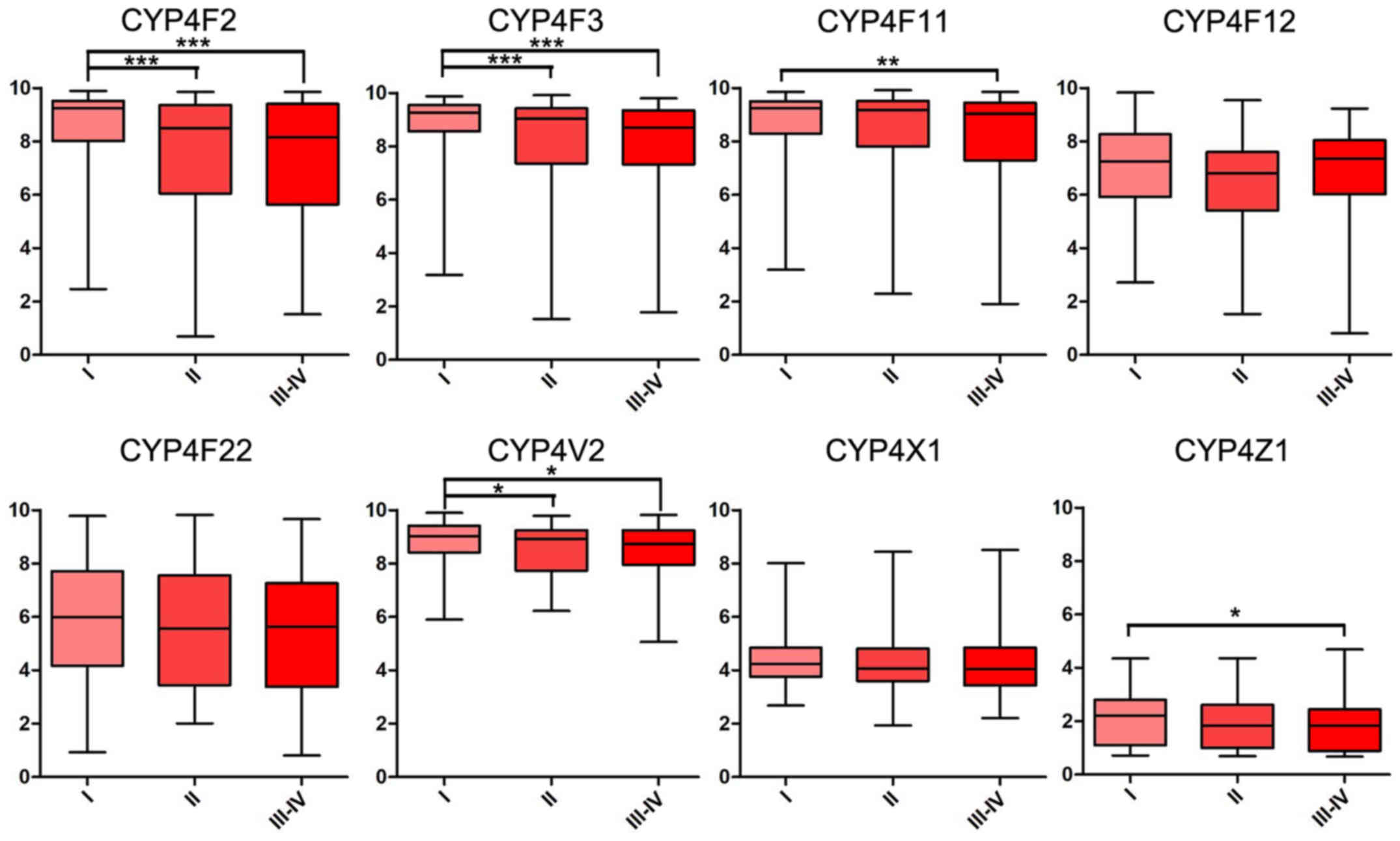
levels of CYP4F2, CYP4F3, CYP4F11 and CYP4V2 were significantly
decreased in patients with higher histological and TNM stages
(Figs. 2 and 3).
