Introduction
Bladder cancer (BC) is the fourth most common type
of cancer in men in the United States and is responsible for
~150,000 cases of mortality worldwide; therefore, it is considered
an important health problem (1,2).
Approximately 75% of patients have non-muscle-invasive BC (NMIBC)
(Ta, T1) and 25% have muscle-invasive BC (≥T2). An important
clinical issue is the high rate of recurrence associated with
NMIBC. Although radical cystectomy and pelvic lymphadenectomy can
be performed, a high percentage of patients progress to advanced
invasive tumors. Tumors at a high risk for recurrence and
progression require neoadjuvant treatment (3).
Chemokine (C-C motif) ligand (CCL)18 is a C-C
chemokine that is highly expressed in human lung tissues and
plasma; CCL18 serves numerous functions in immune modulation and
cancer progression (4). Recent
studies have reported that increased expression levels of CCL18 are
observed in various malignancies, including ovarian cancer, lung
cancer, oral squamous cell cancer and breast cancer (5–8). It
has also been suggested that CCL18 acts as a potential urinary
biomarker in BC (9,10); however, the molecular mechanisms
underlying the effects of CCL18 on BC remain unknown. C-C motif
receptor 8 (CCR8) is a G protein-coupled receptor, which is known
to be expressed by immune cells, including T-helper 2 lymphocytes,
natural killer cells, and monocytes (11). In humans, CCR8 is selectively
activated by CCL1/CC chemokine I-309 (12). Recently, Islam et al
reported that CCR8 is a functional receptor for CCL18 (13).
The present study revealed that CCL18 promoted the
migration and invasion of BC cells. However, the underlying
molecular mechanisms by which CCL18 induces epithelial-mesenchymal
transition (EMT) in BC cells and contributes to cancer cell
invasion have not been identified. In addition, the present study
demonstrated that excessive expression of CCL18 in tumor tissues
was associated with tumor stage and poor prognosis in patients with
BC. Furthermore, the present data indicated that CCL18 may promote
migration, invasion and EMT through the G protein-coupled receptor
chemokine CCR8.
Materials and methods
Gene expression profiling
Expression of CCL18 in BC (BLCA dataset) was checked
using The Cancer Genome Atlas (TCGA) data. TCGA, launched by the
National Institutes of Health (NIH, Bethesda, MD, USA), is a
publicly funded project that comprises a comprehensive ‘atlas’ of
cancer genomic profiles. The gene expression levels of CCL18 in 404
patients with BC compared with 28 normal samples from TCGA database
and GTEx were analyzed using the GEPIA web server (14). The Pathology Atlas from the Human
Protein Atlas (www.proteinatlas.org/pathology) was used to perform
analyses based on the mRNA expression levels of CCL18 in BC tissue
and the clinical outcomes (survival). The data in the Pathology
Atlas are based on the integration of publicly available data from
TCGA. In addition, gene expression profiling studies involving
several clinical samples were performed to analyze the expression
of CCL18 in a dataset available through the Gene Expression Omnibus
(GSE31684; http://www.ncbi.nlm.nih.gov/geo/) (15).
Patients and tissue samples
The present study was approved by the Research
Ethics Committee of the First Affiliated Hospital, Nanchang
University (Nanchang, China). Informed consent was obtained from
all patients. All specimens were handled and anonymized according
to ethical and legal standards. Immunohistochemical analysis of
CCL18 was conducted using 64 primary BC tissues and 30 adjacent
noncancerous bladder tissues. Detailed information on the clinical
features of all patients in this study is presented in Table I.
 | Table I.Association of CCL18 expression with
the clinicopathological characteristics of bladder cancer. |
Table I.
Association of CCL18 expression with
the clinicopathological characteristics of bladder cancer.
|
| CCL18 expression
levels in tumor tissues |
|
|---|
|
|
|
|
|---|
| Parameter | High (Grade ≥2)
(n=37) | Low (Grade <1)
(n=27) | P-value |
|---|
| Agea, mean ± SD | 62.7±9.1 | 64.6±8.8 | 0.403 |
| Sex, n
(%)b |
|
|
|
|
Female | 6
(54.5) | 5
(45.5) | 0.809 |
|
Male | 31 (58.5) | 22 (41.5) |
|
| Tumor stage, n
(%)c |
|
| 0.018 |
| T1 | 10 (47.6) | 11 (52.4) |
|
| T2 | 10 (43.5) | 13 (56.5) |
|
| T3 | 9
(75.0) | 3
(25.0) |
|
| T4 | 8
(100.0) | 0 (0.0) |
|
| Tumor grade, n
(%)a |
|
| 0.447 |
| Low
grade | 12 (48.0) | 13 (52.0) |
|
| High
grade | 25 (64.1) | 14 (35.9) |
|
Cell culture and treatment
The 5637 human BC cell line was purchased from the
Cell Bank of Type Culture Collection of Chinese Academy of
Sciences, Shanghai Institute of Cell Biology (Shanghai, China) and
was maintained in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Media were supplemented with
10% heat-inactivated fetal bovine serum (FBS; HyClone; GE
Healthcare Life Sciences, Logan, UT, USA), 100 U/ml penicillin and
100 µg/ml streptomycin at 37°C in a humidified incubator containing
5% CO2. The small molecule CCR8 inhibitor R243 (cat. no.
AOB2014) was purchased from AOBIOUS, Inc. (Gloucester, MA, USA).
For chemokine treatment, 5637 cells pretreated with or without R243
(5 µM) for 6 h at 37°C/5% carbon dioxide (16), were exposed to 50 or 100 ng/ml
CCL18 (PeproTech, Inc., Rockville, MD, USA) for 36 h.
Short hairpin (sh)RNA
transfection
For transfection, 5637 cells were plated in 6-well
plates at 2×105/well. The human CCR8 shRNA plasmid and
non-target shRNA (NT shRNA) were obtained from Invitrogen; Thermo
Fisher Scientific, Inc. The targeted sequences were: Human CCR8
(5′-CCGGGGATTATACACTTGACCTCAGTGTCGACACTGAGGTCAAGTGTATAATCCTTTTTTG-3′)
and non-target (NT) shRNA
(5′-CCGGCAACAAGATGAAGAGCACAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT-3′).
