Introduction
Liver cirrhosis, caused by progressive hepatocyte
injury-induced conversion of normal liver architecture into
abnormal nodules, is a common chronic liver disease characterized
by hepatic fibrosis. There are a number of factors that may lead to
cirrhosis, including alcohol abuse, viral infection, administration
of drugs or chemicals (1). In
total, one million people succumb to liver cirrhosis every year,
accounting for 2% of the total global mortality in 2010 (2,3).
Therefore, liver cirrhosis is a significant global health burden.
Although the survival of patients with liver cirrhosis has been
improved with the development of medicine and surgical procedures
(including liver transplantation), developing effective therapeutic
agents to prevent the progression of early-stage cirrhosis remains
crucial.
Autophagy is a metabolic process occurring in all
cell types. Under conditions of cellular stress, defective
organelles and excessive components are eliminated to maintain cell
survival and cellular activities through autophagy (4,5).
Previous studies demonstrated that autophagy may drive the
activation of hepatic stellate cells (HSCs) (6,7), a
critical event in liver fibrogenesis (8). Furthermore, inhibition of autophagy
may reduce the expression of fibrosis-associated genes, including
COL1, α-SMA, β-platelet derived growth factor receptor and matrix
metalloproteinase-2 (7).
Therefore, selective suppression of autophagic activity may be a
promising therapeutic strategy for the prevention of early fibrosis
in liver cirrhosis.
α-lipoic acid (ALA) is a naturally occurring thiol
antioxidant that protects against acetaminophen-induced liver
damage (9), high-fat diet
induced-fatty liver (10),
concanavalin A-induced hepatitis (11), lipopolysaccharide induced-acute
liver injury (12) and liver
cirrhosis (13). ALA was able to
regulate autophagic activity in certain cases (14,15).
However, whether ALA may protect the liver through the regulation
of autophagy remains unclear. In the present study, a carbon
tetrachloride (CCl4)-induced liver cirrhosis rat model
was established and the mechanism of the hepatoprotective effect of
ALA was investigated.
Materials and methods
Animals and experimental design
A total of 30 Sprague-Dawley rats (male; 6 week old;
weight, 180–200 g) obtained from the Liaoning Chang Sheng
Biotechnology Co., Ltd. (Benxi, China) were used in the experiment.
After adapting for 1 week under standard animal laboratory
conditions (25±2°C; air humidity 50±10%; 12-h light/dark cycle)
with free access to food and water, the rats were randomly divided
into five groups (6/group): Control group, 100 mg/kg ALA group,
CCl4-treated group, CCl4+50 mg/kg ALA group
and CCl4+100 mg/kg ALA group. The animal raising and
handling procedures were performed in accordance with the Guide for
the Care and Use of Laboratory Animals (16) and approved by the Animal
Experimental Ethics Committee of the Henan University of Chinese
Medicine (Zhengzhou, China).
CCl4-induced liver
cirrhosis model and treatments
A CCl4-induced liver cirrhosis model was
established as previously described (17,18).
Rats were intraperitoneally injected with 50% CCl4
(purchased from Shanghai Aladdin Bio-Chem Technology Co. Ltd.,
Shanghai, China; 1:1 diluted with olive oil) three times a week for
8 consecutive weeks. In addition to treatment with CCl4,
rats in the CCl4+50 mg/kg ALA or CCl4+100
mg/kg ALA groups received 50 or 100 mg/kg ALA (Sangon Biotech Co.
Ltd., Shanghai, China) orally every day for 8 weeks. Rats in the
control group were intraperitoneally injected with an equal volume
of olive oil instead of CCl4 3 times a week and received
0.5% sodium carboxymethylcellulose (the solvent of ALA) 0.5 ml
orally every day. Rats in the 100 mg/kg ALA group were
intraperitoneally injected with an equal volume of olive oil
instead of CCl4 three times a week and received 100
mg/kg ALA orally every day for 8 weeks. At the end of treatment,
rats were anesthetized with isoflurane (2.5–3%), subsequently,
blood was collected from the orbital sinus as Parasuraman et
al (19) described previously.
The rats were sacrificed with 200 mg/kg pentobarbital sodium by
intraperitoneal injection and the liver tissues were harvested.
Histopathological and
immunohistochemical analysis
Liver tissues were fixed in 4% paraformaldehyde for
48 h at 4°C, and were subsequently embedded in paraffin and cut
into 5-µm thick sections. Following washing with xylene and
hydrating in graded ethanol, the sections were stained with
hematoxylin and eosin (H&E) or Masson's trichrome, according to
standard procedures (20). For
immunohistochemical analysis, the deparaffinized liver sections
were incubated with rabbit anti-α-smooth muscle actin (α-SMA)
antibody or rabbit anti-transforming growth factor (TGF)-β antibody
(both 1:50; α-SMA, cat. no. 55135-1-AP; TGF-β, cat. no. 21898-1-AP;
Wuhan Sanying Biotechnology, Wuhan, China) at 4°C overnight.
Subsequently, the specific proteins were detected with biotinylated
goat anti-rabbit immunoglobulin G antibody (1:200; cat. no. A0208;
Beyotime Institute of Biotechnology, Haimen, China) at 37°C for 30
min. The reactions were finally analyzed with horseradish
peroxidase-conjugated streptavidin (Beyotime Institute of
Biotechnology). Following visualization using a diaminobenzidine
substrate kit (Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China), the sections were imaged under a light microscope
(BX53; Olympus Corporation, Tokyo, Japan; magnification,
×100-200).