Effect of CYP4 mRNA expression on HCC
patient survival
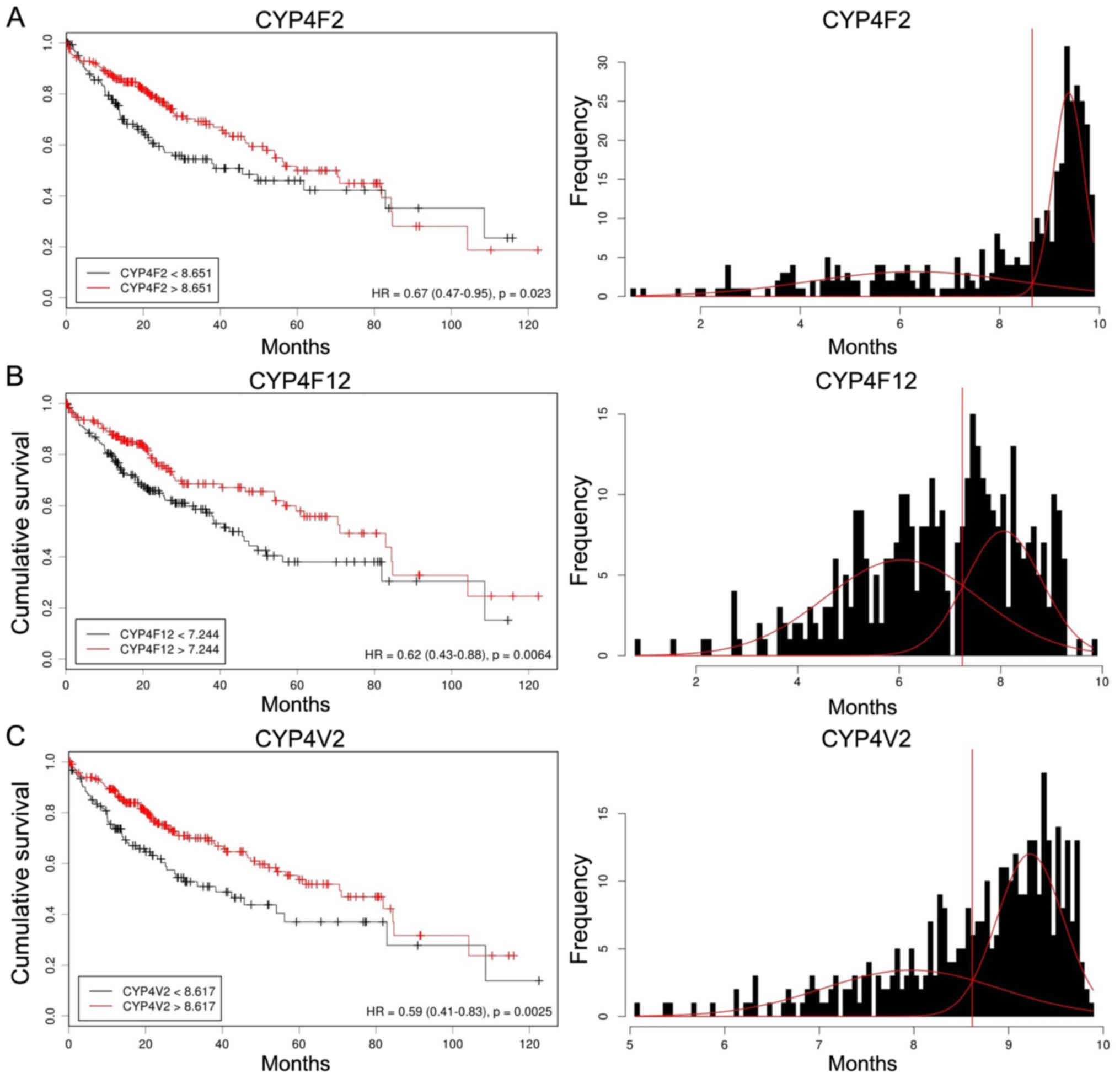
To determine the prognostic significance of CYP4
gene expression in patients with HCC, we examined correlations
between CYP4 gene expression and overall survival. Initially,
Kaplan-Meier curves were used to plot overall survival with mRNA
expression using Cutoff Finder (molpath.charite.de/cutoff) (Fig. 4). High expression levels of CYP4F2,
CYP4F12 and CYP4V2 were significantly associated with a better
prognosis [HR: CYP4F2, 0.67 (95% CI, 0.47–0.95); CYP4F12, 0.62 (95%
CI, 0.43–0.88); CYP4V2, 0.59 (95% CI, 0.41–0.83)].
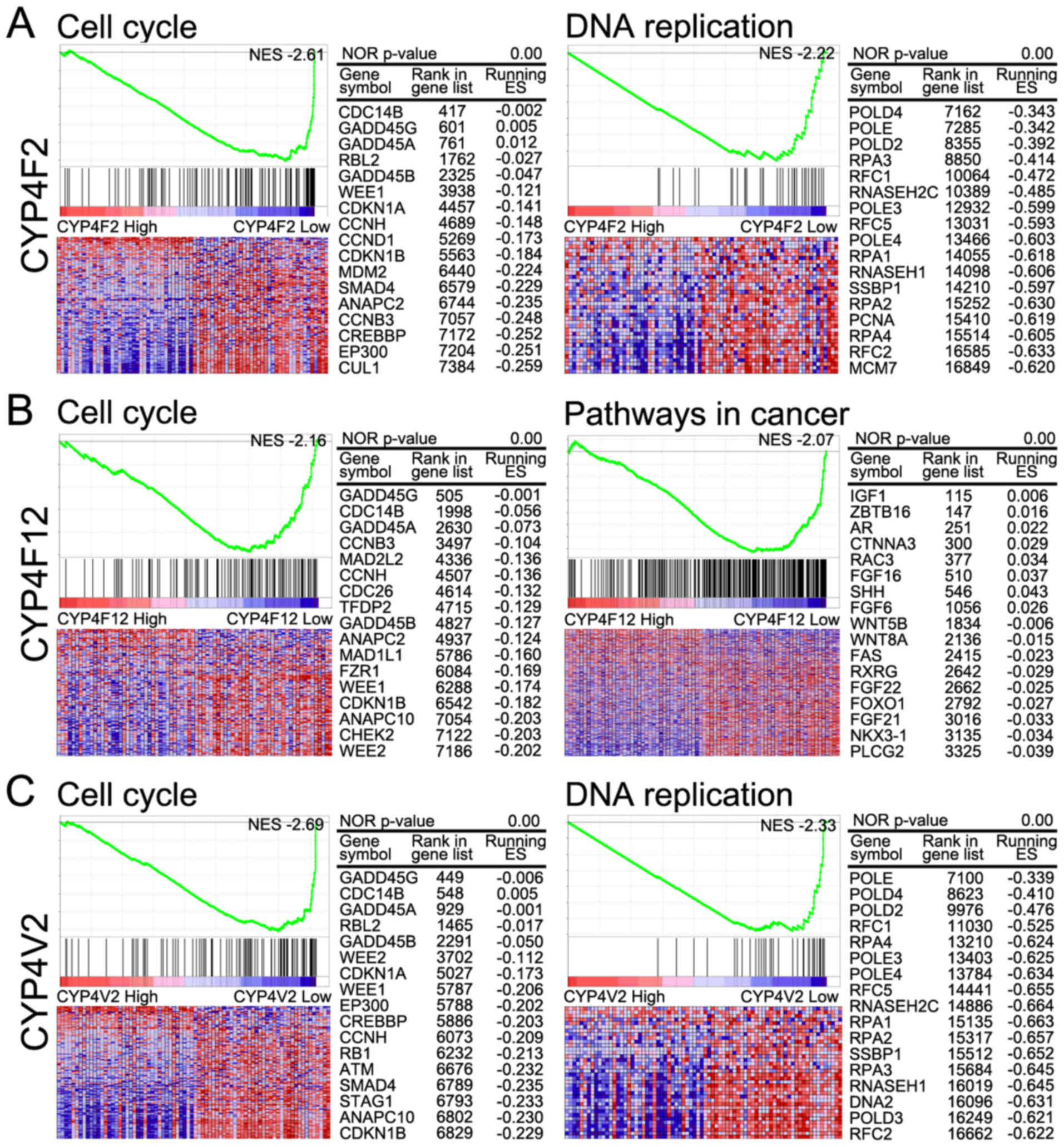
GSEA of CYP4 mRNA expression in
HCC
GSEA was performed to identify significantly
enriched pathways differing between high (top 10%) and low (bottom
