Introduction
Primary liver cancer is one of the most common types
of malignant tumor. Hepatocellular carcinoma (HCC) is the principal
pathological type of liver cancer and is a leading cause of
cancer-associated mortalities worldwide (1). HCC is characterized by high
malignancy and is generally diagnosed at the middle to late stage.
As a result, the prognosis of patients with HCC remains poor. The
majority of patients are treated surgically; however, 60–70% of all
HCC cases relapse within five years (2). Therefore, it is critical to identify
novel molecular markers in order to improve the prognoses of the
patients. Additionally, identification of the molecular mechanisms
underlying HCC progression may help in developing novel targeted
strategies to treat HCC.
In recent years, an increasing number of studies on
the roles of microRNAs (miRNAs) and long noncoding RNAs (lncRNAs)
have been published. The regulatory network modulating miRNAs,
lncRNAs and their target genes represents a popular topic in cancer
research. miRNAs are involved in key biological processes,
including cell growth, differentiation and apoptosis, and are able
to induce mRNA degradation or to inhibit protein translation,
altering the expression levels of the downstream proteins (3,4).
lncRNAs represent a group of noncoding RNA molecules, that, in
contrast to miRNAs, inhibit or activate gene expression by
interacting with transcription factors or by binding to specific
regions of the target mRNAs, modulating their translation (5). Via various mechanisms, lncRNAs serve
important roles in numerous biological processes, including cell
differentiation, cell apoptosis and the heat shock response
(6). A previous study demonstrated
that lncRNAs are a novel class of regulators involved in multiple
human diseases by modulating gene expression at the
transcriptional, post-transcriptional or epigenetic level (7). Therefore, the regulatory association
between lncRNA, miRNA and mRNA, and tumor pathogenesis requires
further investigation.
Genetic mutations are one of the principal factors
involved in tumor occurrence and may lead to the activation of
proto-oncogenes or the loss of tumor suppressor genes. Furthermore,
accumulating evidence demonstrated that genetic modifications are
associated with tumorigenesis (8–11).
DNA methylation is a mechanism underlying epigenetic modifications,
and is able to affect the DNA conformation, the chromatin structure
and the stability of DNA, altering the interactions between DNA and
proteins regulating gene expression (12). Epigenetic modifications, including
DNA methylation and histone modifications, are used as biomarkers
to study tumorigenesis and tumor progression, helping clinical
diagnosis (11). Furthermore, the
investigation of epigenetic modifications may contribute to the
development of targeted treatments aimed to inactivate tumor
suppressor genes (13). Therefore,
examining the associations between genetic mutations and DNA
methylation, and the occurrence, development and prognosis of HCC
may improve the understanding of HCC pathogenesis and may provide
novel strategies to prevent, diagnose and treat this fatal
disease.
In the present study, a prognostic model was
constructed and the genes involved in HCC were investigated by
analyzing biological datasets from online databases. The potential
interactions among lncRNAs, miRNAs and mRNAs involved in the
development of HCC were investigated. Additionally, the association
between RNA expression levels, and the occurrence of genetic
mutations and epigenetic modifications were examined in order to
identify novel potential biomarkers.
Materials and methods
Database screening
Gene expression data from HCC, relative to 374 tumor
samples and 50 normal samples, were downloaded from the The Cancer
Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/) using the key words
‘transcriptome profiling’, ‘HTSeq-Counts’ and ‘TCGA-LIHC’. The data
were combined and the gene identification numbers were converted
using Perl (https://www.perl.org/) in order to
obtain a gene expression matrix.
Identification of differentially
expressed (DE) genes
The expression matrix of miRNAs, lncRNAs and mRNAs
was analyzed using the edgeR package (version 3.22.3; http://www.bioconductor.org/packages/release/bioc/html/edgeR.html)
of R software (version 3.4.2) (http://www.r-project.org/). The correlations between
DElncRNA-DEmiRNA pairs and DEmiRNA-DEmRNA pairs were matched using
Perl. The pairs used the Spearman's test with a coefficient
>0.95 were considered as co-expressed. The lncRNAs-miRNAs-mRNAs
regulatory interactions were identified based on the
DElncRNA-DEmiRNA and DEmiRNA-DEmRNA regulation pairs. The
regulatory associations between DElncRNAs and DEmiRNAs were
investigated using miRcode (http://www.mircode.org). The regulatory associations
between DEmiRNAs and DEmRNAs were investigated using miRDB
(http://www.mirdb.org/), miRTarBase (release 7.0)
(http://mirtarbase.mbc.nctu.edu.tw/php/index.php) and
TargetScan (release 7.2) (http://www.targetscan.org/). The negatively associated
DEmiRNA-DEmRNA and DElncRNA-DEmiRNA regulatory pairs were selected
to construct regulatory networks. The VennDiagram package (version
1.6.20; http://cran.r-project.org/web/packages/VennDiagram/index.html)
of R software was used to draw Venn diagrams.
Multivariable Cox regression analysis
of independent prognostic factors in HCC
Univariate analysis was performed for DElncRNAs,
DEmRNAs and DEmiRNAs. The DE-RNAs with P<0.001 were selected for
further multivariate Cox regression analysis. A prognostic model of
HCC was established, and Receiver Operating Characteristic (ROC)
curves were used to test the reliability of the constructed
model.
Construction of a competing endogenous
(ceRNA) regulatory network and survival analysis
The integrated co-expression network of
DElncRNA-DEmiRNA-DEmRNA regulatory associations was visualized
using Cytoscape software (version 3.6.1) (https://cytoscape.org/). Furthermore, prognostic
DEmRNAs, DElncRNAs and DEmiRNAs in the ceRNA network were
identified, and Kaplan-Meier survival plots were drawn using R
survival package (version 2.42–6) (https://cran.r-project.org/web/packages/survival/index.html).
Functional enrichment analysis
The BINGO plugin (version 3.0.3) (https://www.psb.ugent.be/cbd/papers/BiNGO/Tutorial.html)
of Cytoscape software was used for Gene Ontology (GO) analysis
(https://david.ncifcrf.gov/). KOBAS 3.0
(http://kobas.cbi.pku.edu.cn/) was used
for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
analysis.
Construction of protein-protein
interaction (PPI) networks
PPI networks were constructed using Search Tool for
the Retrieval of Interacting Genes/Proteins (https://string-db.org/). In order to model the
network, the minimum required interaction score was set to 0.4. In
analyzing the six genes associated with miRNA-424, the minimum
required interaction score was set to 0.4.
Association between genetic mutations
and DNA methylation
cBioPortal database (http://www.cbioportal.org/) was used to investigate
the association between the genetic mutations and DNA methylation
of six target genes regulated by miRNA-424 in HCC. Their
association with the overall survival of the patients with HCC was
further assessed by Kaplan-Meier analysis. P<0.05 was considered
to indicate a statistically significant difference.