Biochemical measurement
Serum was isolated from blood samples by
centrifugation at 1,100 × g for 10 min at 4°C. The levels of
alanine transaminase (ALT) and aspartate transaminase (AST) in the
serum were detected using commercial kits (ALT, cat. no. C009-2;
AST, cat. no. C010-2; Nanjing Jiancheng Bioengineering Institute,
Nanjing, China), according to the manufacturer's protocol. The
hydroxyproline level in liver tissues was measured using a
hydroxyproline assay kit (cat. no. A030-2; Nanjing Jiancheng
Bioengineering Institute), according to the manufacturer's
protocol.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from liver tissues was extracted using the
China TRIpure Total RNA Extraction kit (BioTeke Corporation,
Beijing, China). cDNA was synthesized using Super M-MLV Reverse
Transcriptase (BioTeke Corporation) according to the manufacturer's
protocol and the temperature protocol was: 25°C for 10 min, 42°C
for 50 min and 80°C for 10 min. The primer sequences used are
presented in Table I. RT-qPCR
reactions were performed on an Exicycler™ 96 (Bioneer
Corporation, Daejeon, Korea) with the 2X Power Taq PCR Master Mix
(BioTeke Corporation) and SYBR Green (Beijing Solarbio Science
& Technology Co., Ltd.), according to the manufacturer's
protocol. The thermocycling conditions were 94°C for 5 min, 94°C
for 10 sec, 60°C for 20 sec, 72°C for 30 sec, 40 cycles; 72°C for
2.5 min; 40°C for 1.5 min; melting at 60–94°C, every 1°C per
second; incubation at 25°C for 2 min. The relative expression
levels were calculated using the 2−ΔΔCq method (21).
 | Table I.Primer sequences used in the present
study. |
Table I.
Primer sequences used in the present
study.
| Gene | Sequences
(5′-3′) |
|---|
| COL1A1 | Forward:
AGAGGCATAAAGGGTCATCGTG |
|
| Reverse:
CAGGAGAACCAGCAGAGCCA |
| TGF-β | Forward:
CAACAATTCCTGGCGTTACCT |
|
| Reverse:
AGCCCTGTATTCCGTCTCCTT |
| Beclin-1 | Forward:
CAGCAGTTCAAAGAAGAGGTG |
|
| Reverse:
GAGGACACCCAAGCAAGAC |
| p62 | Forward:
AGCGGGTACTGATCCCTGTC |
|
| Reverse:
TCTTCCTCCTTGGCTTTGTC |
| β-actin | Forward:
GGAGATTACTGCCCTGGCTCCTAGC |
|
| Reverse:
GGCCGGACTCATCGTACTCCTGCTT |
Western blot analysis
Total protein was isolated from liver tissues using
radioimmunoprecipitation assay solution (Beyotime Institute of
Biotechnology). Protein quantitation was conducted using the
enhanced Bicinchoninic Acid Protein Assay kit (Beyotime Institute
of Biotechnology). Subsequently, 40 µg protein was separated by
SDS-PAGE on 10–15% gels and electrotransferred onto polyvinylidene
difluoride membranes (EMD Millipore, Bedford, MA, USA). Subsequent
to blocking with 5% nonfat milk at room temperature for 1 h, the
membranes were incubated with specific primary antibodies against
α-SMA (1:1,000; cat. no. 55135-1-AP; Wuhan Sanying Biotechnology),
TGF-β (1:500; cat. no. 21898-1-AP; Wuhan Sanying Biotechnology),
phosphorylated (p)-Smad3 (1:1,000; cat. no. 9520; Cell Signaling
Technology, Inc., Danvers, MA, USA), Smad3 (1:1,000; cat. no. 9523;
Cell Signaling Technology, Inc.,), Beclin-1 (1:1,500; cat. no.
11306-1-AP; Wuhan Sanying Biotechnology), light chain (LC)3II/LC3I
(1:1,000; cat. no. 2775; Cell Signaling Technology, Inc.), p62
(1:1,000; cat. no. 39749; Cell Signaling Technology, Inc.), protein
kinase B (AKT; 1:2,000; cat. no. 9272; Cell Signaling Technology,
Inc.), p-AKT (1:1,000; cat. no. 4060; Cell Signaling Technology,
Inc.), mammalian target of rapamycin (mTOR; 1:1,000; cat. no. 2972;
Cell Signaling Technology, Inc.), p-mTOR (1:1,000; cat. no. 2971;
Cell Signaling Technology, Inc.) or β-actin (1:500; cat. no.
bsm-33139M; BIOSS, Beijing, China) at 4°C overnight. The membranes
were subsequently incubated with horseradish peroxidase
(HRP)-conjugated goat anti-rabbit or goat anti-mouse secondary
antibody (1:5,000; HRP-conjugated goat anti-rabbit antibody, cat.
no. A0208; HRP-conjugated goat anti-mouse antibody, cat. no. A0216;
Beyotime Institute of Biotechnology) at 37°C for 45 min. Finally,
the specific proteins were detected using the Beyo Enhanced
Chemiluminescent kit (Beyotime Institute of Biotechnology). The
relative protein expression level was detected by densitometry
using Gel-Pro Analyzer Version 3.0 (Media Cybernetics, Inc.,
Rockville, MD, USA).
Statistical analysis
Data are presented as the mean ± standard deviation
of six independent experiments. Differences between groups were
analyzed using one-way analysis of variance, followed by the
Bonferroni post hoc test using Image-pro plus 6.0 software (Media
Cybernetics, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
ALA alleviates CCl4-induced
liver injury
Histopathological alterations in the liver following
treatment with CCl4 and different doses of ALA were
examined by H&E staining (Fig.