Once the 5637 cells reached 50–60% confluence, 1.25 µg/ml CCR8
shRNA and NT shRNA were transfected using Lipofectamine®
2000 transfection reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and Opti-MEM medium (Gibco; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. A total of 6 h
post-transfection, the medium was replaced with fresh medium
containing 10% FBS. NT shRNA was used as the negative control.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
A total of 24, 48 and 72 h post-transfection, total
RNA was extracted from 5637 cells using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and cDNA was
synthesized using a Takara PrimeScript RT reagent kit (Takara Bio,
Inc., Otsu, Japan) according to manufacturer's protocols. qPCR was
performed using SYBR Premix Ex Taq kit (Takara Bio, Inc., Otsu,
Japan) in the ABI PRISM® 7500 real-time PCR system
(Thermo Fisher Scientific, Inc.). The thermocycling conditions were
initial denaturation (1 cycle, 95°C, 30 sec), PCR reaction (40
cycles, 95°C, 5 sec, and 60°C, 30 sec for annealing and
elongation). The data of RT-qPCR were normalized against an
internal control β-actin and relative expression levels were
evaluated using the 2−∆∆Cq method and then expressed as
fold changes (17). The
oligonucleotide sequences of the RT-qPCR primers are listed in
Table II.
| Table II.Oligonucleotide sequences for
polymerase chain reaction amplification. |
Table II.
Oligonucleotide sequences for
polymerase chain reaction amplification.
| Gene | NCBI no. | Sequence
(5′-3′) | Product size
(bp) |
|---|
| CCR8 | NM_005201.3 | F:
GTGTGACAACAGTGACCGACT | 173 |
|
|
| R:
CTTCTTGCAGACCACAAGGAC |
|
| E-cadherin | NM_001317186.1 | F:
AGCTGCCCAGAAAATGAAAAAGG | 203 |
|
|
| R:
GTGTATGTGGCAATGCGTTCTC |
|
| MMP-2 | NM_001302510.1 | F:
CCTCTCCACTGCCTTCGATA | 129 |
|
|
| R:
TGGGAGGAGTACAGTCAGCA |
|
| VEGF-C | NM_005429.2 | F:
GGCTGGCAACATAACAGAGAA | 159 |
|
|
| R:
CCCCACATCTATACACACCTCC |
|
| β-actin | NM_001101.3 | F:
CATGTACGTTGCTATCCAGGC | 250 |
|
|
| R:
CTCCTTAATGTCACGCACGAT |
|
Western blot analysis
Total proteins were extracted from cells using
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Haimen, China). Proteins were quantified using a
Pierce bicinchoninic acid protein assay kit (Thermo Fisher
Scientific, Inc.), followed by western blot analysis. 40 µg
proteins from cell lysates were subjected to 12% SDS-PAGE. Once
proteins were transferred to nitrocellulose membranes, they were
incubated with antibodies targeting β-actin (cat. no. 58169,
1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA), CCR8
(cat. no. ab32131, 1:500; Abcam, Cambridge, UK), E-cadherin (cat.
no. 3195, 1:1,000; Cell Signaling Technology, Inc., Danvers, MA,
USA), matrix metalloproteinase (MMP)-2 (cat. no. 40994, 1:1,000;
Cell Signaling Technology, Inc.) and vascular endothelial growth
factor (VEGF)-C (cat. no. 2445, 1:1,000; Cell Signaling Technology,
Inc.), followed by incubation with a horseradish peroxidase-labeled
goat anti-rabbit secondary antibody (cat. no. ab6721, 1:3,000;
Abcam) for 1.5 h at room temperature. Protein bands were visualized
by Millipore enhanced chemiluminescence (cat. no. WBKLS0500; EMD
Millipore, Billerica, MA, USA). ImageJ 1.45 software (NIH) was used
to perform densitometric analysis of each band.
Immunohistochemistry
Tissue sections from paraffin- embedded bladder
cancer were dewaxed in xylene and rehydrated in a graded alcohol
series. Following microwave-induced antigen retrieval (Tris-EDTA pH
9.0), the slides were washed with PBS and incubated with primary
antibody against CCL18 (cat. no. ab104867, 1:200; Abcam) overnight
at 4°C. The sections were incubated with secondary antibody at room
temperature. for 30 min after washing with PBST. Color detection
was performed by liquid DAB+ substrate chromogen system
(Dako; Agilent Technologies, Inc., Santa Clara, CA, USA) according
to the manufacturer's protocols. Slides were counterstained with
hematoxylin. Hemalum was used to stain nuclei of cells blue at room
temperature for 20 sec. Immunohistochemical images of CCL18
expression were analyzed according to a previous study (18). Representative areas with 5 or 20
CCL18 cells per ×400 high-power field were chosen and images were
captured randomly as references for grading. The densities of
CCL18+ cells were graded as follows: Grade 1, <5
positive cells/microscopic field; grade 2, 5–20 cells/field; and
grade 3, >20 cells/field. Grade 1 was considered to be low-level
CCL18+, and grades 2 and 3 were considered to be
high-level CCL18+.
Cell migration and Transwell
assays
Cell migration was determined using a wound-healing
assay. Briefly, 5637 cells were seeded in six-well culture dishes
and cultured at 37°C to form a confluent monolayer. After
transfection with or without CCR8 shRNA for 24 h, and a wound was
made by scratching the monolayer with a 10 µl pipette tip. The
wounded monolayer was then washed three times with PBS to remove
cell debris and treated with CCL18 for 24 h at 37°C. After
scratching, the area of the cell-free scratch was imaged under an
Olympus CKX41 inverted microscope (Olympus Corporation, Tokyo,
Japan) at 0 and 24 h. Cell invasive capacities were measured using
a Transwell assay (Corning Incorporated, Corning, NY, USA).