Results
Identification of DElncRNAs, DEmiRNAs
and DEmRNAs associated with HCC survival
A total of 1,077 DElncRNAs, 122 DEmiRNAs and 1,985
DEmRNAs were identified by analyzing RNA-sequencing (seq) and
miRNA-seq data downloaded from TCGA, and derived from 374 HCC and
50 normal liver samples. Target prediction analysis, performed
using miRDB, miRTarBase and TargetScan, suggested that 747 of the
DEmRNAs were potential targets of lncRNAs and miRNAs. Among the 747
DEmRNAs, 35 were identified as potential targets of 77 DElncRNAs
and 16 DEmiRNAs (data not shown).
Kaplan-Meier survival analysis suggested that 13
DElncRNAs and 19 DEmRNAs were associated with the 5-year survival
rate of patients with HCC (data not shown). In contrast, no miRNAs
were associated with the 5-year survival rate.
Univariate analysis of DERNAs suggested that nine
DElncRNAs, one DEmiRNA and 14 DEmRNAs were risk factors of HCC
development (data not shown. Multivariable Cox regression analysis
suggested that 12 DERNAs were associated with HCC, whereas, long
intergenic non-protein coding RNA 200, miRNA-137, PDZ binding
kinase (PBK) and DNA polymerase θ (POLQ) were independent
prognostic factors (Table I).
These 12 DERNAs were used to construct a prognostic model, and the
overall survival rate of patients with a low risk score was
significantly increased compared with patients with a high risk
score. Furthermore, the ROC analysis performed resulted in an area
under the curve of 0.756, indicating the reliability of the model
(data not shown). A heat map of the 12 DERNAs suggested that their
expression patterns exhibited consistency among 374 tumor samples
compared with 50 normal liver samples (data not shown).
 | Table I.Multivariate Cox regression analysis
for differentially expressed RNAs. |
Table I.
Multivariate Cox regression analysis
for differentially expressed RNAs.
| Type of RNA | Gene name | Coefficient | exp(coef) | se(coef) | Z score | P-value |
|---|
| lncRNA | AC114489.1 | 0.1136 | 1.1203 | 0.0725 | 1.57 | 0.1170 |
| lncRNA | AP002478.1 | 0.0938 | 1.0984 | 0.0605 | 1.55 | 0.1210 |
| lncRNA | C10orf91 | 0.0742 | 1.0770 | 0.0482 | 1.54 | 0.1237 |
| lncRNA | LINC00200 | 0.1568 | 1.1698 | 0.0497 | 3.16 | 0.0016a |
| lncRNA | LINC00462 | 0.1377 | 1.1477 | 0.0731 | 1.88 | 0.0595 |
| miRNA | miRNA-137 | 0.1135 | 1.1202 | 0.0571 | 1.99 | 0.0467a |
| mRNA | CBX2 | 0.1408 | 1.1512 | 0.0770 | 1.83 | 0.0675 |
| mRNA | CLSPN | 0.1861 | 1.2046 | 0.1113 | 1.67 | 0.0945 |
| mRNA | E2F2 | −0.2127 | 0.8084 | 0.1295 | −1.64 | 0.1004 |