1). The livers of healthy and 100 mg/kg ALA-treated rats
exhibited normal liver architecture. Treatment with CCl4
resulted in visible lesions with the formation of fibrotic septa,
the congestion of cytoplasmic vacuolation and hepatocyte necrosis.
However, these histopathological alterations induced by
CCl4 were alleviated in the liver of 50- or 100
mg/ml-ALA treated rats. To further examine liver function, the
activity of two key enzymes, ALT and AST, in the serum were
determined. As presented in Fig. 1B
and C, the activity of ALT and AST in CCl4-treated
rats were significantly increased 2.58- and 3.83-fold,
respectively, compared with the control rats (P<0.01). By
contrast, the activity of ALT and AST was reduced in a
dose-dependent manner following treatment with ALA. These results
suggested that ALA attenuated CCl4-induced liver
injury.
ALA attenuates CCl4-induced
hepatic fibrosis
The collagen deposition of livers was determined
using Masson's trichrome staining (Fig. 2A). Liver tissues from normal rats
and 100 mg/kg ALA-treated rats demonstrated little collagen
deposition, whereas those from CCl4-treated rats
demonstrated dense bundles of collagen fibers around lobules with
disordered liver structure. The liver of CCl4-exposed
rats treated with 50 or 100 mg/ml demonstrated decreased collagen
deposition. Furthermore, the marker of collagen deposition,
hydroxyproline, was additionally significantly increased in the
liver of CCl4-treated rats and decreased in ALA-treated
liver-cirrhosis-rats (P<0.01; Fig.
2B). Consistent with these alterations, the mRNA expression
level of collagen α-1(I) chain (COL1A1) was significantly
upregulated in the liver of rats exposed to CCl4 and
significantly reduced following treatment with 50 or 100 mg/ml ALA
compared with the CCl4 group (P<0.01; Fig. 2C). These data indicated that
CCl4-induced severe fibrosis was improved by treatment
with ALA.
ALA suppresses CCl4-induced
activation of HSCs
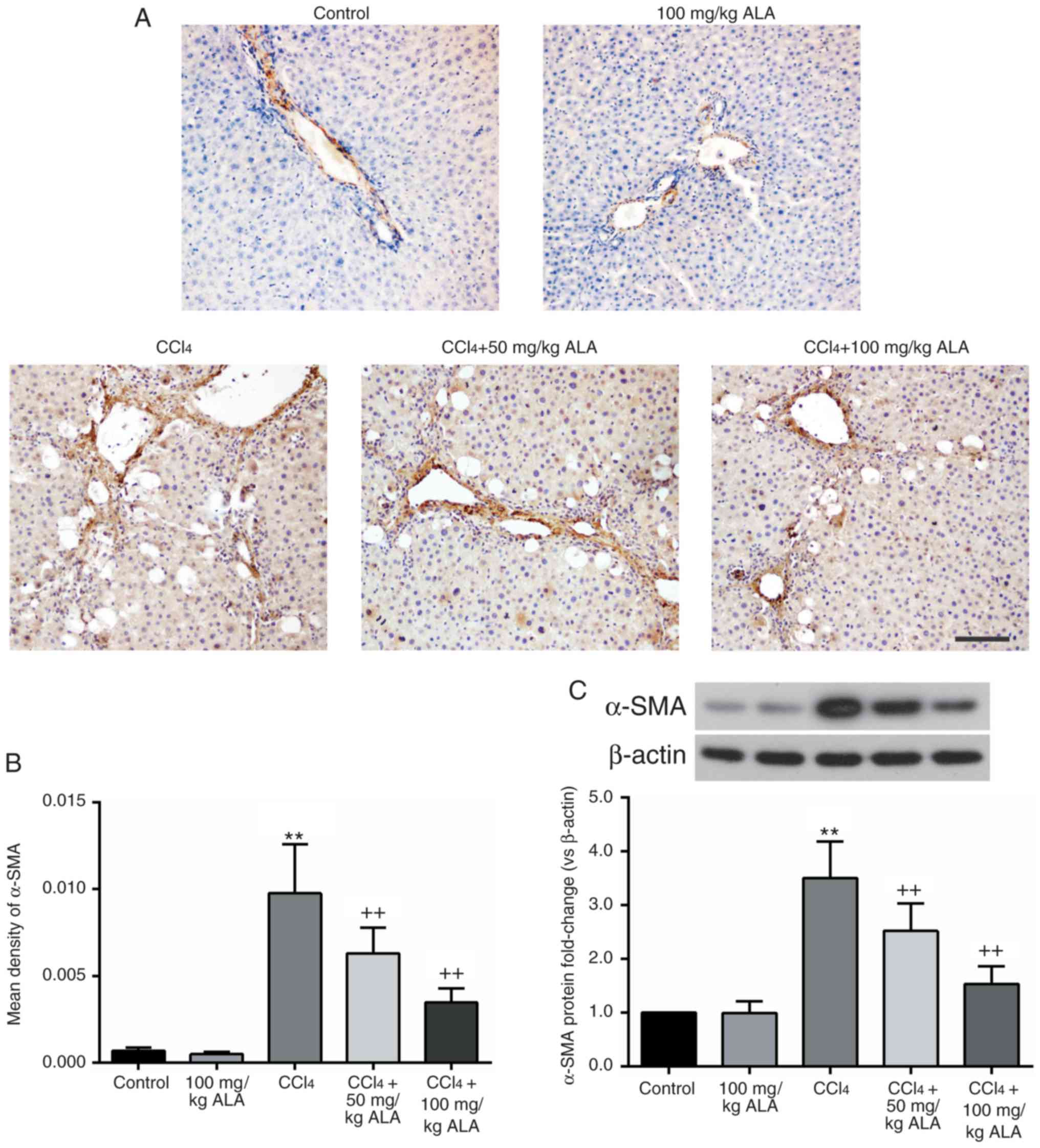
The marker of activated HSCs, α-SMA, was detected
for the purpose of evaluating the effect of ALA on HSCs.