Briefly, 5637 cells were treated with CCL18 and/or transfected with
CCR8 shRNA for 24 h, and were then seeded in the upper chamber of
the Transwell system. The upper chamber of the insert was precoated
with 0.1 ml (300 µg/ml) Matrigel matrix (Corning Incorporated) for
the invasion assay. The invaded cells on the underside of the
membrane were counted. In this assay, prepared cells were seeded in
the upper chamber with serum-free medium and the medium of the
lower chamber was supplemented with 10% FBS as a chemoattractant.
Following incubation for 24 h, the cells were fixed with 4%
formaldehyde at 37°C for 30 min. Cells that did not invade through
the pores were removed with a cotton swab. Cells that had invaded
to the lower surface of the membrane were stained with crystal
violet at 37°C for 15 min. Finally, images of five representative
fields at ×100 magnification were randomly captured using a light
microscope (Carl Zeiss AG, Oberkochen, Germany) and the number of
cells in each well was semi-quantified.
Statistical analysis
All data are presented as the means ± standard
deviation in at least three replicates per group. Statistical
analysis was performed to determine the significance of the
differences between groups using one-way analysis of variance (with
post hoc Turkey's honest significant difference test) or Student's
t-test. χ2 analysis and Fisher's exact test were used to
examine the association between CCL18 expression and the
clinicopathological features of patients with BC. The effects of
CCL18 expression levels on survival were estimated using the
Kaplan-Meier method and were compared by the log-rank test.
Wilcoxon-signed rank test was used to compare CCL18 expression
between 30 paired cancerous and noncancerous tissues of patients
with BC. All statistical analyses were performed using GraphPad
Prism 7.00 software for Windows (GraphPad Software, Inc., La Jolla,
CA, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Association of CCL18 expression with
the clinicopathological characteristics of BC
To assess whether CCL18 may be involved in the
development of BC, this study first investigated the mRNA
expression levels of CCL18 using TCGA dataset. As illustrated in
Fig. 1A, the mRNA expression
levels of CCL18 were significantly higher in BC tissues compared
with in nontumor tissues. In addition, CCL18 mRNA expression was
higher in BC tissues at stages III and IV compared with at stage II
(Fig. 1B and C). Kaplan-Meier
analysis indicated that higher CCL18 expression was associated with
worse patient survival (log-rank test, P=0.0487; Fig. 1D). Subsequently, the expression of
CCL18 was investigated in BC tissues using immunohistochemistry. As
reported in a previous study (10), there was no epithelial staining for
CCL18; however, inflammatory cells in the stroma were
CCL18-positive. It was revealed that CCL18 expression was higher in
the majority of cancerous samples compared with in noncancerous
tissues (Fig. 1E). Furthermore,
the protein expression levels of CCL18 were detected in tissues
obtained from 30 patients with BC by immunohistochemical staining.
The results demonstrated that CCL18 expression was significantly
stronger in the majority of BC tissues compared with in paired
normal tissues (Fig. 1F). As shown
in Fig. 1G, CCL18 was weakly
detected in stage I and II BC tissues, but was strongly detected in
stage III and IV BC tissues in tissue samples employed in the
present study. Subsequently, the association between CCL18
expression in BC cases and various clinical outcomes was analyzed.
As shown in Table I, higher CCL18
expression was associated with higher pathological stages
(P<0.05). These data suggested that CCL18 was elevated in BC
tissues and may be associated with the progression of BC.
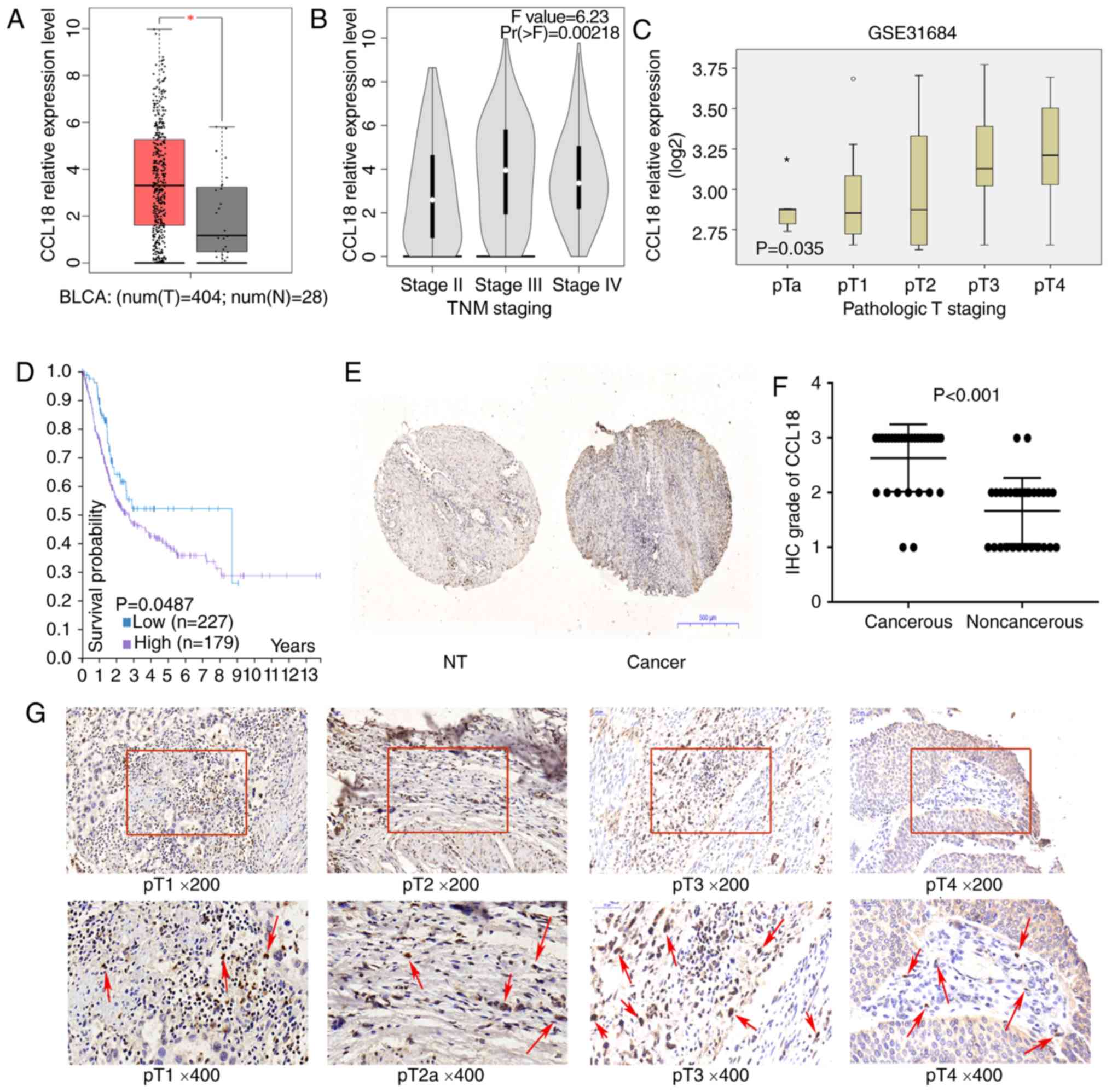 | Figure 1.CCL18 expression in bladder cancer
tissues. (A) Expression of CCL18 in bladder cancer samples (red
box) and adjacent tissues (grey box), *P<0.001. (B) Expression
of CCL18 in TNM stage II, III and IV bladder cancer samples in The
Cancer Genome Atlas BLCA dataset. (C) Expression of CCL18 in
bladder cancer samples, according to the pathological stage, in the
GSE31684 dataset. (D) Results of overall survival analysis in
patients stratified according to CCL18 mRNA expression
(Kaplan-Meier test) in the Human Protein Atlas. (E and F) IHC
staining of CCL18 in 30 paired noncancerous and cancerous tissues
obtained from patients with bladder cancer (magnification, ×40).
