Introduction
X-linked hydrocephalus due to aqueductal stenosis
[HSAS, Online Mendelian Inheritance in Man (OMIM) #307000],
affecting ~1 in 30,000 live male births (1), is the most common of the inherited
form of hydrocephalus. It is caused by mutations in the L1 cell
adhesion molecule (L1CAM) gene; >280 different mutations
have been reported, and ~50% of them are missense mutations
(2). As the majority of the
mutations occur only in one family, they are considered to be
private mutations. Genotype-phenotype correlation has been
described and L1CAM mutations have been divided into four
classes: i) Frameshift or nonsense mutations (Class I) lying in the
extracellular L1 protein resulting in loss of L1 protein function
and severe phenotypes; ii) missense mutations (Class II) in the
extracellular of L1 resulting in partial conservation of L1 protein
function and causing mild or severe phenotypes; iii) mutations in
the cytoplasmic domain of L1 (Class III) which tend to affect
signal transduction and cause mild phenotypes; and iv)
extracellular mutations (Class IV) associated with the aberrant
splicing of pre-mRNA, although no corresponding phenotypes have
been reported (3).
Apart from HSAS, mutations in L1CAM cause
three other associated neurological disorders, known collectively
as L1 syndrome: Mental retardation, aphasia, shuffling gait,
adducted thumbs (MASA) syndrome (OMIM no: 303350); corpus callosum
hypoplasia, retardation, adducted thumbs, spastic paraplegia,
hydrocephalus (CRASH) syndrome (OMIM no: 303350) and X-linked
partial agenesis of corpus callosum (OMIM no: 304100) (4). These allelic disorders are different
presentations of L1CAM mutations.
At present, mutations in L1CAM in the Chinese
population have rarely been reported (5,6). In
the present study, two fetuses with isolated hydrocephaly from two
Chinese families with a history of X-linked hydrocephalus and the
molecular findings were reported.
Case report
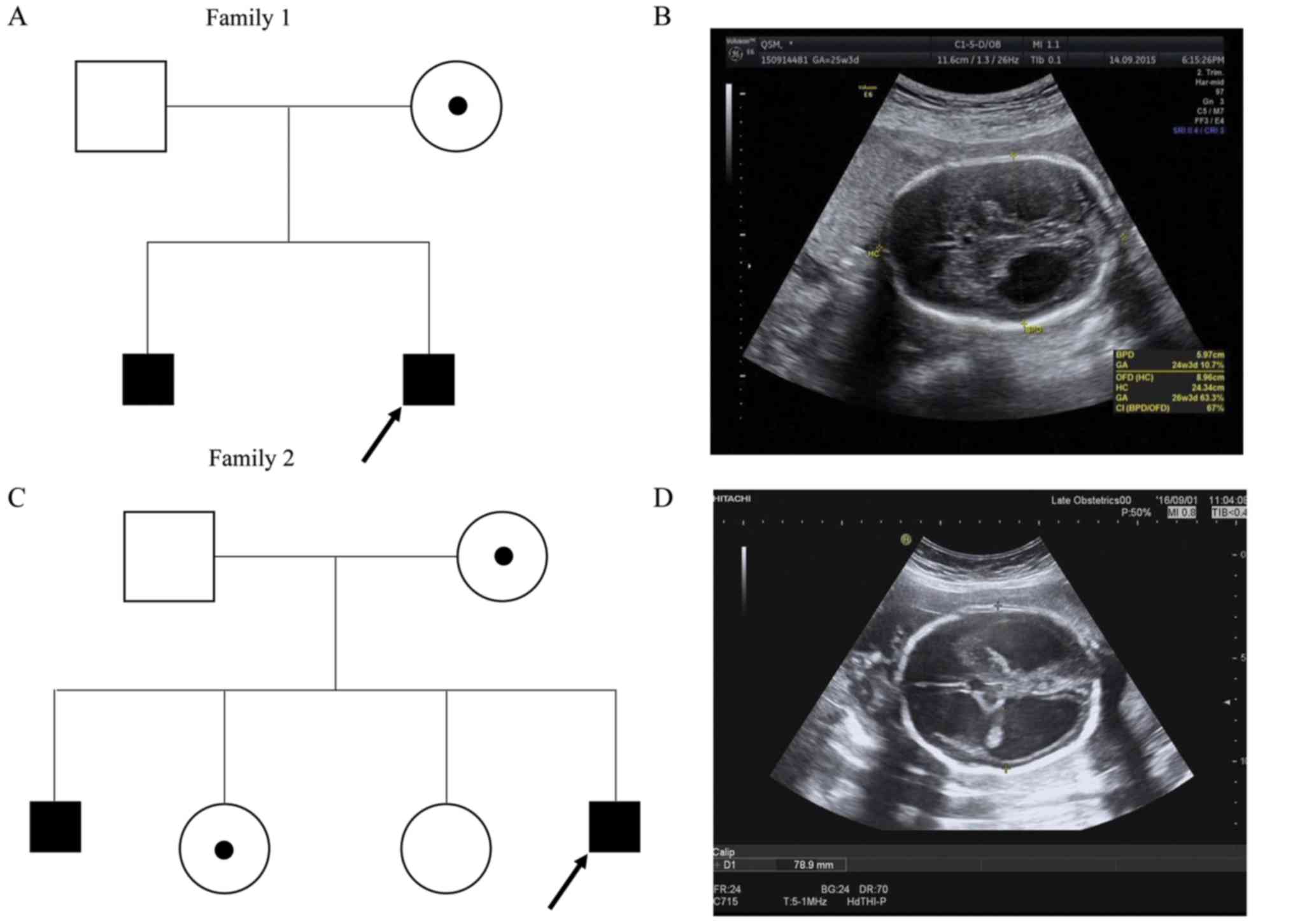
Fetus 1, family 1
A 20-year-old woman, gravida 2 para 0, was referred
to the Guangxi Maternal and Child Health Hospital (Nanning, China)