Immunohistochemical staining results demonstrated that the
expression level of α-SMA was significantly increased in the liver
of CCl4-treated rats and was significantly reduced
following treatment with ALA (P<0.01; Fig. 3A and B). These alterations in the
α-SMA level were additionally confirmed by western blot analysis
(Fig. 3C). Therefore, the
administration of ALA was demonstrated to result in a decrease in
the degree of HSC activation in the liver of
CCl4-treated rats.
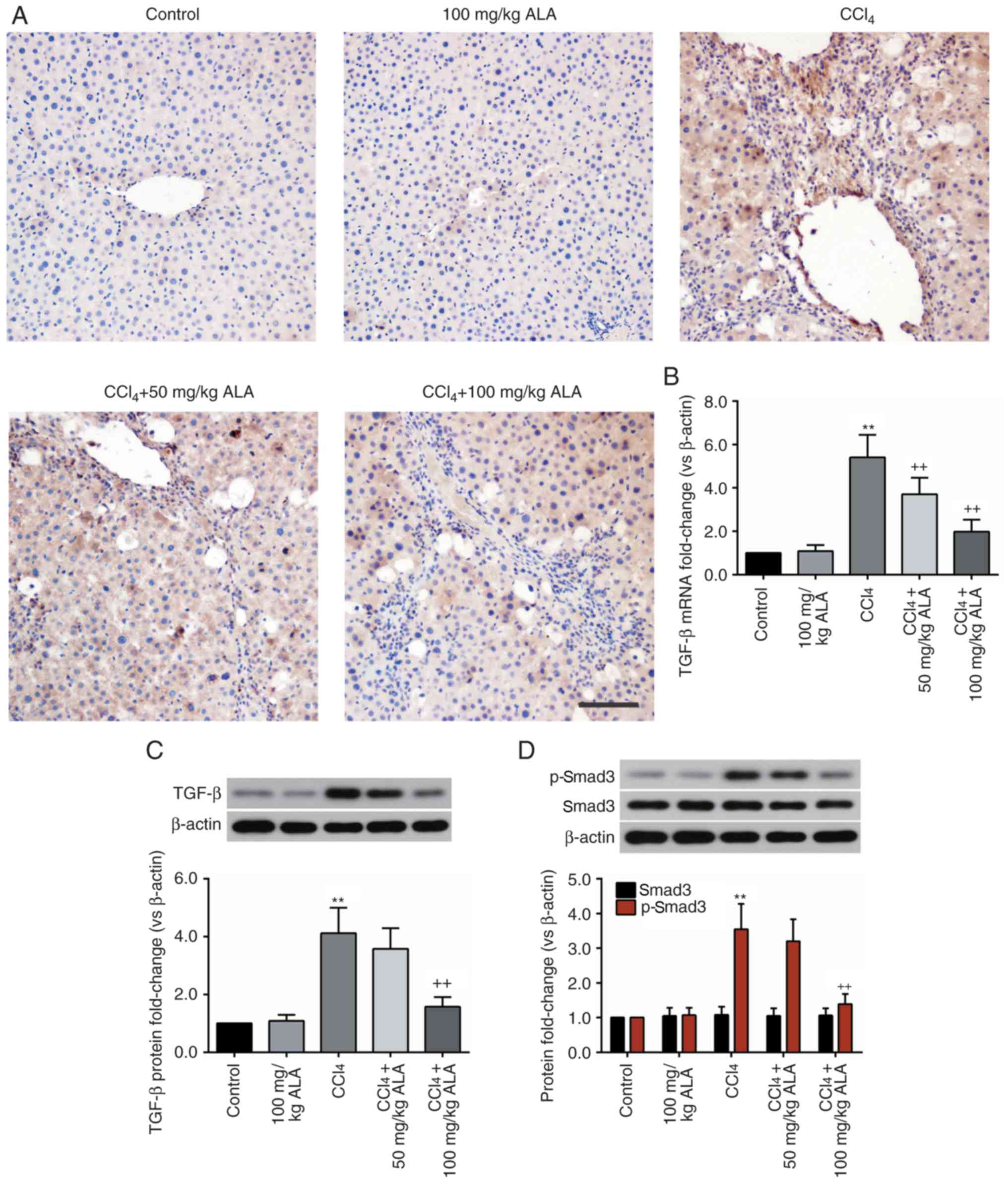
ALA inhibits CCl4-induced
activation of the TGF-β/Smad3 pathway
To investigate the mechanism behind the
hepatoprotective effect of ALA, the activation of the TGF-β/Smad3
pathway, an important mediator of liver fibrosis, was detected
(22). As presented in Fig. 4A, immunohistochemical analysis
demonstrated an increased TGF-β level in the liver tissue of
CCl4-treated rats, when compared with that in the liver
tissue of the control rats, while the elevation of TGF-β was
decreased by ALA. These alterations in the TGF-β levels were
additionally confirmed by RT-qPCR and western blot analysis
(Fig. 4B and C). Consistent with
the results of TGF-β, the protein level of p-Smad3 in the liver was
significantly elevated by CCl4 (P<0.01) and decreased
by ALA. However, the level of Smad3 remained unaltered (Fig. 4D). These results demonstrated that
ALA inhibited the activation of the TGF-β/Smad3 pathway in the
liver of CCl4-treated rats.