10%) CYP4F2-, CYP4F12- and CYP4V2-expression groups based on
pathways provided in curated gene set enrichment analysis and KEGG
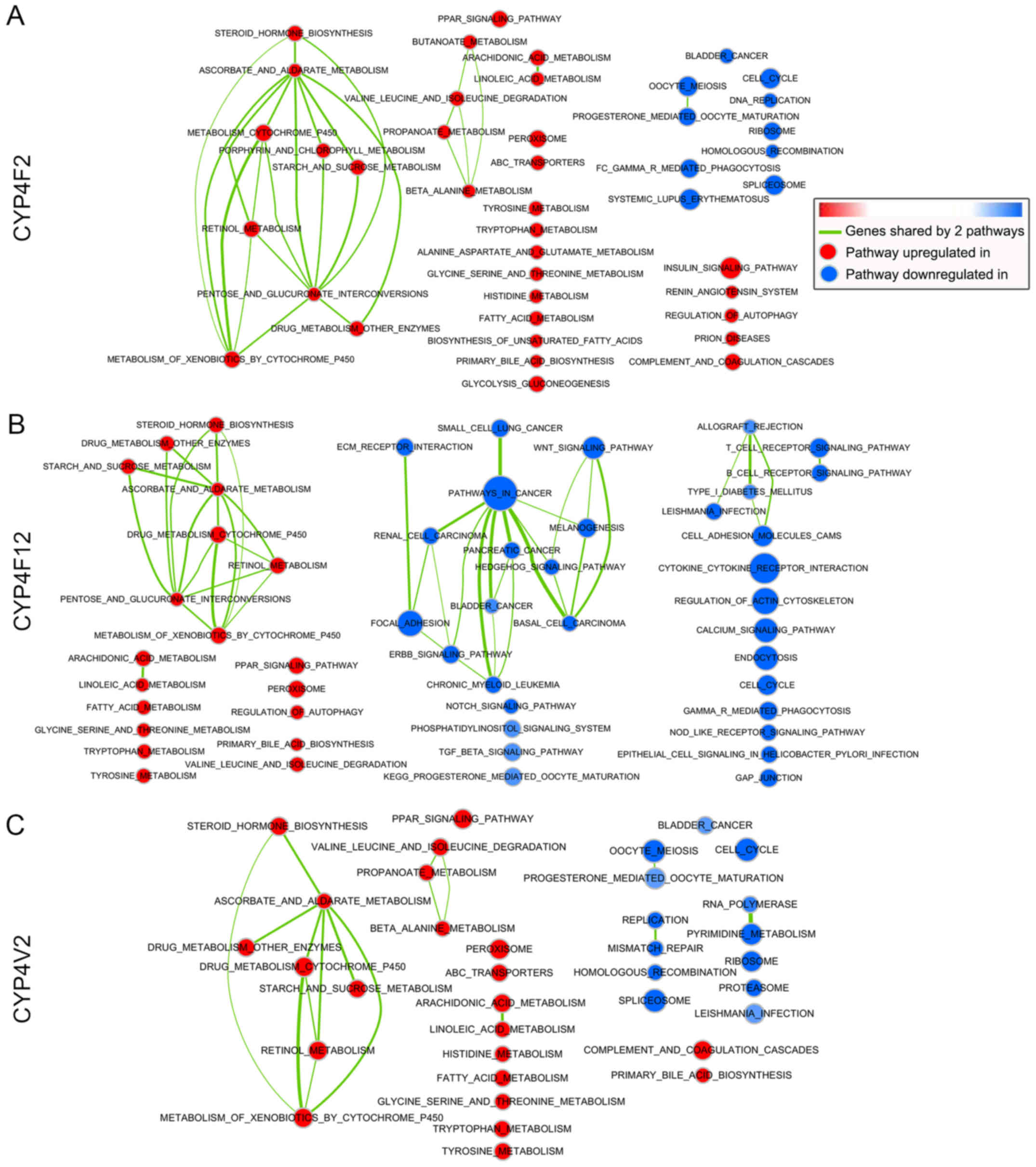
(Figs. 5 and 6). The networks of the GSEA results for
CYP4F2, CYP4F12 and CYPV2 according to Cytoscape are shown in
Fig. 7.
In the high CYPF2 group, significantly positively
correlated pathways included metabolism-related pathways (retinol
metabolism, drug metabolism, steroid biosynthesis, amino acid
metabolism, and fatty acid metabolism) and the PPAR signaling
pathway. Significantly negatively correlated pathways included the
cell cycle, DNA replication, spliceosome, cancer-related pathways
(bladder cancer, pathways in cancer, small cell lung cancer,
pancreatic cancer and renal cell carcinoma) and the Notch signaling
pathway.