on March 14, 2017 at 24 weeks of gestation due to sonographic
detection of fetal hydrocephaly. The family history was notable; a
son with congenital hydrocephaly had been terminated at 28 weeks of
gestation (Fig. 1A). A 3D
ultrasound examination confirmed a fetus with bilateral dilatation
of the cerebral posterior horns [2.54 cm (left), 2.16 cm (right)],
consistent with hydrocephaly (Fig.
1B).
Fetus 2, family 2
A 27-year-old healthy woman, gravida 4 para 2, was
referred to the Guangxi Maternal and Child Health Hospital
(Nanning, China) on April 8, 2017 at 30 weeks of gestation due to
sonographic detection of fetal hydrocephaly. The pregnancy
progressed uneventfully, without exposure to any known teratogens.
The family history was notable; a son had been terminated due to
severe hydrocephalus. There were two healthy daughters (Fig. 1C). A standard second trimester
ultrasound screening, performed at the 28th week of gestation,
revealed that the width of the left lateral brain ventricle was 3.8
cm and the right 4.0 cm, consistent with severe hydrocephaly
(Fig. 1D). No other fetal
structural abnormalities were identified.
A total of 100 healthy individuals of Chinese
ethnicity (50 males and 50 females, aged 1–60 years) were recruited
at the Guangxi Maternal and Child Health Hospital between May 2017
to December 2017. With informed consent from all participants and
approval from the Guangxi Maternal and Child Health Hospital
Medical Ethics Committee [approval no. (2017) Lun Han Shen
(3–10)], cordocentesis was performed for the
two fetuses, and peripheral blood was obtained from the fetuses'
family members and the healthy individuals.
Molecular analysis
Genomic DNA was extracted from the cord blood of the
fetuses and the peripheral blood of both parents and family members
according to the standard protocols of the QIAamp DNA Blood Mini
kit (Qiagen GmbH, Hilden, Germany). Fetal DNA was analyzed for
L1CAM mutations by Sanger sequencing. The primers of the 28
exons with coding region and splicing junctions of L1CAM
used for amplification and sequencing were designed by Primer3 web
(http://primer3.ut.ee; version 4.0.0). The primer
sequences are shown in Table I.