ALA suppresses CCl4-induced
autophagy in the liver
Autophagy has been implicated in
CCl4-induced liver cirrhosis (18) and thus, the expression of
autophagy-associated factors was examined in the present study.
RT-qPCR and western blot analysis demonstrated that the mammalian
autophagy protein, Beclin-1, was significantly increased in the
liver of CCl4-treated rats (P<0.01), and was
decreased by ALA in a dose-dependent manner (Fig. 5A). In addition, the marker of
autophagosome formation, LC3II, which was significantly upregulated
following treatment with CCl4, was additionally
significantly reduced following treatment with ALA (P<0.01;
Fig. 5B). Furthermore, the
expression of the autophagy substrate p62 was significantly
decreased following CCl4 treatment (P<0.01) and
increased following the administration of ALA, in a dose-dependent
manner (Fig. 5C). In combination,
these results suggested that treatment with ALA reversed
CCl4-induced hepatocyte autophagy.
ALA activates the AKT/mTOR pathway in
the liver of CCl4-treated rats
The upstream signaling pathway of autophagy, the
AKT/mTOR pathway, was further examined (23). As expected, the p-AKT and p-mTOR
expression levels were significantly reduced in the liver of
CCl4-treated rats (P<0.01), and alterations were
prominently restored by the administration of ALA (Fig. 6). These results indicated that ALA
was able to activate the AKT/mTOR pathway in the liver, which had
previously been repressed by treatment with CCl4.
Discussion
The results of the present study demonstrated that
treatment with ALA markedly alleviated CCl4-induced
liver cirrhosis, which became evident through positive
histopathological alterations, reduced liver fibrosis and decreased
ALT, AST and HSCs activity. Furthermore, it was demonstrated that
ALA inhibited the activation of the TGF-β/Smad3 pathway in the
liver of CCl4-treated rats. CCl4-induced
hepatocellular autophagy was suppressed by ALA. The administration
of ALA additionally promoted the activation of the AKT/mTOR pathway
in the liver of CCl4-treated rats. These results
suggested that the inhibition of autophagy through the regulation
of the AKT/mTOR pathway and the repression of the TGF-β/Smad3
pathway may be a mechanism potentially responsible for the
protective effect of ALA against CCl4-induced liver
cirrhosis.
Fibrosis is the final stage of chronic liver injury,
characterized by excessive extracellular matrix (ECM) deposition,
particularly that of the fibrous protein collagen I. The excessive
ECM may destroy normal liver architecture and cause hepatic
dysfunction (24). TGF-β is the
most powerful cytokine triggering fibrosis-associated signaling in
the liver. TGF-β initiates intracellular signaling through the
phosphorylation of Smad2/3, which serve as transcription factors
and mediate the transcription of COL1A1 (25). Previous studies identified
abnormally elevated TGF-β and p-Smad3 expression levels in chronic
fibrotic liver disease (26–28).
Accordingly, interfering with the TGF-β/Smad3 pathway results in
reduced collagen I and ECM production in livers that have been
exposed to CCl4 (27,29,30).
Min et al (31) identified
that ALA disrupted the TGF-β/Smad3 pathway in the bile duct
ligation-induced hepatic fibrosis mouse model. ALA additionally
decreased the TGF-β expression level in the liver that had been
exposed to thioacetamide (32).
Similar results were obtained in the present study; the
administration of ALA suppressed the TGF-β/Smad3 pathway in the
liver of CCl4-treated rats. In addition,
CCl4-induced collagen deposition and pathological liver
injuries were improved by ALA. These results indicated that ALA was
able to protect the liver against cirrhosis through the regulation
of the TGF-β/Smad3 pathway.
Insulin plus ALA reduced the mRNA expression level
of TGF-β in peripheral blood mononuclear cells of patients with
type 1 diabetes, compared with the patients in the insulin
treatment group (33).
Administration of ALA attenuates glomerular injury in diabetes
mellitus by the reduction of nephrogenous TGF-β (34,35).
Although the inhibitory effects of ALA on TGF-β expression have
been widely reported, the precise mechanisms by which ALA regulate
the TGF-β signaling are still not fully understood. Min et
al (31) demonstrated that ALA
was able to suppress the activity of Smad3 and reduce the binding
activity of AP-1 and Sp1, which are downstream mediators of TGF-β.
Therefore, it was hypothesized that ALA may decrease the TGF-β
expression level through negative feedback regulation. Considering
that ALA additionally reduced the mRNA expression level of TGF-β
(32), the modulation appears to
occur on the transcriptional level.
Under normal circumstances, there is a basal level
of autophagy in approximately all cell types; however, when cells
were threatened by cellar stress (including cytotoxins and
hypoxia), autophagy was rapidly enhanced to provide intracellular
nutrients and energy to maintain cell survival (36,37).
In the liver, intensive autophagy may drive the activation of HSCs
(6,7), which serve a critical role in liver
fibrosis. Blocking autophagy was able to inhibit HSC activation and
fibrogenesis in cultured cells. Furthermore, the knockout of the
HSC-specific Atg7 attenuated CCl4/thioacetamide-induced
liver fibrosis in mice (6). When
autophagy is activated, LC3 transforms from soluble LC3 I to
lipid-bound LC3 II, indicating the formation of autophagosomes
(38). Beclin-1 is another
autophagy marker involved in the formation and maturation of
autophagosomes. p62 is a scaffolding protein that may be degraded
by autophagy. In the present study, treatment with ALA suppressed
CCl4-induced autophagy flow, as demonstrated by the
decreased Beclin-1 and LC3II/LC3I and increased p62 expression.