| mRNA | EZH2 | 0.3413 | 1.4068 | 0.1907 | 1.79 | 0.0734 |
| mRNA | PBK | 0.2564 | 1.2923 | 0.1205 | 2.13 | 0.0333a |
| mRNA | POLQ | −0.3420 | 0.7104 | 0.1503 | −2.28 | 0.0228a |
Construction of a ceRNA regulatory
network for HCC
The regulatory associations among DElncRNAs,
DEmiRNAs and DEmRNAs were analyzed by constructing a ceRNA network.
A total of 290 lncRNA-miRNA pairs, 16 DEmiRNAs and 35 DEmRNAs were
used to construct the ceRNA network (data not shown). The
lncRNA-miRNA-mRNA regulatory network included 128 nodes and 331
edges. Among the DEmiRNAs, only miRNA-424 was downregulated. In a
secondary regulatory network centered on miRNA-424, 13 DEmRNAs were
identified: One was downregulated and 12 were upregulated (data not
shown). Among these 13 DEmRNAs, the following six genes were
associated with the overall survival of patients with HCC: E2F
transcription factor 7 (E2F7), cytoplasmic polyadenylation element
binding protein 3 (CPEB3), claspin (CLSPN), centrosomal protein 55
(CEP55), cell division cycle 25A (CDC25A) and cyclin E1 (CCNE1;
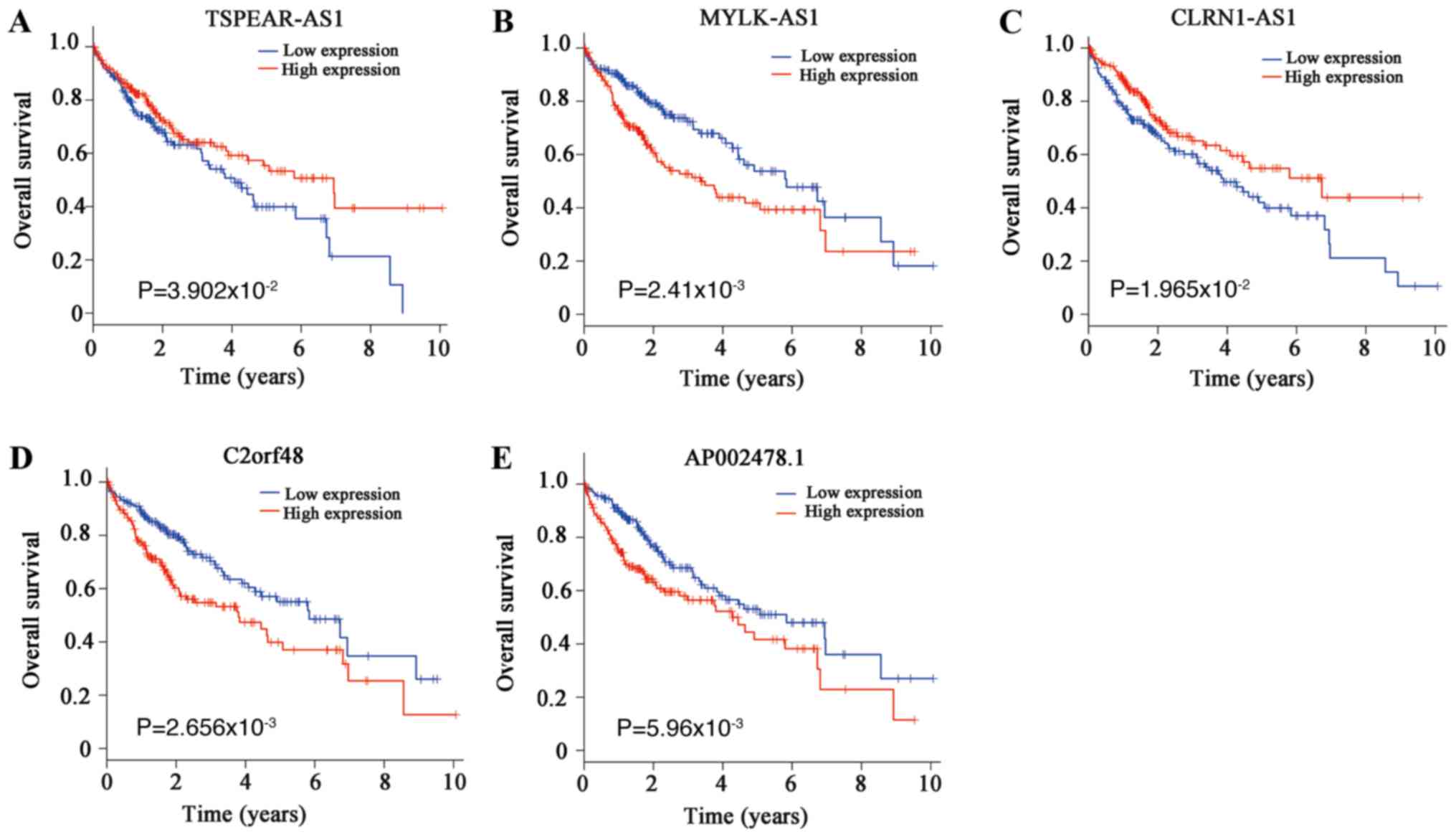
Fig. 1). Furthermore, 29 DElncRNAS
were identified, and five of them, including TSPEAR antisense RNA
1, MYLK antisense RNA 1, CLRN1 antisense RNA 1, chromosome 2 open
reading frame 48 and AP002478.1, were associated with the survival
rate of patients with HCC (Fig.
2). Therefore, the regulatory network of miRNA-424 was further
investigated in the present study.
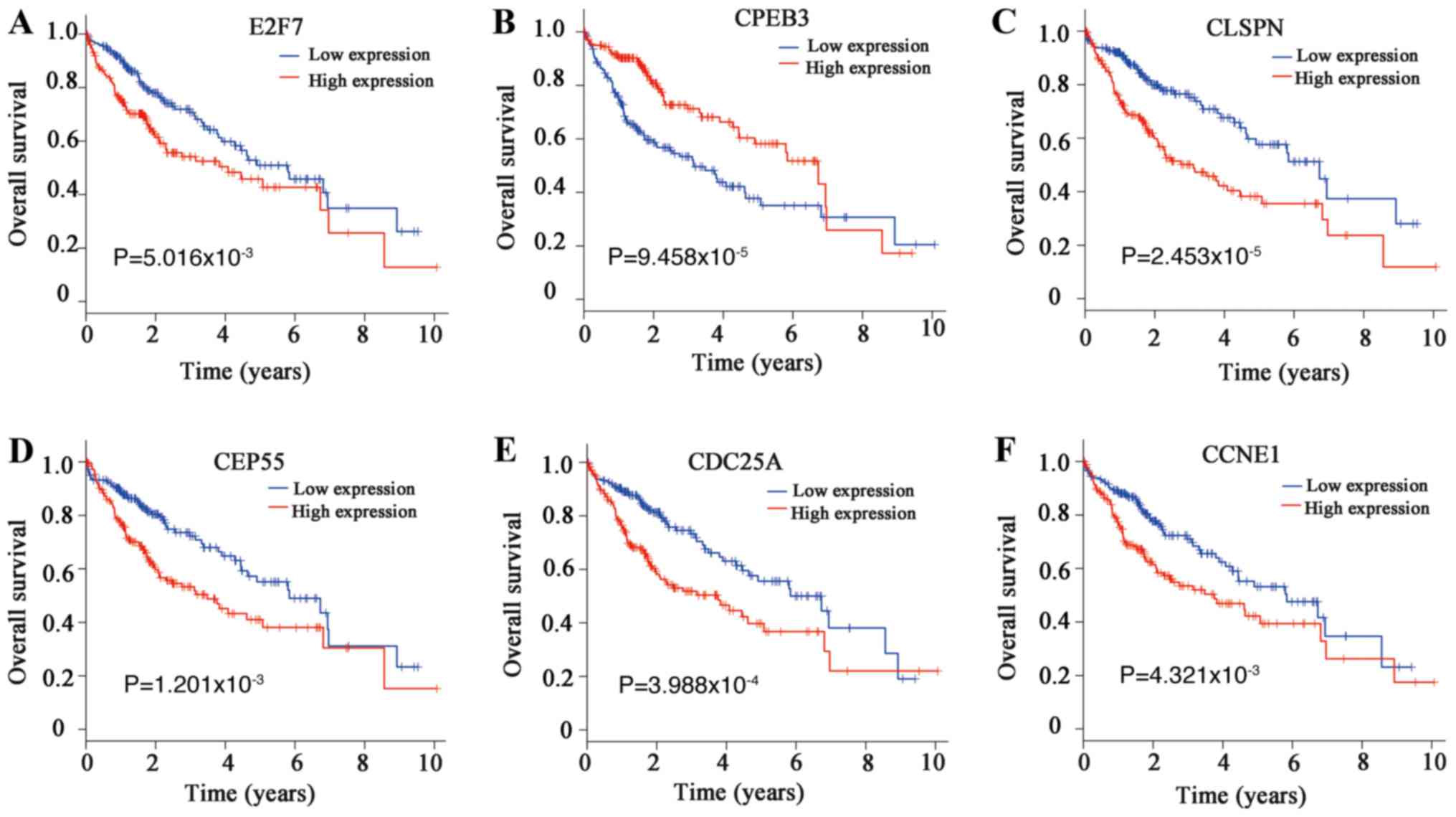 | Figure 1.Survival analysis of differentially
expressed mRNAs in a competing endogenous RNA regulatory network in
hepatocellular carcinoma. Association between the overall survival
rate and the expression levels of (A) E2F7, (B) CPEB3, (C) CLSPN,
(D) CEP55, (E) CDC25A and (F) CCNE1. CCNE1, cyclin E1; CDC25A, cell
division cycle 25A; CEP55, centrosomal protein 55; CLSPN, claspin;
CPEB3, cytoplasmic polyadenylation element binding protein 3; E2F7,
E2F transcription factor 7. |
GO and KEGG functional enrichment
analyses in HCC
Functional enrichment analysis of the mRNAs
regulated by miRNA-424 resulted in five enriched GO terms. The GO
terms are categorized into ‘biological process’, ‘molecular
function’ and ‘cellular component’. A total of 108 nodes and 166
edges constituted a functional enrichment regulatory network (data
not shown). Additionally, 13 enriched signaling pathways were
identified by KEGG analysis and the most significant enriched
pathways were ‘progesterone-mediated oocyte maturation’, ‘oocyte
meiosis’, ‘cell cycle’ and ‘microRNAs in cancer’ (Fig. 3A).