Polymerase chain reaction was conducted in a 25 µl reaction volume
using 12.5 µl Premix Taq™ (cat. no. RR901A; Takara Biotechnology
Co., Ltd., Dalian, China), 9.5 µl distilled water, 200 nmol/l
primer and 100 ng/µl DNA. The thermocycling conditions were as
follows: Initial denaturation for 5 min at 95°C, followed by 35
cycles of 30 sec at 95°C, 30 sec at 60°C, 60 or 90 sec at 72°C, and
final extension for 7 min at 72°C. DNA sequencing was performed
using an ABI3730XL Genetic Analyzer (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) to detect mutations in
the entire coding region of L1CAM. Fetus sequence data was
aligned with the reference sequence obtained from the University of
California, Santa Cruz (Santa Cruz, CA, USA) Genome Browser
(https://genome.ucsc.edu/; L1CAM,
GRCh37/hg19, NM_000425.4) using Mutation Surveyor software
(https://softgenetics.com/mutationSurveyor.php; version
4.0.4.0). Variants detected in the fetuses, but absent in the
ClinVar (www.ncbi.nlm.nih.gov/clinvar/), Human Gene Mutation
Database (www.hgmd.cf.ac.uk/ac/), L1CAM Mutation Database
(http://www.l1cammutationdatabase.info/mutations.aspx),
Single Nucleotide Polymorphism Database (www.ncbi.nlm.nih.gov/SNP), Ensembl (http://grch37.ensembl.org/index.html)
and Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/) were considered novel
variants, and were further examined in the DNA samples of 100
healthy individuals, to exclude the possibility of
ethnicity-specific polymorphisms.
 | Table I.Polymerase chain reaction primers for
molecular analysis of the L1CAM gene. |
Table I.
Polymerase chain reaction primers for
molecular analysis of the L1CAM gene.
| Exon | Forward (5′-3′) | Reverse (5′-3′) | Amplified product
size (bp) |
|---|
| 1 |
GTCCTTCCGTTCTCTCGTCT |
AACGTCCTGGCTATCTCCTG | 397 |
| 2–4 |
AGCTTACTATGTCCCCTGCC |
ATGAGATGACCGGAAGTGCA | 1401 |
| 5–6 |
CTGGCCTCCCTAAGTGCTAG |
GCCTCTGTGTCTTCCTCCAT | 794 |
| 7–10 |
AATTCTGGGGTGGAGGGAAG |
AGGGTGTCAGCAAGGAGAAA | 1178 |
| 11–15 |
AACCAAGATTTGCAGGGCTC |
CCGACAATGGAGTGATCAGC | 1500 |
| 16–18 |
GCTGATCACTCCATTGTCGG |
AATGCTGAGAGGTGTGGACA | 1020 |
| 19–23 |
TCTCTGTGTGTAGGGGCTTG |
ACATACTGTGGCGAAAGGGA | 1461 |
| 24–27 |
TCCCTTTCGCCACAGTATGT |
TGTTGGCCTCTCCCTGAAAT | 1250 |
| 28 |
AGATGACAGCTCCAGACCTG |
AAGTTCTCCTCTGCCCCAAC | 494 |
In silico prediction and pathogenicity
assessment
The amino acid sequences of human neural cell
adhesion molecule L1CAM, obtained from the UniProt database
(http://www.uniprot.org/), were aligned with
orthologs from 6 different eukaryotic species by DNAMAN software
(https://www.lynnon.com/; version 8.0), including
Mus musculus, Bos taurus, Gallus gallus, Danio rerio, Xenopus
tropicalis and Drosophila melanogaster. The
pathogenicity of the identified variants was assessed using SIFT
(http://sift.jcvi.org/), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/),
and MutationTaster (http://www.mutationtaster.org/) prediction tools, and
further classified following the guidelines of American College of
Medical Genetics and Genomics (ACMG) /the Association for Molecular
Pathology (AMP) (7).