Therefore, the hepatoprotective effect of ALA appears to be
associated with the inhibition of autophagy. The
autophagy-inhibitory effect of ALA was additionally reported in
3T3-L1 pre-adipocytes (14),
Parkinson's cellular models (39)
and hypoxia/reoxygenation-treated H9c2 cells (15).
mTOR, which consists of the two complexes mTORC1 and
mTORC2, is known to be a principal negative regulator of autophagy
(40). mTORC1 is sensitive to
rapamycin and serves as a pivotal checkpoint in modulating the
balance between cell growth and autophagy. The PI3K/AKT pathway,
which is involved in cell survival, is an upstream modulator of
mTORC1 (41). Morales-Ruiz et
al (42) suggested that the
activation of the PI3K/AKT pathway was disturbed in the livers that
had been exposed to CCl4. In accordance with previous
results, it was demonstrated in the present study that the p-AKT
expression level in the liver of CCl4-treated rats was
reduced compared with the control rats. It was previously
demonstrated that ALA was able to protect nerve cells from
H2O2-induced cell death through the
activation of the PI3K/AKT/mTOR pathway (43). Pre- or post-treatment with ALA was
demonstrated to upregulate p-AKT and p-mTOR in
ischemia/reperfusion-injured cerebral endothelial cells (44). Herein, treatment with ALA reversed
the CCl4-induced inhibition of the AKT/mTOR pathway in
the liver, suggesting that ALA may suppress autophagy through the
regulation of the AKT/mTOR pathway.
Liver cirrhosis is frequently accompanied by
enhanced oxidative stress that is demonstrated by the increase of
lipid peroxidation marker, malondialdehyde (MDA) and the decrease
of the most studied antioxidant, glutathione (GSH) (45,46).
Previously, accumulating evidence suggested that reactive oxygen
species (ROS) are the primary intracellular signal transducer in
inducing and sustaining autophagy (47–49).
Furthermore, ROS was able to inhibit the activation of the AKT/mTOR
pathway in vascular smooth muscle cells (50) and human hepatoma cells (51). ALA was originally used as an
antioxidant, treatment with ALA significantly restored GSH and
antioxidant capacity levels in liver fibrosis rat models (52). Supplementation with ALA
additionally reduced MDA level, total oxidant status and lipid
peroxides concentration in liver homogenates from high-fat diet
rats (53). Therefore, as Rudich
et al (54) suggested, ALA
may activate the AKT/mTOR pathway and inhibit autophagy by the
suppression of oxidative stress.
In conclusion, the present study demonstrated that
ALA effectively attenuates liver cirrhosis in
CCl4-poisoned rats. Additionally, ALA administration
suppressed CCl4-induced autophagy in the liver.
CCl4-induced dysregulation of the TGF-β/Smad3 and
AKT/mTOR pathways was restored by ALA. These data suggested that
ALA may alleviate liver cirrhosis through the inhibition of the
TGF-β/Smad3 pathway and autophagy.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81573933).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GL designed the present study, performed the animal
experiments, analyzed the data and wrote the manuscript. JL and LP
performed the animal experiments, the physiological test and the
pathological experiments. SG analyzed the data, organized the
images and improved the language. BL performed the molecular and
protein detection. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The animal raising and handling procedures were
performed in accordance with the Guide for the Care and Use of
Laboratory Animals and approved by the Animal Experimental Ethics
Committee of the Henan University of Chinese Medicine (Zhengzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murray CJ, Vos T, Lozano R, Naghavi M,
Flaxman AD, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S,
et al: Disability-adjusted life years (DALYs) for 291 diseases and
injuries in 21 regions, 1990–2010: A systematic analysis for the
Global Burden of Disease Study 2010. Lancet. 380:2197–2223. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lozano R, Naghavi M, Foreman K, Lim S,
Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et
al: Global and regional mortality from 235 causes of death for 20
age groups in 1990 and 2010: A systematic analysis for the Global
Burden of Disease Study 2010. Lancet. 380:2095–2128. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cecconi F and Levine B: The role of
autophagy in mammalian development: Cell makeover rather than cell
death. Dev Cell. 15:344–357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thoen LF, Guimarães EL, Dollé L, Mannaerts
I, Najimi M, Sokal E and van Grunsven LA: A role for autophagy
during hepatic stellate cell activation. J Hepatol. 55:1353–1360.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hernández-Gea V, Ghiassi-Nejad Z,
Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ and Friedman SL:
Autophagy releases lipid that promotes fibrogenesis by activated
hepatic stellate cells in mice and in human tissues.
Gastroenterology. 142:938–946. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mahmoud YI, Mahmoud AA and Nassar G:
Alpha-lipoic acid treatment of acetaminophen-induced rat liver
damage. Biotech Histochem. 90:594–600. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang Y, Li W, Liu Y, Sun Y, Li Y, Yao Q,
Li J, Zhang Q, Gao Y, Gao L and Zhao J: Alpha-lipoic acid improves
high-fat diet-induced hepatic steatosis by modulating the
transcription factors SREBP-1, FoxO1 and Nrf2 via the
SIRT1/LKB1/AMPK pathway. J Nutr Biochem. 25:1207–1217. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fei M, Xie Q, Zou Y, He R, Zhang Y, Wang
J, Bo L, Li J and Deng X: Alpha-lipoic acid protects mice against
concanavalin A-induced hepatitis by modulating cytokine secretion
and reducing reactive oxygen species generation. Int
Immunopharmacol. 35:53–60. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tanaka Y, Kaibori M, Miki H, Nakatake R,
Tokuhara K, Nishizawa M, Okumura T and Kwon AH: Alpha-lipoic acid
exerts a liver-protective effect in acute liver injury rats. J Surg
Res. 193:675–683. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Morsy MA, Abdalla AM, Mahmoud AM,
Abdelwahab SA and Mahmoud ME: Protective effects of curcumin,
α-lipoic acid, and N-acetylcysteine against carbon
tetrachloride-induced liver fibrosis in rats. J Physiol Biochem.