(G) Protein expression levels of CCL18 in bladder tumor tissues, as
observed by IHC staining. Red arrows indicate CCL18-positive cells
in the stroma. Hemalum was used to stain nuclei of cells blue.
CCL18 immunoreactivity in stromal cells were located in cytoplasm;
no CCL18 staining was detected in the nuclei of cells. BLCA,
bladder cancer; CCL18, chemokine (C-C motif) ligand 18; IHC,
immunohistochemistry; NT, nontumorous. |
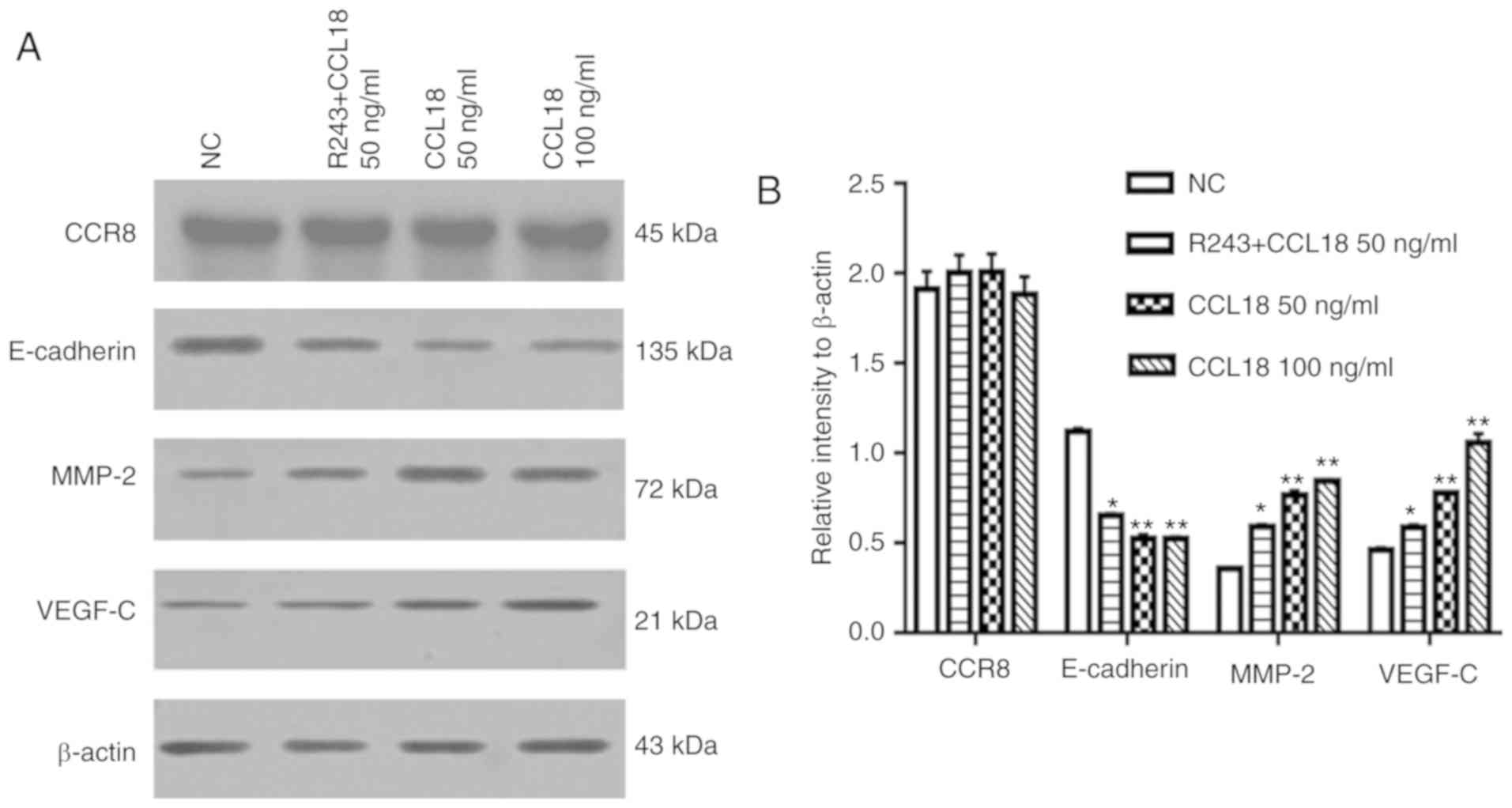
CCL18 regulates the expression of
E-cadherin, MMP2 and VEGF-C in 5637 BC cells
EMT is thought to serve a key role in the invasion
and metastasis of numerous types of tumor (19). EMT is an evolutionarily conserved
developmental program, which is believed to have a critical role in
carcinogenesis and tumor metastasis by enhancing mobility and
invasion. Our previous study revealed that VEGF-C, MMP-2 and MMP-9
are immunosuppressive factors, which serve important roles in BC
invasion and metastasis (20). To
determine whether CCL18 enhanced BC invasion and metastasis through
EMT, and by altering MMP-2 and VEGF-C expression, 5637 BC cells
were treated with rCCL18. Following treatment of 5637 BC cells with
50 or 100 ng/ml CCL18 for 36 h, the expression levels of E-cadherin
were significantly lower, and the expression levels of MMP-2 and
VEGF-C were higher compared with in the control group (Fig. 2).