Significantly positively correlated pathways in the
high CYP4F12 group included metabolism-related pathways (retinol
metabolism, drug metabolism, steroid biosynthesis, amino acid
metabolism, and fatty acid metabolism) and the PPAR signaling
pathway. Significantly negatively correlated pathways included
cancer-related pathways (small cell lung cancer, pathways in
cancer, renal cell carcinoma, pancreatic cancer, basal cell
carcinoma, bladder cancer, colorectal cancer, melanoma and
non-small cell cancer), Wnt signaling, Hedgehog signaling, TGF beta
signaling, MAPK signaling, ERBB signaling and Notch signaling.
As with the high CYP4F12 group, significantly
positively correlated pathways in the high CYP4V2 group, included
metabolism-related pathways (retinol metabolism, drug metabolism,
steroid biosynthesis, amino acid metabolism and fatty acids
metabolism) and the PPAR signaling pathway. Significantly
negatively correlated pathways in this group included cell cycle,
spliceosome, DNA replication, homologous replication and
cancer-related pathways (bladder cancer, pathways in cancer, small
cell lung cancer and pancreatic cancer).
Association between the level of
CYP4F2 protein expression and the clinicopathological features of
113 HCC cases
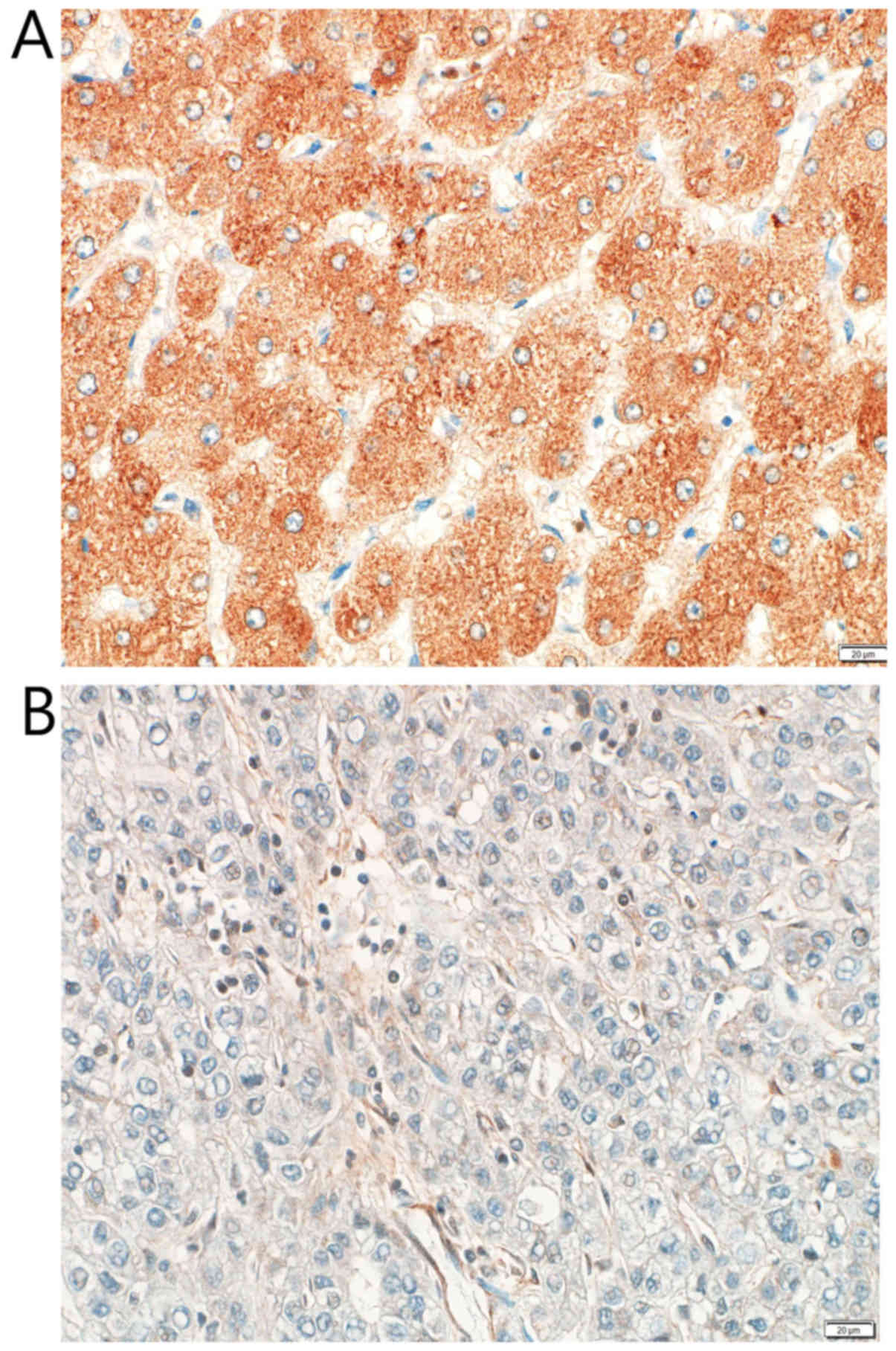
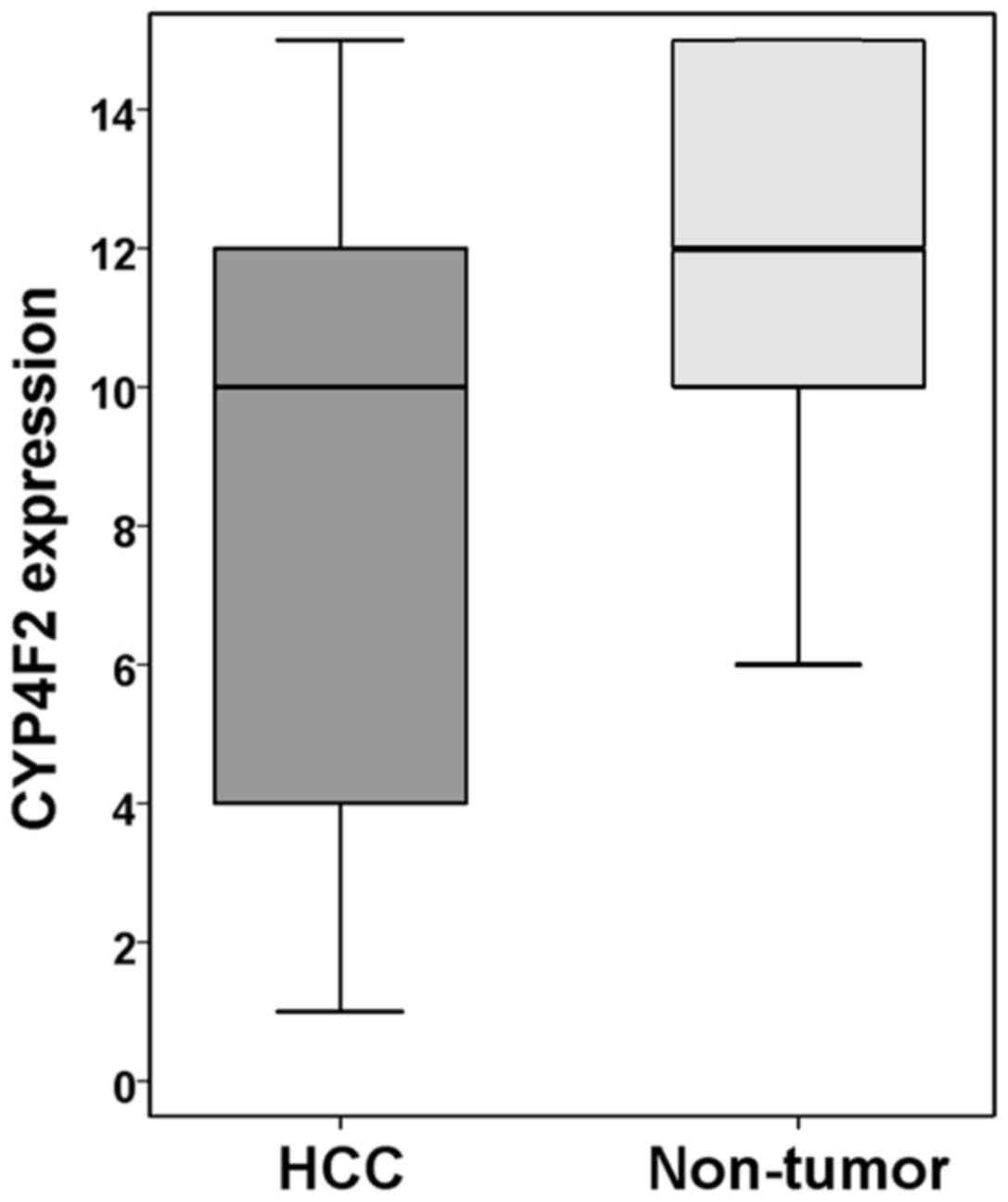
The 113 HCC cases were immunohistochemically
evaluated for CYP4F2 expression in tumor cells and in
non-neoplastic hepatocytes. Most non-neoplastic hepatocytes were
strongly and diffusely positive for CYP4F2 expression and scored
higher than did the HCC cells (P<0.001; Figs. 8 and 9).
The clinicopathological characteristics of the 113
HCC patients in association with CYP4F2 protein expression by
immunohistochemical staining are presented in Table II (14,15).
High CYP4F2 expression positively correlated with a favorable
pathological TNM stage (stage I vs. stages II–IV) (P=0.022).
Univariate and multivariate analyses using Cox's proportional