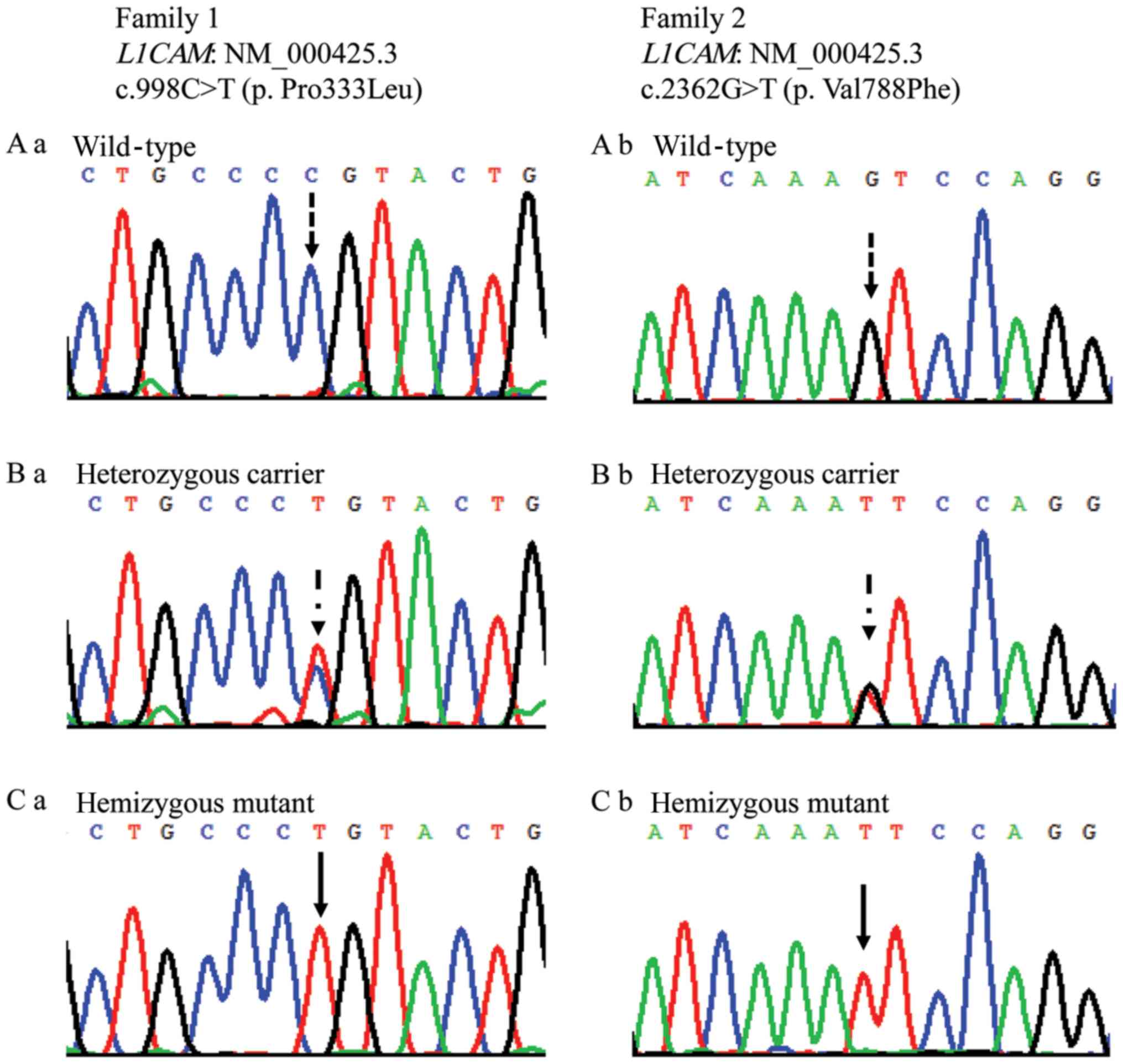
Chromosome analysis revealed a normal male karyotype
for the two fetuses, and molecular analysis identified two novel
missense variants in L1CAM from two families:
c.998C>T(p.Pro333Leu) in family 1, and c.2362G>T(p.Val788Phe)
in family 2 (Fig. 2). The affected
fetuses were hemizygous, and the mothers of the fetuses were
heterozygous; fathers were wild type for the identified variants.