CCR8 is required for migration and
invasion of BC cells via CCL18
CCL18 has numerous receptors, including PITPNM
family member 3 (PITPNM3) and G protein-coupled estrogen receptor 1
(GPR30), which mediate CCL18-induced migration of cancer cells, and
promote tumor invasion and metastasis (8,21).
Recently, Islam et al reported that CCR8 is a functional
receptor for CCL18 (13). To
identify whether CCR8 was associated with CCL18-induced tumor
invasion and EMT, this study examined the expression levels of
E-cadherin, MMP-2 and VEGF-C in 5637 BC cells treated with a small
molecule CCR8 inhibitor, R243, or transfected with CCR8 shRNA,
followed by CCL18 treatment (Figs.
2 and 3). As shown in Fig. 2, R243 reversed the decreased levels
of E-cadherin and increased levels of MMP-2 and VEGF-C caused by
CCL18, without affecting CCR8 levels. The results of RT-qPCR
confirmed that the mRNA expression levels of CCR8 were
downregulated by CCR8 shRNA (Fig.
3A); CCR8 protein expression was also reduced in response to
CCR8 shRNA (Fig. 3D). RT-qPCR
analysis revealed that the mRNA expression levels of E-cadherin
were increased, whereas those of MMP-2 and VEGF-C were decreased
compared with in cells transfected with NT shRNA (Fig. 3B). Similar results were revealed by
western blot analysis for the protein expression levels of
E-cadherin, MMP-2 and VEGF-C (Fig. 3C
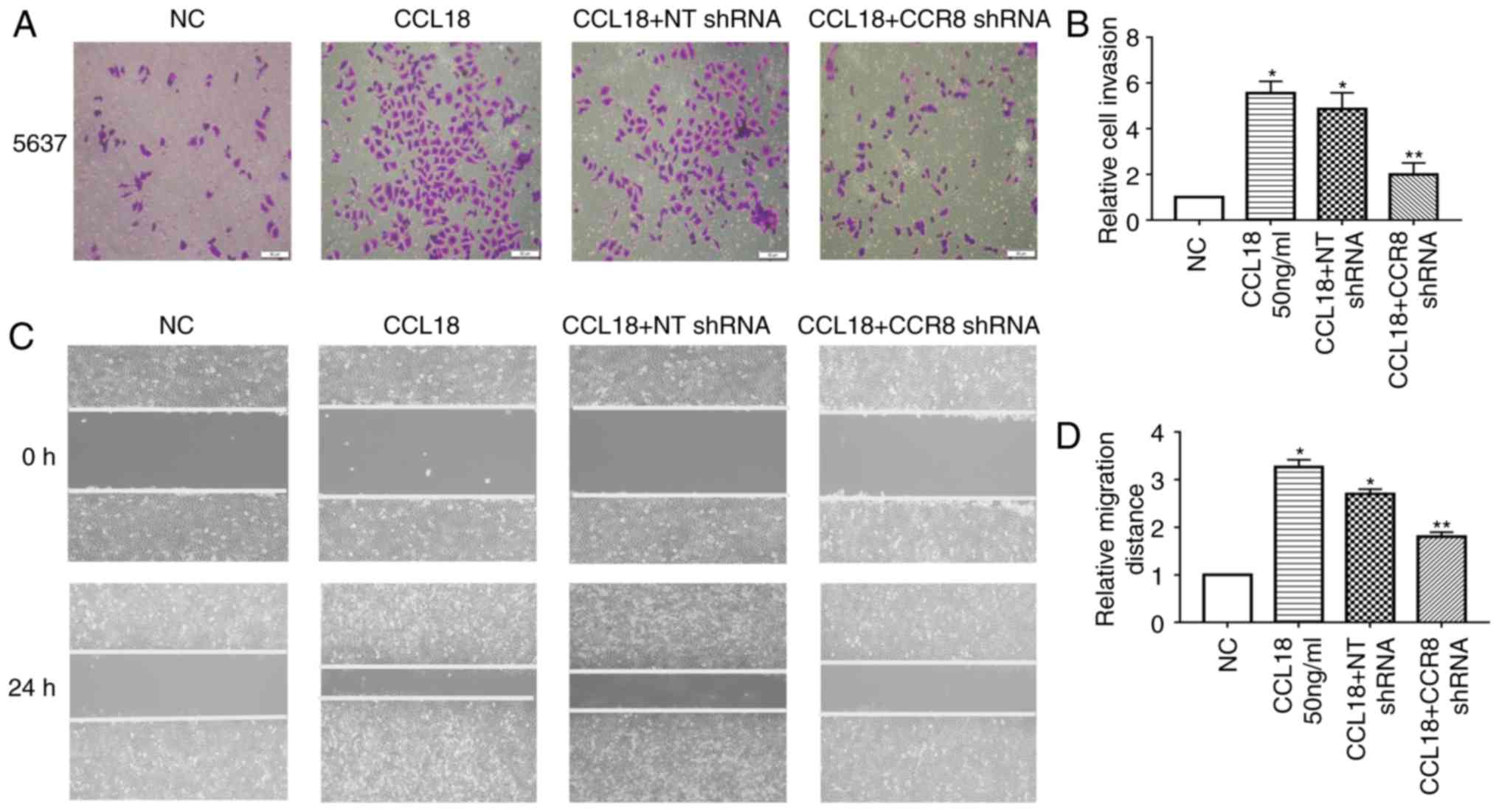
and D). Subsequently, the migratory and invasive abilities of
BC cells were reduced in CCR8-knockdown cells compared with in
control cells (Fig. 4A-D).