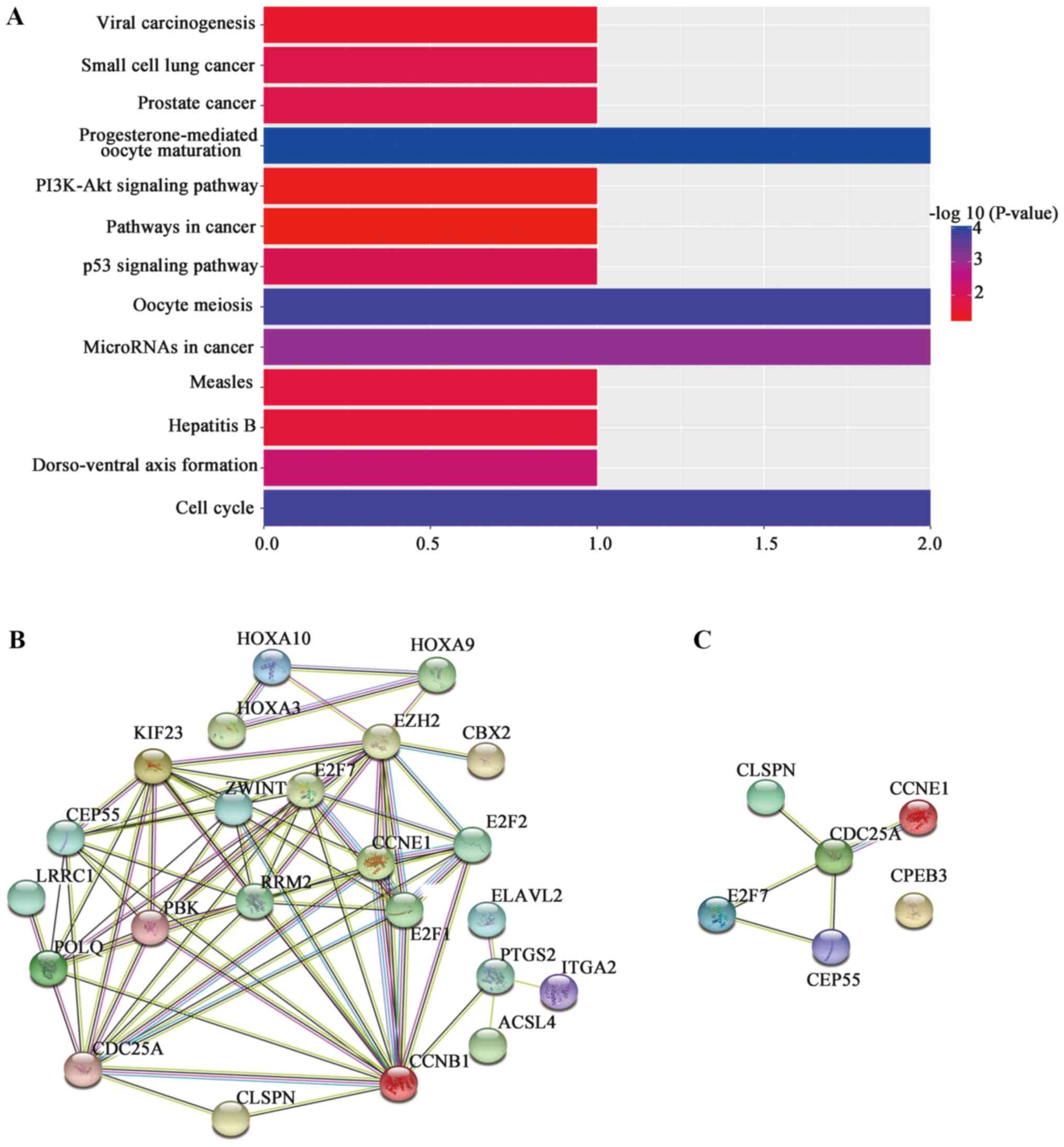 | Figure 3.KEGG and PPI analyses. (A) KEGG
analysis of genes regulated by microRNA-424. (B) PPI analysis of
genes that are differentially expressed in HCC. (C) PPI analysis of
the predicted target genes of microRNA-424 that are associated with
the survival rate of patients with HCC. The nodes represent
proteins and each color corresponds to a cluster. The edges
indicate the functional associations and are colored according to
the type of functional association. Red, green, blue, purple,
yellow, light blue and black lines indicate fusion evidence,
neighborhood evidence, co-occurrence evidence, experimental
evidence, text-mining evidence, database evidence and co-expression
evidence, respectively. The line thickness indicates the degree of
confidence for the prediction of the interaction. KEGG, Kyoto
Encyclopedia of Genes and Genomes; PPI, protein-protein
interaction; HCC, hepatocellular carcinoma. |
PPI network analysis in HCC
The functional interactions were examined at the
protein level by constructing PPI networks based on the DEmRNAs
(Fig. 3B) and the six target genes
of miRNA-424 (Fig. 3C). CDC25A
presented multiple interactions in the two networks and was the
central node in the miRNA-424-regulated network (Fig. 3C). Furthermore, CEP55, CLSPN, E2F7
and CCNE1 were associated with CDC25A, although no interaction with
CPEB3 was identified (Fig. 3C).
The interaction between CDC25A and CCNE1 presented the highest
combined score, 0.981.