The older sister of family 2 was heterozygous and the younger
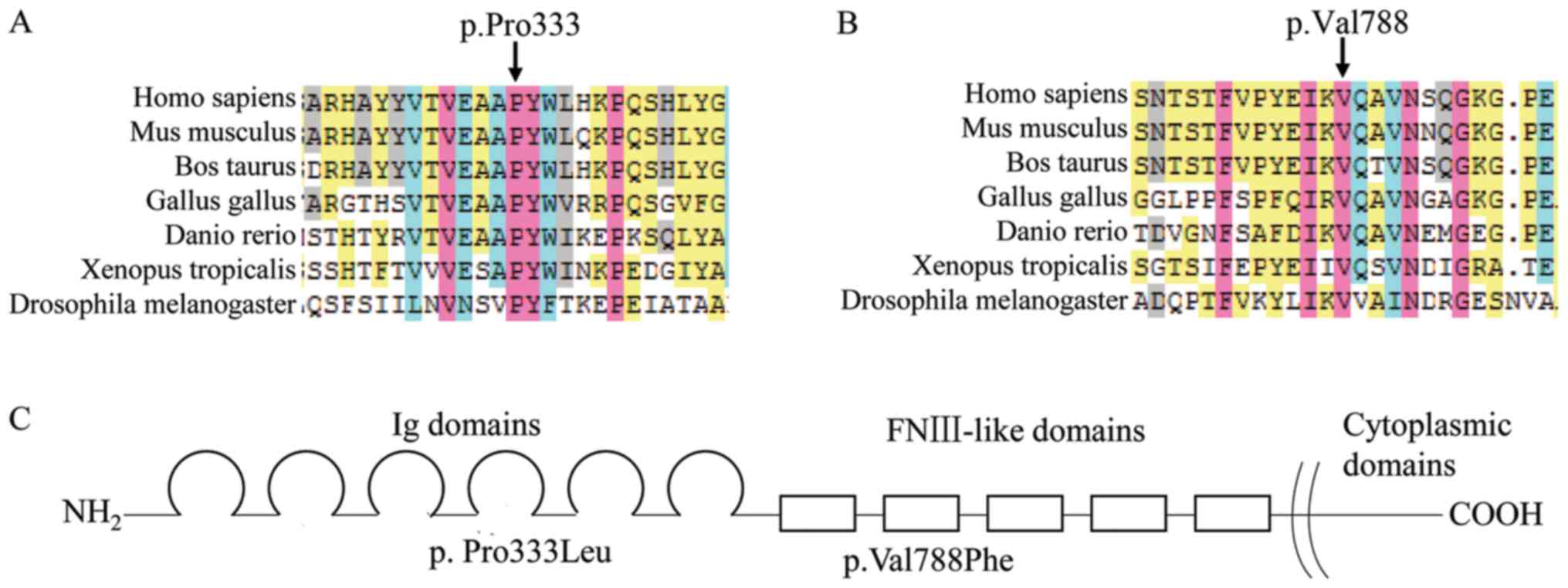
sister was wild type. The proline residue at position 333 and the
valine residue at position 788 of L1CAM are highly conserved across
7 eukaryotic species. The two variants were predicted to be
‘disease causing’ based on SIFT, PolyPhen2 and MutationTaster
prediction tools. The residues were absent in the dbSNP, Ensembl,
ExAC and the 100 normal controls. Alterations in amino acids at
position 333 (Fig. 3A) had been
reported as pathogenic in patients with hydrocephaly, and so,
according to the ACMG/AMP guidelines, PM1, PM2 and PM5, in addition
to PP3 and PP4, apply to this variant and is classified as likely
pathogenic. The variant at position 788 (Fig. 3B), PM1, PM2, PP3 and PP4 also
apply, and can also be classified as likely pathogenic.