68:29–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hahm JR, Noh HS, Ha JH, Roh GS and Kim DR:
Alpha-lipoic acid attenuates adipocyte differentiation and lipid
accumulation in 3T3-L1 cells via AMPK-dependent autophagy. Life
Sci. 100:125–132. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cao X, Chen A, Yang P, Song X, Liu Y, Li
Z, Wang X, Wang L and Li Y: Alpha-lipoic acid protects
cardiomyocytes against hypoxia/reoxygenation injury by inhibiting
autophagy. Biochem Biophys Res Commun. 441:935–940. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
National Research Council (U.S.).
Committee for the Update of the Guide for the Care and Use of
Laboratory Animals., Institute for Laboratory Animal Research
(U.S.) and National Academies Press (U.S.): Guide for the Care and
Use of Laboratory Animals. National Academies Press; Washington,
DC: 2011
|
|
17
|
Chen Q, Zhang H, Cao Y, Li Y, Sun S, Zhang
J and Zhang G: Schisandrin B attenuates CCl4-induced liver fibrosis
in rats by regulation of Nrf2-ARE and TGF-β/Smad signaling
pathways. Drug Des Devel Ther. 11:2179–2191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hung TM, Yuan RH, Huang WP, Chen YH, Lin
YC, Lin CW, Lai HS and Lee PH: Increased autophagy markers are
associated with ductular reaction during the development of
cirrhosis. Am J Pathol. 185:2454–2467. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Parasuraman S, Raveendran R and Kesavan R:
Blood sample collection in small laboratory animals. J Pharmacol
Pharmacother. 1:87–93. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang Y, Wang S, Zhao Y, Lin C, Zhong F,
Jin L, He F and Wang H: Histone H3K9 demethylase JMJD1A modulates
hepatic stellate cells activation and liver fibrosis by
epigenetically regulating peroxisome proliferator-activated
receptor γ. FASEB J. 29:1830–1841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gressner AM, Weiskirchen R, Breitkopf K
and Dooley S: Roles of TGF-beta in hepatic fibrosis. Front Biosci.
7:d793–d807. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dazert E and Hall MN: mTOR signaling in
disease. Curr Opin Cell Biol. 23:744–755. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu F, Liu C, Zhou D and Zhang L:
TGF-β/SMAD pathway and its regulation in hepatic fibrosis. J
Histochem Cytochem. 64:157–167. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Breitkopf K, Godoy P, Ciuclan L, Singer MV
and Dooley S: TGF-beta/Smad signaling in the injured liver. Z
Gastroenterol. 44:57–66. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Inagaki Y, Mamura M, Kanamaru Y, Greenwel
P, Nemoto T, Takehara K, Ten Dijke P and Nakao A: Constitutive
phosphorylation and nuclear localization of Smad3 are correlated
with increased collagen gene transcription in activated hepatic
stellate cells. J Cell Physiol. 187:117–123. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pathil A, Mueller J, Ludwig JM, Wang J,
Warth A, Chamulitrat W and Stremmel W: Ursodeoxycholyl
lysophosphatidylethanolamide attenuates hepatofibrogenesis by
impairment of TGF-β1/Smad2/3 signalling. Br J Pharmacol.
171:5113–5126. 2014.PubMed/NCBI
|
|
28
|
Perumal N, Perumal M, Halagowder D and
Sivasithamparam N: Morin attenuates diethylnitrosamine-induced rat
liver fibrosis and hepatic stellate cell activation by co-ordinated
regulation of Hippo/Yap and TGF-β1/Smad signaling. Biochimie.
140:10–19. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ganai AA and Husain M: Genistein
attenuates D-GalN induced liver fibrosis/chronic liver damage in
rats by blocking the TGF-β/Smad signaling pathways. Chem Biol
Interact. 261:80–85. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Iordanskaia T, Hubal MJ, Koeck E, Rossi C,
Schwarz K and Nadler EP: Dysregulation of upstream and downstream
transforming growth factor-β transcripts in livers of children with
biliary atresia and fibrogenic gene signatures. J Pediatr Surg.
48:2047–2053. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Min AK, Kim MK, Seo HY, Kim HS, Jang BK,
Hwang JS, Choi HS, Lee KU, Park KG and Lee IK: Alpha-lipoic acid
inhibits hepatic PAI-1 expression and fibrosis by inhibiting the
TGF-beta signaling pathway. Biochem Biophys Res Commun.
393:536–541. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Foo NP, Lin SH, Lee YH, Wu MJ and Wang YJ:
α-lipoic acid inhibits liver fibrosis through the attenuation of
ROS-triggered signaling in hepatic stellate cells activated by PDGF
and TGF-β. Toxicology. 282:39–46. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hegazy SK, Tolba OA, Mostafa TM, Eid MA
and El-Afify DR: Alpha-lipoic acid improves subclinical left
ventricular dysfunction in asymptomatic patients with type 1
diabetes. Rev Diabet Stud. 10:58–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Melhem MF, Craven PA, Liachenko J and
DeRubertis FR: Alpha-lipoic acid attenuates hyperglycemia and
prevents glomerular mesangial matrix expansion in diabetes. J Am
Soc Nephrol. 13:108–116. 2002.PubMed/NCBI
|
|
35
|
Melhem MF, Craven PA and Derubertis FR:
Effects of dietary supplementation of alpha-lipoic acid on early
glomerular injury in diabetes mellitus. J Am Soc Nephrol.