Analysis of genetic mutations of
miRNA-424 target genes in HCC
The mutation rate in HCC was 5% for CDC25A, CEP55,
CCNE1 and CPEB3, and 6% for CLSPN and E2F7, and mutations in these
genes were significantly associated with vascular invasion (data
not shown). The associations among the genetic mutations of the six
genes were analyzed using the cBioPortal database. The results
suggested that mutations in CDC25A were positively associated with
mutations in CEP55, CLSPN and E2F7; however, no significant
association was identified with mutations in CCNE1 or CPEB3
(Table II). Furthermore, the
associations between the mutations of the six genes and the
survival rates of patients with HCC were assessed by Kaplan-Meier
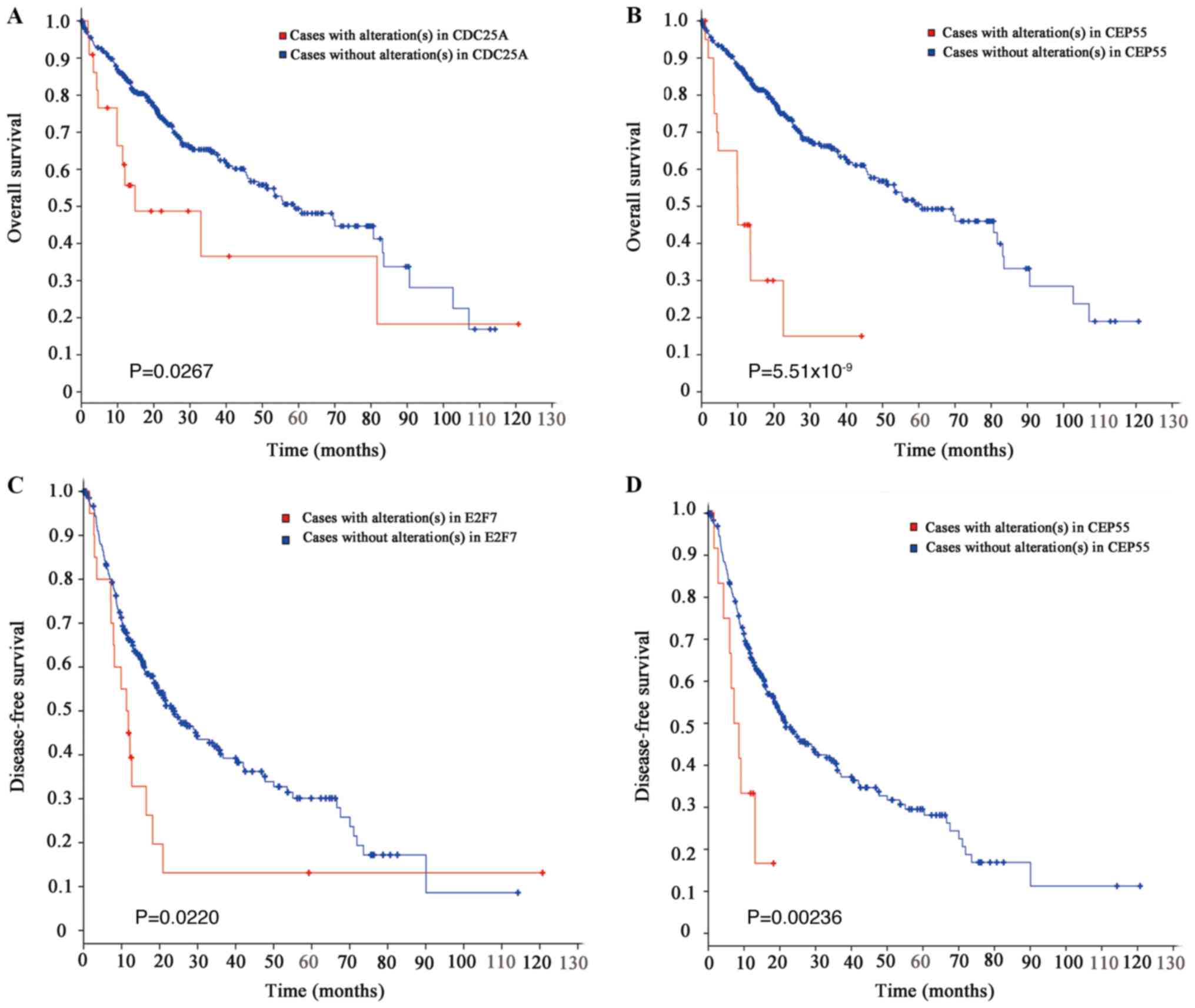
analysis (Fig. 4). Mutations in
CEP55 were associated with the overall survival and the
disease-free survival rates of patients with HCC (Fig. 4B and D). CDC25A mutations were
associated with the overall survival rate (Fig. 4A); however, they were not
significantly associated with the disease-free survival rate (data
not shown). In contrast, mutations in E2F7 were associated with the
disease-free survival (Fig. 4C)
and not with the overall survival rate (data not shown). Mutations
in CCNE1, CLSPN and CPEB3 were not significantly associated with
the survival rates (data not shown).
| Table II.Association between genetic
mutations. |
Table II.
Association between genetic
mutations.
| A, Tendency toward
co-occurrence |
|---|
|
|---|
| Gene A | Gene B | P-value |
|---|
| CLSPN | E2F7 | <0.001 |
| CLSPN | CEP55 | <0.001 |
| CDC25A | CLSPN | <0.001 |
| E2F7 | CEP55 | <0.001 |
| CDC25A | CEP55 | <0.001 |
| CDC25A | E2F7 | 0.002 |
| CDC25A | CCNE1 | 0.087 |
| CLSPN | CCNE1 | 0.115 |
| E2F7 | CCNE1 | 0.115 |
| CCNE1 | CPEB3 | 0.612 |
| CEP55 | CCNE1 | 0.631 |
|
| B, Tendency
toward mutual exclusivity |
|
| Gene A | Gene B | P-value |
|
| CLSPN | CPEB3 | 0.275 |
| CDC25A | CPEB3 | 0.319 |
| CEP55 | CPEB3 | 0.369 |
| E2F7 | CPEB3 | 0.651 |
DNA methylation affects the biological
features of HCC
The association between the levels of DNA
methylation of the miRNA-424 target genes and the biological
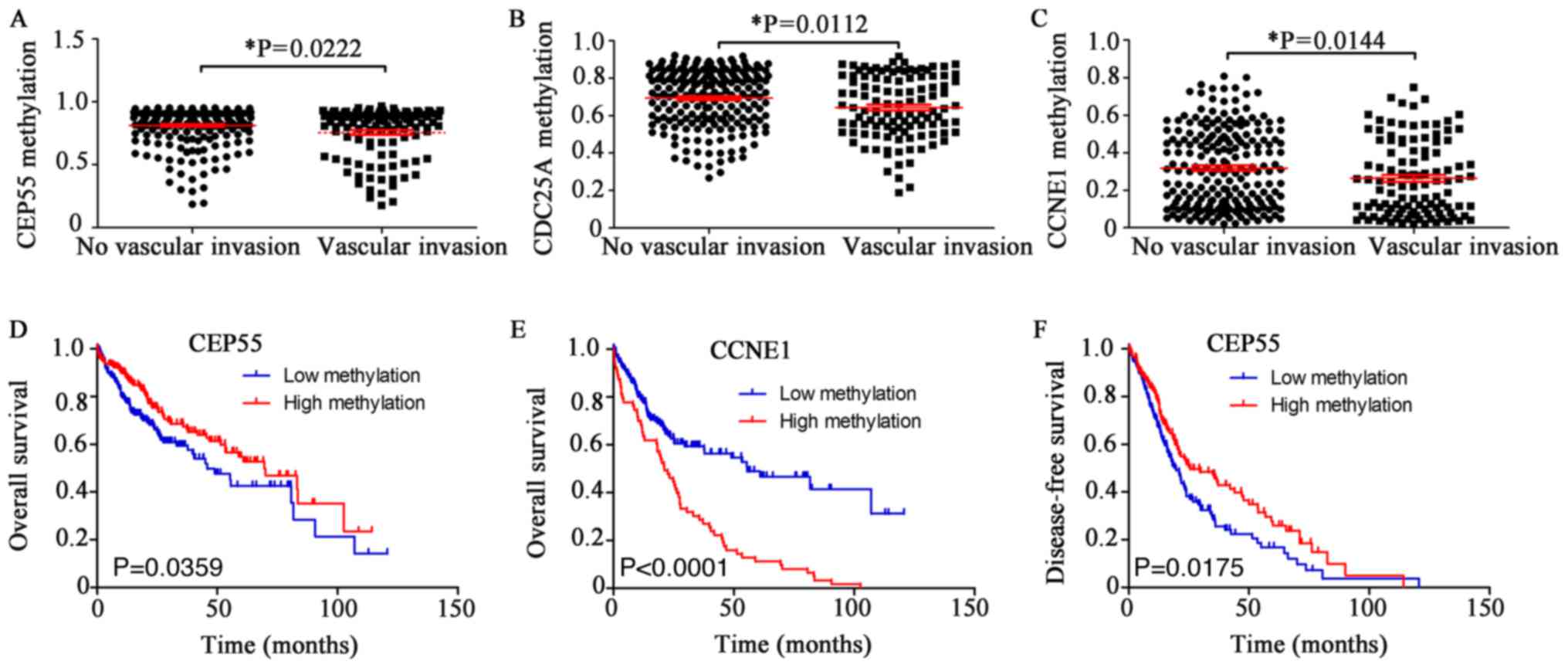
features of HCC samples were investigated (Fig. 5). The DNA methylation levels of
CEP55, CDC25A and CCNE1 were negatively associated with vascular
invasion. HCC samples presenting decreased levels of methylation
exhibited increased vascular invasion (Fig. 5A-C). The DNA methylation levels of
E2F7, CLSPN and CPEB3 were not associated with vascular invasion
(data not shown). To investigate the association between DNA
methylation levels and prognosis of patients with HCC, a
Kaplan-Meier analysis was performed. The overall survival was
increased when the methylation level of CEP55 was increased
(Fig. 5D). The overall survival
was decreased when the methylation level of CCNE1 was high
(Fig. 5E). The disease-free
survival was higher when CEP55 methylation was higher (Fig. 5F). Disease-free survival relative
to CCNE1 methylation is not presented. No significant associations
were identified between the DNA methylation levels of the other
four genes and survival rates (data not shown).
Discussion
HCC is one of the most common types of tumor,
exhibiting high morbidity, malignancy and mortality rates. The
occurrence and development of HCC are regulated by complex
multi-factorial processes involving multiple genes, and gene