Discussion
Congenital hydrocephaly is a common medical
condition with complex and multifactorial causes. L1 syndrome
accounts for ~5–10% of cases of congenital hydrocephaly in males.
Molecular testing of L1CAM for fetuses with isolated
hydrocephaly may have limited usefulness (6). Additional clinical signs and family
history can improve the diagnostic yield significantly (8). In the present study, it was
demonstrated that L1CAM testing for isolated hydrocephaly in
a fetus with family history could lead to confirmatory molecular
findings. Prior to the present study, there were only two
L1CAM variants reported in Chinese patients (fetuses). The
findings of the present study expand the mutational spectrum of the
L1CAM gene in the Chinese population, and the information
can be used in genetic counseling, carrier testing of female
relatives and prenatal, as well as preimplantation genetic
diagnosis.
HSAS exhibits a variable phenotype within and
between families. Female carriers exhibit no evident clinical
symptoms or signs. The milder phenotype is characterized by
low-grade hydrocephalus, whereas the severe phenotype is
characterized by stillbirth or infant mortality (9). The L1CAM protein consists of six
immunoglobulin (Ig) and five fibronectin type III (FnIII) domains
in the extracellular region, a transmembrane domain and a short
cytoplasmic C terminal domain (Fig.
3C) (10). Mutations occurring
in the key residues are mostly considered to be pathogenic.
Missense mutations in the extracellular domains may affect the
integrity of the domains and disturb the binding activities of
L1CAM to itself (homophilic) and to the other cell adhesion
molecules including integrins and neurocan, causing mild or severe
phenotypes, whereas mutations occurring in the cytoplasmic domain
tend to exhibit the least severe effects, since the extracellular
transmembrane domains remain intact (11).
Fetus 1 possessed a missense variant (c.998C>T;
p.Pro333Leu) impairing the fourth Ig domain of the extracellular
domains. Proline 333 is a key residue that is buried in a close
packed cavity and is important for the conformation of L1CAM
(12). As a result of replacing an
internal residue by a larger charged residue, the change of a
proline to leucine likely results in the disruption of steric
strain, thereby possibly reducing homophilic binding. This variant
belongs to the class II of mutations, according to the
classification by Fransen et al (3). A different variant located in the
same position (c.998C>G; p.Pro333Arg) has been reported in a
patient with hydrocephalus and adducted thumbs (13). Fetus 1 only demonstrated isolated
hydrocephalus. The proline and leucine residues are hydrophobic
uncharged amino acids, but arginine is a hydrophilic basic amino
acid. The difference in the nature of the amino acids could lead to
varying severity of the phenotype.
Fetus 2 possessed a missense class II variant
(c.2362G>T; p.Val788Phe) lying in the extracellular second FnIII
domain. Valine 788 is involved in hydrogen bonding with another
residue at 799–803. The introduction of a phenylalanine residue may
disrupt the β-sheet hydrogen bonds and steric strain, affecting the
structure and function of L1CAM protein.
In conclusion, the present study identified two
novel pathogenic class II variants in L1CAM in two Chinese
families with a history of hydrocephaly. The fetuses in these two
families presented with isolated X-linked hydrocephaly,
highlighting the importance of prenatal ultrasonography and
L1CAM mutation testing in the diagnosis of HSAS.
Acknowledgements
Not applicable.
Funding
The present study was supported by the project of
science and technology of Guangxi Zhuang Autonomous Region (grant
no. gui-ke-gong 14124004-1-8), and the Open Project Program of the
Shanghai Key Laboratory of Birth Defect, Children's Hospital of
Fudan University (grant no. 16DZKF1014).