hazard regression model were performed for age, sex, hepatitis B or
C viral infection, liver cirrhosis, histologic grade, pathologic
tumor stage and CYP4F2 expression in the 113 HCC cases (Tables III and IV). The univariate analyses showed
decreased CYP4F2 expression to be a prognostic factor indicating
shorter disease-free and overall survival (P=0.043 and P=0.008,
respectively). The multivariate analysis revealed that CYP4F2
expression was an independent favorable prognostic factor for
overall survival (P=0.004).
| Table II.Patient characteristics according to
the immunohistochemical expression of CYP4F2 in hepatocellular
carcinoma (n=113). |
Table II.
Patient characteristics according to
the immunohistochemical expression of CYP4F2 in hepatocellular
carcinoma (n=113).
|
|
| CYP4F2
expression |
|---|
|
|
|
|
|---|
|
Characteristics | Total n (%) | Low (%) [80
(100.0)] | High (%) [33
(100.0)] | P-value |
|---|
| Age (years) | 113 (100.0) | 57.53±9.467 | 59.48±10.028 | 0.376 |
| Gender |
|
|
| 0.932 |
|
Male | 85 (75.2) | 60 (75.0) | 25 (75.8) |
|
|
Female | 28 (24.8) | 20 (25.0) | 8 (24.2) |
|
| HBV or HCV |
|
|
| 0.241 |
|
Negative | 23 (20.4) | 14 (17.5) | 9 (27.3) |
|
|
Positive | 90 (79.6) | 66 (82.5) | 24 (72.7) |
|
| Liver
cirrhosis |
|
|
| 0.932 |
|
Negative | 28 (24.8) | 20 (25.0) | 8 (24.2) |
|
|
Positive | 85 (75.2) | 60 (75.0) | 25 (75.8) |
|
| Histologic
grade |
|
|
| 0.321a |
| 1
(well) | 16 (14.2) | 12 (15.0) | 4 (12.1) |
|
| 2
(moderate) | 76 (67.3) | 55 (68.8) | 21 (63.6) |
|
| 3
(poorly) | 21 (18.6) | 13 (16.3) | 8 (24.2) |
|
| 4
(undifferentiated) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
|
| Pathologic
stage |
|
|
| 0.022b |
| I | 31 (27.4) | 17 (21.3) | 14 (42.4) |
|
| II | 70 (61.9) | 51 (63.8) | 19 (57.6) |
|
|
III | 11 (9.7) | 11 (13.8) | 0 (0.0) |
|
| IV | 1 (0.9) | 1 (1.3) | 0 (0.0) |
|
| Table III.Univariate analysis results of
overall survival and disease-free survival in 113 patients with
hepatocellular carcinoma. |
Table III.