regulation may be influenced by lncRNAs, miRNAs, genetic mutations
and epigenetic modifications (14,15).
Therefore, investigating a single factor associated with gene
regulation is not sufficient to assess the prognosis of patients
with HCC. Previous studies have identified genes that regulate HCC
progression; however, the majority of these studies focused on
single genes or investigated only one of the factors influencing
gene regulation (16–18), without considering their
combinatorial effect. To understand the complex molecular
biological characteristics of HCC, differentially expressed
lncRNAs, miRNAs and mRNAs were identified by analyzing biological
datasets from TCGA database. Subsequently, lncRNA-miRNA-mRNA gene
regulatory networks were constructed. To identify the independent
prognostic factors that affected the biological features of HCC,
the functions and the signaling pathways associated with the genes
identified were investigated. Additionally, genetic mutations and
epigenetic modifications were examined, and an association between
DNA methylation level and tumor vascularization was identified.
Furthermore, the epigenetic states of the genes examined were
identified to be associated with patient survival.
Accumulating evidence has demonstrated the
regulatory association between miRNAs and mRNAs, although the roles
of lncRNAs have frequently been ignored. lncRNAs serve numerous
roles in tumor occurrence and progression (15,19,20),
whereas, miRNAs have been identified to exert their biological
functions primarily by regulating gene expression. Furthermore,
previous studies demonstrated that lncRNAs, miRNAs and mRNAs are
able to serve important roles in tumor progression by regulating
each other (21–23). Zhang et al (24) performed a comprehensive analysis of
the lncRNA-miRNA-mRNA regulatory network in breast cancer by
analyzing the TCGA database. The previous study by Zhang et
al (24) provided important
insight for future breast cancer studies, and laid the foundation
for the development of novel strategies for targeting breast
cancer. Previous studies investigated the role of lncRNA and miRNA
regulation in HCC (14,25); however, to the best of the authors'
knowledge, the lncRNA-miRNA-mRNA network in HCC has not been
examined. Therefore, the present systematic analysis of the
lncRNA-miRNA-mRNA regulatory network provides novel insight into
the complex regulation of HCC development and progression. In the
present study, univariate and multivariate Cox regression analysis
based on genomic datasets from TCGA database suggested that
miRNA-137, PBK, LINC00200 and POLQ are independent prognostic
factors of HCC. Furthermore, Kaplan-Meier survival analyses
suggested that PBK and POLQ were associated with the 5-year
survival rate of patients with HCC (data not shown), and that these
two genes may serve a role in carcinogenesis. He et al
(26) demonstrated, by analyzing
the Gene Expression Omnibus database, that PBK was associated with
the prognosis of patients with HCC; however, to the best of the
authors' knowledge, experimental studies of POLQ in HCC have not
been conducted. Therefore, the present results provide an in
silico analysis that may represent the basis for future
experimental and theoretical studies aiming to investigate the
prognosis of HCC. The present prognosis model of HCC may help in
performing individualized diagnoses and developing targeted
treatments for HCC.