Availability of data and materials
The data used and/or analyzed during the current
study are available from the corresponding author or first author
on reasonable request.
Authors' contributions
XF and BX designed the study and experiments. JL
analyzed the clinical phenotype and followed up the patients. HW
performed cordocentesis and induced abortion. ZY performed prenatal
ultrasonography examination. SL and ZQ prepared reagents and
designed the primers, QY and ML performed the PCR and Sanger
sequencing experiments. SY and JW conducted the bioinformatic
analysis. BX and YL made substantial contributions to the collation
of data and wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by Guangxi Maternal
and Child Health Hospital Medical Ethics Committee [approval no.
(2017) Lun Han Shen (3–10)] and written informed consent was
obtained from parents and patients.
Patient consent for publication
Written informed consent was obtained from fetuses'
parents for the publication of this study including images and
data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rosenthal A, Jouet M and Kenwrick S:
Aberrant splicing of neural cell adhesion molecule L1 mRNA in a
family with X-linked hydrocephalus. Nat Genet. 2:107–112. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yamasaki M and Kanemura Y: Molecular
biology of pediatric hydrocephalus and hydrocephalus-related
diseases. Neurol Med Chir (Tokyo). 55:640–646. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fransen E, Van Camp G, D'Hooge R, Vits L
and Willems PJ: Genotype-phenotype correlation in L1 associated
diseases. J Med Genet. 35:399–404. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Muñoz A, Cabrera-López JC,
Santana-Rodríguez A, de Laguna Toledo-Bravo L, Santana-Artiles A
and Sebastián-García I: X-linked hereditary spastic paraplegia due
to mutation in the L1CAM gene: Three cases reports of CRASH
syndrome. Rev Neurol. 62:218–222. 2016.(In Spanish). PubMed/NCBI
|
|
5
|
Yao F, Zhang W, Gao J, Chen W and Liu X:
Detection of L1CAM mutation and prenatal diagnosis of X-linked
hydrocephalus. Sheng Zhi Yi Xue Za Zhi. 25:347–352. 2016.(In
Chinese).
|
|
6
|
Duan H, Zhao G, Wang Y, Zhu X and Li J:
Novel missense mutation of L1CAM in a fetus with isolated
hydrocephalus. Congenit Anom (Kyoto). 58:176–177. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Adle-Biassette H, Saugier-Veber P,
Fallet-Bianco C, Delezoide AL, Razavi F, Drouot N, Bazin A,
Beaufrère AM, Bessières B, Blesson S, et al: Neuropathological
review of 138 cases genetically tested for X-linked hydrocephalus:
Evidence for closely related clinical entities of unknown molecular
bases. Acta Neuropathol. 126:427–442. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chidsey BA, Baldwin EE, Toydemir R, Ahles
L, Hanson H and Stevenson DA: L1CAM whole gene deletion in a child
with L1 syndrome. Am J Med Genet A. 164A:1555–1558. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bateman A, Jouet M, MacFarlane J, Du JS,
Kenwrick S and Chothia C: Outline structure of the human L1 cell
adhesion molecule and the sites where mutations cause neurological
disorders. EMBO J. 15:6050–6059. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shieh C, Moser F, Graham JM Jr, Watiker V
and Pierson TM: Mutation in the sixth immunoglobulin domain of
L1CAM is associated with migrational brain anomalies. Neurol Genet.
1:e342015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
De Angelis E, Watkins A, Schäfer M,
Brümmendorf T and Kenwrick S: Disease-associated mutations in L1
CAM interfere with ligand interactions and cell-surface expression.
Hum Mol Genet. 11:1–12. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
De Angelis E, MacFarlane J, Du JS, Yeo G,
Hicks R, Rathjen FG, Kenwrick S and Brümmendorf T: Pathological
missense mutations of neural cell adhesion molecule L1 affect
homophilic and heterophilic binding activities. EMBO J.
18:4744–4753. 1999. View Article : Google Scholar : PubMed/NCBI
|