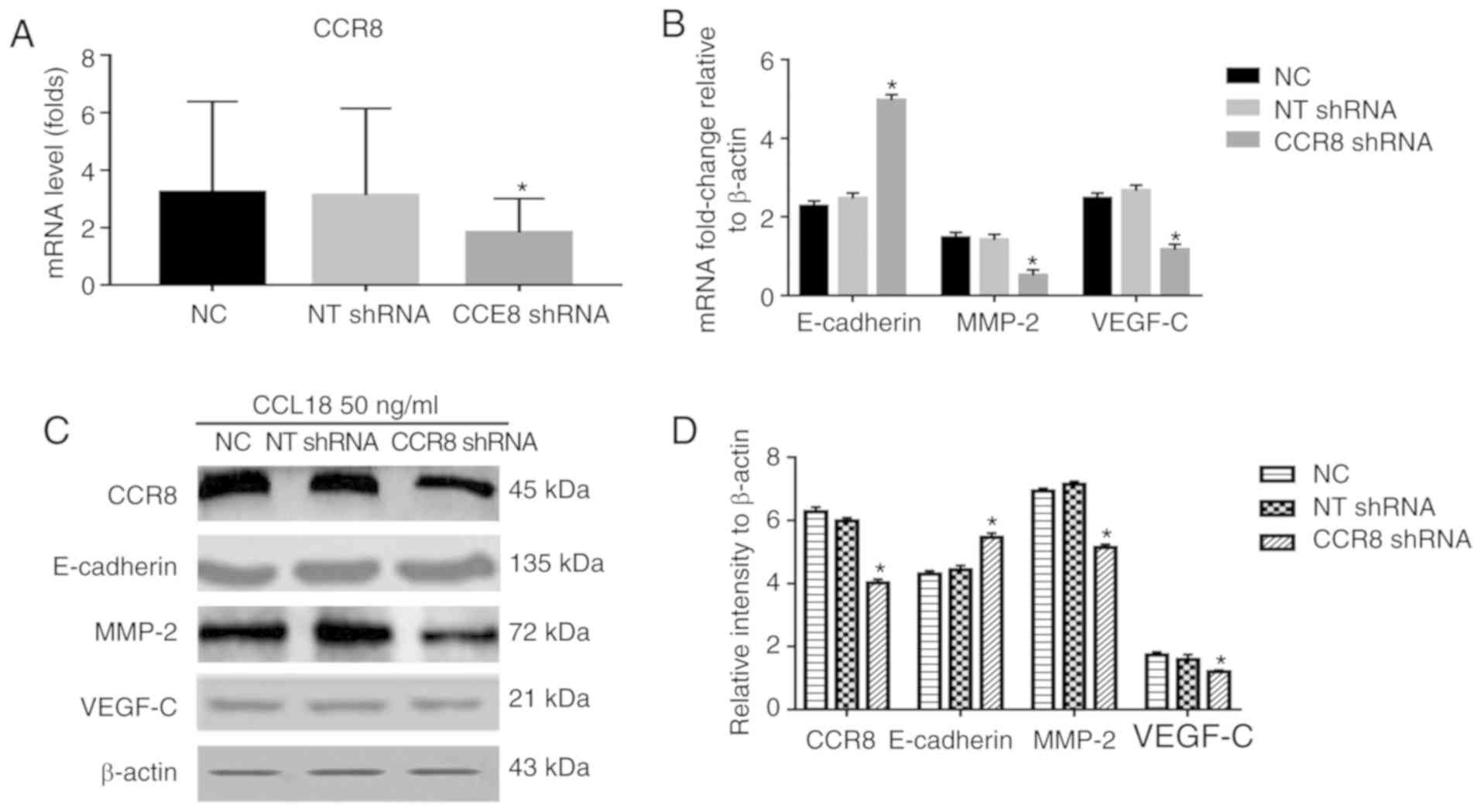 | Figure 3.Effects of CCR8 shRNA on
CCL18-induced expression of E-cadherin, MMP-2 and VEGF-C. 5637
bladder cancer cells were treated with CCL18 (50 ng/ml), CCL18 + NT
shRNA or CCL18 + CCR8 shRNA for 36 h. (A) mRNA expression levels of
CCR8 after being transfected with CCR8 shRNA was examined by
reverse transcription-quantitative polymerase chain reaction.
β-actin was used as an internal control. (B) mRNA expression levels
of CCR8, E-cadherin, MMP-2 and VEGF-C were examined by reverse
transcription-quantitative polymerase chain reaction. β-actin was
used as an internal control. (C and D) Protein expression levels of
CCR8, E-cadherin, MMP-2 and VEGF-C in 5637 cells were detected by
western blotting. β-actin was used as an internal control.
*P<0.05 compared with the NC and NT shRNA groups. CCL18,
chemokine (C-C motif) ligand 18; CCR8, chemokine (C-C motif)
receptor 8; MMP-2, matrix metalloproteinase-2; NC, normal control;
NT, non-target; shRNA, short hairpin RNA; VEGF-C, vascular
endothelial growth factor-C. |
Discussion
To the best of our knowledge, the present study is
the first to demonstrate that CCL18 may be frequently overexpressed
in BC. Furthermore, it was revealed that CCL18 enhanced bladder
cancer cell invasion, migration and EMT. CCL18 has numerous
receptors, including PITPNM3 (8)
and GPR30 (21), which mediate
CCL18-induced migration of cancer cells, and promote tumor invasion
and metastasis. CCL18 activates proline-rich tyrosine kinase 2 and
the Src kinase PITPNM3 by binding to PITPNM3, which is expressed in
breast cancer cells, thereby promoting metastasis of breast cancer
(8). However, the functional
receptor for CCL18 that mediates BC cell invasion and migration
remains unknown. Recently, Islam et al reported that CCR8 is
a functional receptor for CCL18 (13). When CCL18 binds to CCR8, CCR8 is
activated through G-protein signaling, inducing CCR8
internalization and activating cell chemotaxis and calcium flux in
human CCR8-transfected cells. In the present study, it was further
deduced that the G-protein coupled receptor CCR8 may mediate
signaling by CCL18, and a potential mechanism underlying tumor
regulation in BC was determined.