A total of 77 lncRNAs, 16 DEmiRNAs and 35 DEmRNAs
were identified to be involved in the constructed lncRNA-miRNA-mRNA
regulation network. In total, 12 DEmiRNAs were upregulated and only
one DEmiRNA, miRNA-424, was downregulated. The majority of the
predicted target genes of miRNA-424, which were additionally
differentially expressed in HCC, were associated with the 5-year
survival rate of patients with HCC. Therefore, the miRNA-424
regulation network was selected for subsequent analyses. To assess
the biological effects of the miRNA-424 regulatory network in HCC,
functional enrichment analyses using GO and KEGG were performed. GO
analysis suggested that the examined genes were associated with a
number of ‘biological process’, ‘molecular function’ and ‘cellular
component’ terms, whereas, KEGG analysis suggested that the
signaling pathways ‘progesterone-mediated oocyte maturation’,
‘oocyte meiosis’, ‘cell cycle’ and ‘microRNAs in cancer’ were
significantly enriched. Notably, these pathways have been
previously identified to be associated with the occurrence and
development of cancer (27–29).
The present results suggested that the prognosis of HCC may be
improved by testing the identified regulatory genes, although
further experimental studies are required to verify this
hypothesis.
PPI analysis identified that CDC25A, CEP55, CLSPN,
CCNE1 and E2F7 were associated with the prognosis of HCC, further
suggesting that the occurrence and development of HCC is a
multi-factor process involving numerous genes and proteins. CDC25A
was identified to be associated with CCNE1, in agreement with
previous studies (30–33). Notably, CDC25A and CCNE1 were
identified to be associated with the development of HCC.
Accumulating evidence demonstrated that genetic
mutations are associated with the occurrence and prognosis of
tumors (34–36), including HCC (37–39).
In the present study, it was identified that mutations in miRNA-424
target genes were associated with biological features of HCC.
Mutations in CEP55, CDC25A and E2F7 were identified to affect
overall survival and disease-free survival. Notably, these genes
may represent potential novel biomarkers for assessing the
prognosis of patients with HCC, and may be considered potential
targets for treating HCC. Epigenetic analysis suggested that
differential methylation states of CDC25A, CCNE1 and CEP55 were
associated with the occurrence, prognosis and biological features
of HCC. Previous studies demonstrated that DNA methylation serves
an important role in cancer (10,11,40).
In the present study, the level of DNA methylation of CDC25A, CCNE1
and CEP55 was negatively associated with vascular invasion in HCC.
Furthermore, the methylation state of CCNE1 and CEP55 was
associated with the prognosis of HCC. The present findings
identified a number of novel genes potentially involved in HCC
development and progression that may represent novel biomarkers and
novel targets for gene therapy, resulting in an improved prognosis
of HCC.
Acknowledgements
The authors would like to thank Dr Li Ting
(Department of Clinical Medical Oncology, Fujian Provincial
Hospital, Fuzhou, China) and Dr Wang Yan (Department of
Hepatobiliary Surgery, Guilin Medical University, Guilin, China)
for their expert technical assistance.
Funding
The present study was supported in part by The
National Natural Science Foundation of China (grant nos. 81430014
and 81771674; China) and by The 111 Project (grant no. D17011.
Availability of data and materials
The genetic transcriptome of hepatocellular
carcinoma and the methylation data were downloaded from the TCGA
data portal (https://portal.gdc.cancer.gov/). All datasets used
and/or analyzed during the current study are available from the
corresponding author on reasonable request.
Authors' contributions
SH designed and conducted the study. CL performed
all bioinformatics analysis and wrote the manuscript. GY analyzed
the data and performed the statistical analyses. ZH and YZ
participated in literature retrieval. XQ and HY made contributions
to the design of this study and revised the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tang Z: Treatment strategies for cancer.
Chin J Clin Hepatol. 27:337–339. 2011.(In Chinese).
|
|
3
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krol J, Loedige I and Filipowicz W: The
widespread regulation of microRNA biogenesis, function and decay.
Nat Rev Genet. 11:597–610. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mercer TR and Mattick JS: Structure and
function of long noncoding RNAs in epigenetic regulation. Nat
Struct Mol Biol. 20:300–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Quinn JJ and Chang HY: Unique features of
long non-coding RNA biogenesis and function. Nat Rev Genet.
17:47–62. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wapinski O and Chang HY: Long noncoding
RNAs and human disease. Trends Cell Biol. 21:354–361. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rozalski R, Gackowski D, Siomek-Gorecka A,
Banaszkiewicz Z and Olinski R: Urinary measurement of epigenetic
DNA modifications: A non-invasive assessment of the whole-body
epigenetic status in healthy subjects and colorectal cancer
patients. ChemistryOpen. 5:550–553. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shakeri H, Fakhrjou A, Nikanfar A and
Mohaddes-Ardebili SM: Methylation analysis of BRCA1 and APC in
breast cancer and it's relationship to clinicopathological
features. Clin Lab. 62:2333–2337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peng D, Zhang H and Sun G: The
relationship between P16 gene promoter methylation and gastric
cancer: A meta-analysis based on Chinese patients. J Cancer Res
Ther. 10 Suppl:S292–S295. 2014. View Article : Google Scholar
|
|
11
|
Stephen JK, Chen KM, Merritt J, Chitale D,
Divine G and Worsham MJ: Methylation markers for early detection
and differentiation of follicular thyroid cancer subtypes. Cancer
Clin Oncol. 4:1–12. 2015.PubMed/NCBI
|
|
12
|
Furukawa R, Hachiya T, Ohmomo H, Shiwa Y,
Ono K, Suzuki S, Satoh M, Hitomi J, Sobue K and Shimizu A:
Intraindividual dynamics of transcriptome and genome-wide stability
of DNA methylation. Sci Rep. 6:264242016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu XN, Liu XH, Yu XX, et al: DNA
methylation of tumor suppressor genes and hepatocellular carcinoma.
China Cancer. 26:727–732. 2017.(In Chinese).
|
|
14
|
Wang F, Ying HQ, He BS, Pan YQ, Deng QW,
Sun HL, Chen J, Liu X and Wang SK: Upregulated lncRNA-UCA1
contributes to progression of hepatocellular carcinoma through
inhibition of miR-216b and activation of FGFR1/ERK signaling
pathway. Oncotarget. 6:7899–7917. 2015.PubMed/NCBI
|
|
15
|
Zhang Z, Weaver DL, Olsen D, deKay J, Peng
Z, Ashikaga T and Evans MF: Long non-coding RNA chromogenic in situ
hybridisation signal pattern correlation with breast tumour
pathology. J Clin Pathol. 69:76–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
He JH, Han ZP, Liu JM, Zhou JB, Zou MX, Lv
YB, Li YG and Cao MR: Overexpression of long non-coding RNA MEG3
inhibits proliferation of hepatocellular carcinoma Huh7 cells via
negative modulation of miRNA-664. J Cell Biochem. 118:3713–3721.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang BP, Lin CS, Wang CJ and Kao SH:
Upregulation of heat shock protein 70 and the differential protein
expression induced by tumor necrosis factor-alpha enhances
migration and inhibits apoptosis of hepatocellular carcinoma cell
HepG2. Int J Med Sci. 14:284–293. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ye Y, Wei Y, Xu Y, Li Y, Wang R, Chen J,
Zhou Y, Fu Z, Chen Y, Wang X, et al: Induced MiR-1249 expression by
aberrant activation of Hedegehog signaling pathway in
hepatocellular carcinoma. Exp Cell Res. 355:9–17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Cao L, Wu J and Wang Q: Long
non-coding RNA SNHG1 regulates NOB1 expression by sponging miR-326
and promotes tumorigenesis in osteosarcoma. Int J Oncol. 52:77–88.