Univariate analysis results of
overall survival and disease-free survival in 113 patients with
hepatocellular carcinoma.
|
| Overall
survival | Disease-free
survival |
|---|
|
|
|
|
|---|
| Prognostic
factor | HR (95% CI) | Pa | HR (95% CI) | Pa |
|---|
| CYP4F2
expression | 0.893
(0.821–0.971) | 0.008 | 0.948
(0.901–0.998) | 0.043 |
| Age at
operation | 1.019
(0.983–1.055) | 0.303 | 0.992
(0.969–1.016) | 0.511 |
| Sex |
|
|
|
|
|
Male | 1 (reference) |
| 1 (reference) |
|
|
Female | 0.631
(0.244–1.636) | 0.344 | 1.002
(0.582–1.725) | 0.996 |
| HBV or HCV |
|
|
|
|
| No | 1 (reference) |
| 1 (reference) |
|
|
Yes | 0.825
(0.388–1.756) | 0.618 | 1.171
(0.665–2.064) | 0.584 |
| Cirrhosis |
|
|
|
|
| No | 1 (reference) |
| 1 (reference) |
|
|
Yes | 1.950
(0.811–4.691) | 0.136 | 2.143
(1.191–3.857) | 0.011 |
| Histologic
grade |
| 0.011 |
| 0.005 |
| 1
(well) | 1 (reference) |
| 1 (reference) |
|
| 2
(moderate) | 1.117
(0.419–2.974) | 0.825 | 1.429
(0.699–2.920) | 0.328 |
| 3
(poorly) | 3.408
(1.151–10.090) | 0.027 | 3.204
(1.418–7.240) | 0.005 |
| 4
(undifferentiated) | NA. | NA. | NA. | NA. |
| Pathologic
stage |
| 0.003 |
| <0.001 |
| I | 1 (reference) |
| 1 (reference) |
|
| II | 0.872
(0.408–1.866) | 0.725 | 1.167
(0.688–1.981) | 0.567 |
|
III | 3.524
(1.356–9.157) | 0.010 | 4.357
(2.001–9.490) | <0.001 |
| IV | 8.942
(1.088–73.469) | 0.041 | 8.873
(1.128–69.803) | 0.038 |
| Table IV.Multivariate analysis results of
overall survival and disease-free survival in 113 patients with
hepatocellular carcinoma. |
Table IV.
Multivariate analysis results of
overall survival and disease-free survival in 113 patients with
hepatocellular carcinoma.
|
| Overall
survival | Disease-free
survival |
|---|
|
|
|
|
|---|
| Prognostic
factor | HR (95% CI) | Pa | HR (95% CI) | Pa |
|---|
| CYP4F2
expression | 0.872
(0.794–0.958) | 0.004 | 0.948
(0.896–1.003) | 0.064 |
| Age at
operation | 1.028
(0.987–1.071) | 0.188 | 0.995
(0.970–1.019) | 0.667 |
| Sex |
|
|
|
|
|
Male | 1 (reference) |
| 1 (reference) |
|
|
Female | 0.886
(0.314–2.504) | 0.820 | 1.205
(0.664–2.189) | 0.540 |
| HBV or HCV |
|
|
|
|
| No | 1 (reference) |
| 1 (reference) |
|
|
Yes | 0.936
(0.395–2.216) | 0.880 | 1.106
(0.601–2.037) | 0.746 |
| Cirrhosis |
|
|
|
|
| No | 1 (reference) |
| 1 (reference) |
|
|
Yes | 1.969
(0.747–5.196) | 0.171 | 2.139
(1.144–3.999) | 0.017 |
| Histologic
grade |
| 0.009 |
|
|
| 1
(well) | 1 (reference) |
| 1 (reference) |
|
| 2
(moderate) | 1.145
(0.371–3.529) | 0.814 | 1.297
(0.599–2.810) | 0.509 |
| 3
(poorly) | 3.856
(1.201–12.380) | 0.023 | 2.940
(1.232–7.016) | 0.015 |
| 4
(undifferentiated) | NA. | NA. | NA. | NA. |
| Pathologic
stage |
| 0.074 |
| 0.113 |
| I | 1 (reference) |
| 1 (reference) |
|
| II | 0.783
(0.331–1.854) | 0.578 | 1.112
(0.624–1.984) | 0.719 |
|
III | 2.808
(0.944–8.355) | 0.064 | 2.734
(1.167–6.400) | 0.021 |
| IV | 1.916
(0.210–17.457) | 0.564 | 3.210
(0.379–27.193) | 0.285 |
Discussion
CYP4 proteins are traditionally known as
ω-hydroxylases responsible for endogenous fatty acid metabolism.
Several previous studies have demonstrated that specific CYP4
subfamilies are selectively associated with certain cancers, such
as thyroid, breast, colon, and ovarian cancers (2,8).