E-cadherin is an epithelial marker, the functional
loss of which is a hallmark of EMT, which is thought to promote BC
progression and metastasis (22).
MMP-2 is considered a mesenchymal marker of EMT (23). In the present study, CCL18 was
revealed to mediate EMT through MMP-2-dependent pathways.
MMPs are best known for their profound role in
malignant transformation and metastasis via extracellular matrix
disruption (24). MMP-2, which is
a member of the MMP family, is associated with tumor invasion
through degradation of the extracellular matrix and basement
membrane. In BC, high MMP2 expression is strongly correlated with
decreased survival (25). In
addition, MMP2 is overexpressed at the invasive front and is
associated with a higher tumor grade in BC (26).
Previous studies have reported that upregulation of
the lymphangiogenic growth factor VEGF-C is positively correlated
with regional lymph node metastasis and poor survival in BC
(27–32). VEGF-C is a protein precursor that
must be activated by a converting enzyme (proprotein convertases).
VEGF-receptor 3 (VEGF-R3) is one of the receptors of VEGF-C, which
promotes lymphangiogenesis, tumor cell migration, invasion and
metastasis. Blocking VEGF-C signaling via either small interfering
RNA or a VEGF-R3 antibody inhibits lymphatic-based metastatic
spread of human malignancies (33).
Immunosuppressive factors can be secreted into the
extracellular environment by tumor cells in an autocrine or
paracrine fashion, resulting in deep immunosuppressive regions that
are formed locally in tumor cells. Our previous study revealed that
VEGF-C, MMP-2 and MMP-9, as immunosuppressive factors, serve
important roles in BC invasion and metastasis (20). In the present study, it was
demonstrated that the expression levels of VEGF-C and MMP-2 were
elevated by CCL18 treatment. Furthermore, the mRNA expression
levels of CCL18 were assessed in BC tissues according to TCGA data,
and protein levels were detected by immunohistochemistry. The mRNA
and protein expression levels of CCL18 were higher compared with in
noncancerous bladder tissues. In the present cohort, CCL18
upregulation was associated with a high pathological grade.
Furthermore, it was revealed that rCCL18 stimulation significantly
enhanced the invasive potential of 5637 cells, and the CCL18
receptor, CCR8, mediated this activity in 5637 cells. Conversely, a
CCR8 inhibitor or CCR8 shRNA abrogated the decreased levels of
E-cadherin, and increased levels of MMP-2 and VEGF-C caused by
CCL18. These data indicated that CCR8 may be associated with the
oncogenic role of CCL18 in 5637 BC cells.
The CCR8 axis is associated with cancer progression
in BC, renal carcinoma (34) and
pancreatic cancer (35). R243 is a
novel small molecule CCR8 inhibitor, which inhibits the effects of
CCR8 in vivo and in vitro (16). CCR8 inhibition by R243 is able to
counteract the phenotypes induced by extracellular vesicles
decorated with CCL18 in glioblastoma cells (36). The present study demonstrated that
the CCR8 inhibitor, R243, abrogated the effects of CCL18 on BC
cells, thus suggesting a potential role for R243 in blocking the
CCL18/CCR8 axis during BC progression. Although R243 is able to
inhibit the effects of CCR8, the expression of CCR8 were unaffected
in present study. Additionally, due to very low signal levels in a
previous Biacore analysis, determination of intact R243 binding to
immobilized CCR8 failed (16).
Binding to the receptor or the site of action for R243 remains
unclear. Therefore, in this study, CCR8 shRNA was selected, rather
than R243, for use in cell migration and invasion experiments.
In conclusion, the present data offered convincing
evidence to suggest that upregulation of CCL18 may be involved in
the aggressive progression of BC partially through the
G-protein-coupled receptor CCR8. These results provided information
regarding the mechanisms underlying prevention of the progression
of BC by inhibiting CCL18 activity via blocking CCR8. However,
further studies are required to explore the detailed mechanisms
underlying the effects of CCL18 and CCR8 on the regulation of BC
progression.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of P.R. China (grant no. 81560419), the Natural
Science Foundation of Jiangxi (grant no. 20151BAB205047) and the
Jiangxi Province Infrastructure Facilities for Scientific Research
Institutes (grant no. 20142BBA13038).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XL, XX, WD, MH, YWu, ZZ and KZ performed the
experiments and generated data. XL, ZZ, YWang, XC, XZ, LC and YL
analyzed the data. GW and BF made substantial contributions to the
design of the experiments. XL, XX and BF wrote the manuscript. All
authors reviewed and approved the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Research
Ethics Committee of the First Affiliated Hospital, Nanchang
University (Nanchang, China). Informed consent was obtained from
all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prasad SM, Decastro GJ, Steinberg GD and
Medscape: Urothelial carcinoma of the bladder: Definition,
treatment and future efforts. Nat Rev Urol. 8:631–642. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chenivesse C and Tsicopoulos A:
CCL18-Beyond chemotaxis. Cytokine. 109:52–56. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang Q, Tang Y, Yu H, Yin Q, Li M, Shi L,
Zhang W, Li D and Li L: CCL18 from tumor-cells promotes epithelial
ovarian cancer metastasis via mTOR signaling pathway. Mol Carcinog.