12:124–133. 2001.PubMed/NCBI
|
|
36
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mehrpour M, Esclatine A, Beau I and
Codogno P: Autophagy in health and disease. 1. Regulation and
significance of autophagy: An overview. Am J Physiol Cell Physiol.
298:C776–C785. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao H, Zhao X, Liu L, Zhang H, Xuan M,
Guo Z, Wang H and Liu C: Neurochemical effects of the R form of
alpha-lipoic acid and its neuroprotective mechanism in cellular
models of Parkinson's disease. Int J Biochem Cell Biol. 87:86–94.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Heras-Sandoval D, Pérez-Rojas JM,
Hernández-Damián J and Pedraza-Chaverri J: The role of
PI3K/AKT/mTOR pathway in the modulation of autophagy and the
clearance of protein aggregates in neurodegeneration. Cell Signal.
26:2694–2701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yap TA, Garrett MD, Walton MI, Raynaud F,
de Bono JS and Workman P: Targeting the PI3K-AKT-mTOR pathway:
Progress, pitfalls, and promises. Curr Opin Pharmacol. 8:393–412.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Morales-Ruiz M, Cejudo-Martín P,
Fernández-Varo G, Tugues S, Ros J, Angeli P, Rivera F, Arroyo V,
Rodés J, Sessa WC and Jiménez W: Transduction of the liver with
activated Akt normalizes portal pressure in cirrhotic rats.
Gastroenterology. 125:522–531. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kamarudin MN, Mohd Raflee NA, Hussein SS,
Lo JY, Supriady H and Abdul Kadir H: (R)-(+)-α-lipoic acid
protected NG108-15 cells against H2O2-induced
cell death through PI3K-Akt/GSK-3β pathway and suppression of
NF-κβ-cytokines. Drug Des Devel Ther. 8:1765–1780. 2014.PubMed/NCBI
|
|
44
|
Xie R, Li X, Ling Y, Shen C, Wu X, Xu W
and Gao X: Alpha-lipoic acid pre- and post-treatments provide
protection against in vitro ischemia-reperfusion injury in cerebral
endothelial cells via Akt/mTOR signaling. Brain Res. 1482:81–90.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gutiérrez R, Alvarado JL, Presno M,
Pérez-Veyna O, Serrano CJ and Yahuaca P: Oxidative stress
modulation by Rosmarinus officinalis in CCl4-induced liver
cirrhosis. Phytother Res. 24:595–601. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Galicia-Moreno M, Rosique-Oramas D,
Medina-Avila Z, Álvarez-Torres T, Falcón D, Higuera-de la Tijera F,
Béjar YL, Cordero-Pérez P, Muñoz-Espinosa L, Pérez-Hernández JL, et
al: Behavior of oxidative stress markers in alcoholic liver
cirrhosis patients. Oxid Med Cell Longev. 2016:93705652016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Filomeni G, Desideri E, Cardaci S, Rotilio
G and Ciriolo MR: Under the ROS…thiol network is the principal
suspect for autophagy commitment. Autophagy. 6:999–1005. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Filomeni G, De Zio D and Cecconi F:
Oxidative stress and autophagy: The clash between damage and
metabolic needs. Cell Death Differ. 22:377–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Petrović A, Bogojević D, Korać A, Golić I,
Jovanović-Stojanov S, Martinović V, Ivanović-Matić S, Stevanović J,
Poznanović G and Grigorov I: Oxidative stress-dependent
contribution of HMGB1 to the interplay between apoptosis and
autophagy in diabetic rat liver. J Physiol Biochem. 73:511–521.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Peng J, He X, Zhang L and Liu P:
MicroRNA26a protects vascular smooth muscle cells against
H2O2-induced injury through activation of the
PTEN/AKT/mTOR pathway. Int J Mol Med. 42:1367–1378. 2018.PubMed/NCBI
|
|
51
|
Marin JJ, Hernandez A, Revuelta IE,
Gonzalez-Sanchez E, Gonzalez-Buitrago JM and Perez MJ:
Mitochondrial genome depletion in human liver cells abolishes bile
acid-induced apoptosis: Role of the Akt/mTOR survival pathway and
Bcl-2 family proteins. Free Radic Biol Med. 61:218–228. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Fayez AM, Zakaria S and Moustafa D: Alpha
lipoic acid exerts antioxidant effect via Nrf2/HO-1 pathway
activation and suppresses hepatic stellate cells activation induced
by methotrexate in rats. Biomed Pharmacother. 105:428–433. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zalejska-Fiolka J, Wielkoszyński T,
Rokicki W Jr, Dąbrowska N, Strzelczyk JK, Kasperczyk A, Owczarek A,
Błaszczyk U, Kasperczyk S, Stawiarska-Pięta B, et al: The influence
of α-lipoic acid and garlic administration on biomarkers of
oxidative stress and inflammation in rabbits exposed to oxidized
nutrition oils. Biomed Res Int. 2015:8278792015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rudich A, Tirosh A, Potashnik R, Khamaisi
M and Bashan N: Lipoic acid protects against oxidative stress
induced impairment in insulin stimulation of protein kinase B and
glucose transport in 3T3-L1 adipocytes. Diabetologia. 42:949–957.
1999. View Article : Google Scholar : PubMed/NCBI
|