2018.PubMed/NCBI
|
|
20
|
Zeng S, Xie X, Xiao YF, Tang B, Hu CJ,
Wang SM, Wu YY, Dong H, Li BS and Yang SM: Long noncoding RNA
LINC00675 enhances phosphorylation of vimentin on Ser83 to suppress
gastric cancer progression. Cancer Lett. 412:179–187. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
He JH, Han ZP, Zou MX, Wang L, Lv YB, Zhou
JB, Cao MR and Li YG: Analyzing the LncRNA, miRNA and mRNA
regulatory network in prostate cancer with bioinformatics software.
J Comput Biol. 25:146–157. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang H, Ma R, Zou S, Wang Y, Li Z and Li
W: Reconstruction and analysis of the lncRNA-miRNA-mRNA network
based on competitive endogenous RNA reveal functional lncRNAs in
rheumatoid arthritis. Mol Biosyst. 13:1182–1192. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li DY, Chen WJ, Luo L, Wang YK, Shang J,
Zhang Y, Chen G and Li SK: Prospective lncRNA-miRNA-mRNA regulatory
network of long non-coding RNA LINC00968 in non-small cell lung
cancer A549 cells: A miRNA microarray and bioinformatics
investigation. Int J Mol Med. 40:1895–1906. 2017.PubMed/NCBI
|
|
24
|
Zhang Y, Li Y, Wang Q, Zhang X, Wang D,
Tang HC, Meng X and Ding X: Identification of an lncRNA-miRNA-mRNA
interaction mechanism in breast cancer based on bioinformatic
analysis. Mol Med Rep. 16:5113–5120. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang J, Fan D, Jian Z, Chen GG and Lai
PB: Cancer specific long noncoding RNAs show differential
expression patterns and competing endogenous RNA potential in
hepatocellular carcinoma. PLoS One. 10:e01410422015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He B, Yin J, Gong S, Gu J, Xiao J, Shi W,
Ding W and He Y: Bioinformatics analysis of key genes and pathways
for hepatocellular carcinoma transformed from cirrhosis. Medicine
(Baltimore). 96:e69382017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kuznetsov VA, Tang Z and Ivshina AV:
Identification of common oncogenic and early developmental pathways
in the ovarian carcinomas controlling by distinct prognostically
significant microRNA subsets. BMC Genomics. 18 Suppl 6:S6922017.
View Article : Google Scholar
|
|
28
|
Fu Z, Han X, Du J, Han X, Liu W, Shao S
and Liu X: Euphorbia lunulata extract acts on multidrug resistant
gastric cancer cells to inhibit cell proliferation, migration and
invasion, arrest cell cycle progression, and induce apoptosis. J
Ethnopharmacol. 212:8–17. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
El-Daly SM, Abba ML and Gamal-Eldeen AM:
The role of microRNAs in photodynamic therapy of cancer. Eur J Med
Chem. 142:550–555. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu X, Yamamoto H, Sakon M, Yasui M, Ngan
CY, Fukunaga H, Morita T, Ogawa M, Nagano H, Nakamori S, et al:
Overexpression of CDC25A phosphatase is associated with hypergrowth
activity and poor prognosis of human hepatocellular carcinomas.
Clin Cancer Res. 9:1764–1772. 2003.PubMed/NCBI
|
|
31
|
Yuan P, Li J, Zhou F, Huang Q, Zhang J,
Guo X, Lyu Z, Zhang H and Xing J: NPAS2 promotes cell survival of
hepatocellular carcinoma by transactivating CDC25A. Cell Death Dis.
8:e27042017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiang W, Huang H, Ding L, Zhu P, Saiyin H,
Ji G, Zuo J, Han D, Pan Y, Ding D, et al: Regulation of cell cycle
of hepatocellular carcinoma by NF90 through modulation of cyclin E1
mRNA stability. Oncogene. 34:4460–4470. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang X, Hu S, Zhang X, Wang L, Zhang X,
Yan B, Zhao J, Yang A and Zhang R: MicroRNA-7 arrests cell cycle in
G1 phase by directly targeting CCNE1 in human hepatocellular
carcinoma cells. Biochem Biophys Res Commun. 443:1078–1084. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bahrami A, Hesari A, Khazaei M, Hassanian
SM, Ferns GA and Avan A: The therapeutic potential of targeting the
BRAF mutation in patients with colorectal cancer. J Cell Physiol.
233:2162–2169. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vietsch EE, Graham GT, McCutcheon JN,
Javaid A, Giaccone G, Marshall JL and Wellstein A: Circulating
cell-free DNA mutation patterns in early and late stage colon and
pancreatic cancer. Cancer Genet. 218–219. 39–50. 2017.
|
|
36
|
Zhang K, Zhou J, Zhu X, Luo M, Xu C, Yu J,
Deng M, Zheng S and Chen Y: Germline mutations of PALB2 gene in a
sequential series of Chinese patients with breast cancer. Breast
Cancer Res Treat. 166:865–873. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hayashi T, Yamashita T, Okada H, Nio K,
Hara Y, Nomura Y, Hayashi T, Asahina Y, Yoshida M, Oishi N, et al:
Sporadic PCDH18 somatic mutations in EpCAM-positive
hepatocellular carcinoma. Cancer Cell Int. 17:942017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jiao J, Niu W, Wang Y, Baggerly K, Ye Y,
Wu X, Davenport D, Almeda JL, Betancourt-Garcia MM, Forse RA, et
al: Prevalence of Aflatoxin-associated TP53R249S mutation in
hepatocellular carcinoma in hispanics in South texas. Cancer Prev
Res (Phila). 11:103–112. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Riordan JD, Feddersen CR, Tschida BR,
Beckmann PJ, Keng VW, Linden MA, Amin K, Stipp CS, Largaespada DA
and Dupuy AJ: Chronic liver injury alters driver mutation profiles
in hepatocellular carcinoma in mice. Hepatology. 67:924–939. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tommasi S and Besaratinia A: A versatile
assay for detection of aberrant DNA methylation in bladder cancer.
Methods Mol Biol. 1655:29–41. 2018. View Article : Google Scholar : PubMed/NCBI
|