However, variation in expression among CYP4 subfamily members in
HCC is unknown. The present study defined the CYP4 gene expression
profile in HCC and correlated the CYP4 mRNA expression levels of
CYP4 subfamilies as well as the CYP4F2 protein expression level
with clinicopathologic values, including prognostic factors. This
study found that the mRNA expression levels of CYP4F2, CYP4F12, and
CYP4V2 were significantly associated with good prognostic factors,
including histologic grade, TNM stage, and overall survival. The
protein level of CYP4F2 based on immunohistochemistry was higher in
non-neoplastic hepatocytes than in HCC cells, and positive
correlations were observed between low CYP4F2 protein expression
and poor prognostic factors, including higher pathologic TNM stage
and shorter overall and disease-free survival.
CYP4F subfamily enzymes are known for catalyzing
ω-hydroxylation of long-chain fatty acids, leukotrienes,
prostaglandins, vitamins with long alkyl side chains, and
hydroxyeicosatetraenoic acid (HETE) (6,16).
Such mammalian CYP4F gene amplification is associated with
increased diversity in the metabolism of both endogenous and
exogenous compounds (5,17,18).
For example, CYP4F2 and −4F3 hydroxylate pro- and anti-inflammatory
leukotrienes, whereas CYP4F11 metabolizes eicosanoids and drugs,
and CYP4F8 and 4F12 metabolize prostaglandins, endoperoxides and
arachidonic acid (5,19). In addition to these metabolic
roles, CYP4F is well known as a biomarker of tumors, such as
thyroid, ovarian, breast, and colon cancers (8). In particular, progression of
HCV-infected liver disease to HCC tends to occur less frequently
patients with lower CYP4F expression (20).
CYP4V2 is predicted to perform fatty acid metabolism
and is associated with Bietti crystalline dystrophy, an autosomal
recessive disorder that causes progressive night blindness and
constriction of vision and is characterized by the presence of
shiny yellow crystals with complex lipid deposits in the cornea and
retina (21). CYP4V2 is expressed
in human THP1 macrophages that exhibit fatty acid metabolism
catalytic activity, and its expression is regulated by peroxisome
proliferator activated receptor gamma (PPARγ) (22). CYP4V2 expression has also been
correlated with lower tumor grades in breast cancer (1,2).
In the present study, CYP4F2, CYP4F12, and CYPV2
were found to be important indicators of cumulative survival
differences based on histologic grade and TNM stage in patients
with HCC. Consistently, the level of CYP4F2 protein expression was
lower in HCC cells than in matched non-neoplastic hepatocytes.
Indeed, the GSEA data support the results of
improved survival in patients with high expression of CYP4F2,
CYP4F12 and CYP4V2 genes. Correlations were observed between these
patients and upregulation of specific metabolic pathways, such as
drug metabolism pathways related to cytochrome P450, fatty acid
metabolism and the PPAR signaling pathway, in HCC. A previous study
showed that CYP4 is associated with PPAR signaling, which is
related to cancer proliferation and metastasis; CYP4F is also
significantly decreased in groups with a low incidence of breast
cancer, though this is not related to HCC (23). Moreover, the CYP4F12 gene has been
linked to malignancy-associated metabolic abnormalities in
cholesterol (fatty acid metabolism) and primary bile acid metabolic
homeostasis (24). Specifically,
the CYP4F12 gene has been connected to genetic and epigenetic
alterations in Sirt6, a member of the sirtuin family of
NAD-dependent deacetylases, which are involved in HCC development
and progression (24).
Furthermore, our study revealed that high expression
of genes CYP4F2, CYP4F12, and CYP4V2 is related to downregulation
of cell cycle pathways. Therefore, HCC with low expression of these
CYP genes is associated with tumor proliferation. In addition,
genes related to the cell cycle, DNA replication, cancer, and Wnt
signaling pathways were significantly downregulated in patients
with high expression levels of CYP4F2 and CYP4F12. These results
indicate higher cancer cell survival in HCC patients with low
expression of CYP4F2, CYP4F12, and CYP4V2 genes, and these factors
may contribute to tumor progression and decreased survival.
Moreover, high expression of these CYP genes was related to
downregulation of several cancer-related pathways. For example,
higher expression of CYP4F12 is related to aberrant Wnt signaling.
In HCC, as in other types of tumors, aberrant activation of the
canonical Wnt/beta-catenin signaling pathway is an important
contributor to tumorigenesis (25).
Taken together, CYP4F2, CYP4F12, and CYP4V2 in HCC
are involved in patient survival via components of various
metabolic pathways. Our study suggests that the gene expression
levels of CYP4F2, CYP4F12, and CYP4V2 may not only serve as
diagnostic markers but may also function as prognostic factors for
HCC. Further clinicopathologic studies are required to verify the
roles of CYP4F2, CYP4F3 and CYP4V2 gene expression in HCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Basic Science Research Program through the National Research
Foundation of Korea (NRF) funded by the Ministry of Education,
Science, and Technology (grant nos. NRF-2016R1D1A1B01014311 and
NRF-2017R1C1B1004924).
Availability of data and materials
The datasets generated and analyzed during the
current study are available in TCGA (cancergenome.nih.gov/) and Firebrowse (firebrowse.org/?cohort=LIHC&download_dialog=true;
the ‘illuminahiseq_rnaseqv2-RSEM_genes_normalized (MD5)
dataset’).
Authors' contributions
KK, HSE and SYC designed the study. KK, HSE, SYC,
BSL and IOS performed the study. KK conducted the pathological
analysis. HSE and SYC drafted the original manuscript, and KK
edited the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of Chungnam National University Hospital (CNUH
2018-02-017). The requirement for written informed consent was
waived due to the retrospective nature of the study; however,
consent was originally obtained at the time of data collection.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Murray GI: The role of cytochrome P450 in
tumour development and progression and its potentil in therapy. J
Pathol. 192:419–426. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murray GI, Patimalla S, Stewart KN, Miller
ID and Heys SD: Profiling the expression of cytochrome P450 in
breast cancer. Histopathology. 57:202–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mackay DS and Halford S: Focus on
molecules: Cytochrome P450 family 4, subfamily V, polypeptide 2
(CYP4V2). Exp Eye Res. 102:111–112. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nelson DR: The cytochrome p450 homepage.
Hum Genomics. 4:59–65. 2009.PubMed/NCBI
|
|
5
|
Hardwick JP: Cytochrome P450 omega
hydroxylase (CYP4) function in fatty acid metabolism and metabolic
diseases. Biochem Pharmacol. 75:2263–2275. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hsu MH, Savas U, Griffin KJ and Johnson
EF: Human cytochrome p450 family 4 enzymes: Function, genetic
variation and regulation. Drug Metab Rev. 39:515–538. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kirischian NL and Wilson JY: Phylogenetic
and functional analyses of the cytochrome P450 family 4. Mol
Phylogenet Evol. 62:458–471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alexanian A, Miller B, Roman RJ and
Sorokin A: 20-HETE-producing enzymes are up-regulated in human
cancers. Cancer Genom Proteom. 9:163–169. 2012.
|
|
9
|
Johnson AL, Edson KZ, Totah RA and Rettie
AE: Cytochrome P450 ω-Hydroxylases in inflammation and cancer. Adv
Pharmacol. 74:223–262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ciriello G, Gatza ML, Beck AH, Wilkerson
MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et
al: Comprehensive molecular portraits of invasive lobular breast
cancer. Cell. 163:506–519. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Budczies J, Klauschen F, Sinn BV, Győrffy
B, Schmitt WD, Darb-Esfahani S and Denkert C: Cutoff Finder: A
comprehensive and straightforward Web application enabling rapid
biomarker cutoff optimization. PLoS One. 7:e518622012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yeo MK, Lee YM, Seong IO, Choi SY, Suh KS,
Song KS, Lee CS, Kim JM and Kim KH: Up-regulation of cytoplasmic
CD24 expression is associated with malignant transformation but
favorable prognosis of colorectal adenocarcinoma. Anticancer Res.
36:6593–6598. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Allred DC, Harvey JM, Berardo M and Clark
GM: Prognostic and predictive factors in breast cancer by
immunohistochemical analysis. Mod Pathol. 11:155–168.
1998.PubMed/NCBI
|
|
14
|
Amin MB, Edge S, Greene F, Byrd DR,
Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR,
Sullivan DC, et al: AJCC cancer staging manual. Eighth edition.
Chicago, IL: Springer; 2017, View Article : Google Scholar
|
|
15
|
Bosman FT, Carneiro F, Hruban RH and
Theise ND: WHO classification of tumours of the digestive system.
4th edition. Lyon: IARC; 2010
|
|
16
|
Kalsotra A and Strobel HW: Cytochrome P450
4F subfamily: At the crossroads of eicosanoid and drug metabolism.
Pharmacol Ther. 112:589–611. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cui X, Kawashima H, Barclay TB, Peters JM,
Gonzalez FJ, Morgan ET and Strobel HW: Molecular cloning and
regulation of expression of two novel mouse CYP4F genes: Expression
in peroxisome proliferator-activated receptor alpha-deficient mice
upon lipopolysaccharide and clofibrate challenges. J Pharmacol Exp
Ther. 296:542–550. 2001.PubMed/NCBI
|
|
18
|
Kalsotra A, Turman CM, Kikuta Y and
Strobel HW: Expression and characterization of human cytochrome
P450 4F11: Putative role in the metabolism of therapeutic drugs and
eicosanoids. Toxicol Appl Pharmacol. 199:295–304. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bylund J, Hidestrand M, Ingelman-Sundberg
M and Oliw EH: Identification of CYP4F8 in human seminal vesicles
as a prominent 19-hydroxylase of prostaglandin endoperoxides. J
Biol Chem. 275:21844–21849. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsunedomi R, Iizuka N, Hamamoto Y,
Uchimura S, Miyamoto T, Tamesa T, Okada T, Takemoto N, Takashima M,
Sakamoto K, et al: Patterns of expression of cytochrome P450 genes
in progression of hepatitis C virus-associated hepatocellular
carcinoma. Int J Oncol. 27:661–667. 2005.PubMed/NCBI
|
|
21
|
Nakano M, Kelly EJ, Wiek C, Hanenberg H
and Rettie AE: CYP4V2 in Bietti's crystalline dystrophy: Ocular
localization, metabolism of ω-3-polyunsaturated fatty acids, and
functional deficit of the p.H331P variant. Mol Pharmacol.
82:679–686. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yi M, Shin JG and Lee SJ: Expression of
CYP4V2 in human THP1 macrophages and its transcriptional regulation
by peroxisome proliferator-activated receptor gamma. Toxicol Appl
Pharmacol. 330:100–106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi Y, Steppi A, Cao Y, Wang J, He MM, Li
L and Zhang J: Integrative comparison of mRNA expression patterns
in breast cancers from Caucasian and Asian Americans with
implications for precision medicine. Cancer Res. 77:423–433. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marquardt JU, Fischer K, Baus K, Kashyap
A, Ma S, Krupp M, Linke M, Teufel A, Zechner U, Strand D, et al:
SIRT6 dependent genetic and epigenetic alterations are associated
with poor clinical outcome in HCC patients. Hepatology.
58:1054–1064. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Takigawa Y and Brown AM: Wnt signaling in
liver cancer. Curr Drug Targets. 9:1013–1024. 2008. View Article : Google Scholar : PubMed/NCBI
|