55:1688–1699. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shi L, Zhang B, Sun X, Zhang X, Lv S, Li
H, Wang X, Zhao C, Zhang H, Xie X, et al: CC chemokine ligand
18(CCL18) promotes migration and invasion of lung cancer cells by
binding to Nir1 through Nir1-ELMO1/DOC180 signaling pathway. Mol
Carcinog. 55:2051–2062. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang H, Liang X, Li M, Tao X, Tai S, Fan
Z, Wang Z, Cheng B and Xia J: Chemokine (CC motif) ligand 18
upregulates Slug expression to promote stem-cell like features by
activating the mammalian target of rapamycin pathway in oral
squamous cell carcinoma. Cancer Sci. 108:1584–1593. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen J, Yao Y, Gong C, Yu F, Su S, Chen J,
Liu B, Deng H, Wang F, Lin L, et al: CCL18 from tumor-associated
macrophages promotes breast cancer metastasis via PITPNM3. Cancer
Cell. 19:541–555. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Urquidi V, Kim J, Chang M, Dai Y, Rosser
CJ and Goodison S: CCL18 in a multiplex urine-based assay for the
detection of bladder cancer. PLoS One. 7:e377972012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miyake M, Ross S, Lawton A, Chang M, Dai
Y, Mengual L, Alcaraz A, Giacoia EG, Goodison S and Rosser CJ:
Investigation of CCL18 and A1AT as potential urinary biomarkers for
bladder cancer detection. BMC Urol. 13:422013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
D'Ambrosio D, Iellem A, Bonecchi R, Mazzeo
D, Sozzani S, Mantovani A and Sinigaglia F: Selective up-regulation
of chemokine receptors CCR4 and CCR8 upon activation of polarized
human type 2 Th cells. J Immunol. 161:5111–5115. 1998.PubMed/NCBI
|
|
12
|
Roos RS, Loetscher M, Legler DF,
Clark-Lewis I, Baggiolini M and Moser B: Identification of CCR8,
the receptor for the human CC chemokine I-309. J Biol Chem.
272:17251–17254. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Islam SA, Ling MF, Leung J, Shreffler WG
and Luster AD: Identification of human CCR8 as a CCL18 receptor. J
Exp Med. 210:1889–1898. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Riester M, Taylor JM, Feifer A, Koppie T,
Rosenberg JE, Downey RJ, Bochner BH and Michor F: Combination of a
novel gene expression signature with a clinical nomogram improves
the prediction of survival in high-risk bladder cancer. Clin Cancer
Res. 18:1323–1333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oshio T, Kawashima R, Kawamura YI,
Hagiwara T, Mizutani N, Okada T, Otsubo T, Inagaki-Ohara K,
Matsukawa A, Haga T, et al: Chemokine receptor CCR8 is required for
lipopolysaccharide-triggered cytokine production in mouse
peritoneal macrophages. PLoS One. 9:e944452014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Leung SY, Yuen ST, Chu KM, Mathy JA, Li R,
Chan AS, Law S, Wong J, Chen X and So S: Expression profiling
identifies chemokine (C-C motif) ligand 18 as an independent
prognostic indicator in gastric cancer. Gastroenterology.
127:457–469. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mittal V: Epithelial mesenchymal
transition in tumor metastasis. Annu Rev Pathol. 13:395–412. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fu B, Wang Y, Zhang X, Lang B, Zhou X, Xu
X, Zeng T, Liu W, Zhang X, Guo J and Wang G: miR-221-induced PUMA
silencing mediates immune evasion of bladder cancer cells. Int J
Oncol. 46:1169–1180. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Catusse J, Wollner S, Leick M, Schröttner
P, Schraufstätter I and Burger M: Attenuation of CXCR4 responses by
CCL18 in acute lymphocytic leukemia B cells. J Cell Physiol.
225:792–800. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McConkey DJ, Choi W, Marquis L, Martin F,
Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, et al: Role
of epithelial-to-mesenchymal transition (EMT) in drug sensitivity
and metastasis in bladder cancer. Cancer Metastasis Rev.
28:335–344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Orlichenko LS and Radisky DC: Matrix
metalloproteinases stimulate epithelial-mesenchymal transition
during tumor development. Clin Exp Metastasis. 25:593–600. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Deryugina EI and Quigley JP: Matrix
metalloproteinases and tumor metastasis. Cancer Metastasis Rev.
25:9–34. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Davies B, Waxman J, Wasan H, Abel P,
Williams G, Krausz T, Neal D, Thomas D, Hanby A and Balkwill F:
Levels of matrix metalloproteases in bladder cancer correlate with
tumor grade and invasion. Cancer Res. 53:5365–5369. 1993.PubMed/NCBI
|
|
26
|
Kanayama HO, Yokota KY, Kurokawa Y,
Murakami Y, Nishitani M and Kagawa S: Prognostic values of matrix
metalloproteinase-2 and tissue inhibitor of metalloproteinase-2
expression in bladder cancer. Cancer. 82:1359–1366. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
He W, Zhong G, Jiang N, Wang B, Fan X,
Chen C, Chen X, Huang J and Lin T: Long noncoding RNA BLACAT2
promotes bladder cancer-associated lymphangiogenesis and lymphatic
metastasis. J Clin Invest. 128:861–875. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mandriota SJ, Jussila L, Jeltsch M,
Compagni A, Baetens D, Prevo R, Banerji S, Huarte J, Montesano R,
Jackson DG, et al: Vascular endothelial growth factor-C-mediated
lymphangiogenesis promotes tumour metastasis. EMBO J. 20:672–682.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Suzuki K, Morita T and Tokue A: Vascular
endothelial growth factor-C (VEGF-C) expression predicts lymph node
metastasis of transitional cell carcinoma of the bladder. Int J
Urol. 12:152–158. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miyata Y, Kanda S, Ohba K, Nomata K,
Hayashida Y, Eguchi J, Hayashi T and Kanetake H: Lymphangiogenesis
and angiogenesis in bladder cancer: Prognostic implications and
regulation by vascular endothelial growth factors-A, -C, and -D.
Clin Cancer Res. 12:800–806. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen JC, Chang YW, Hong CC, Yu YH and Su
JL: The role of the VEGF-C/VEGFRs axis in tumor progression and
therapy. Int J Mol Sci. 14:88–107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Keck B, Wach S, Taubert H, Zeiler S, Ott
OJ, Kunath F, Hartmann A, Bertz S, Weiss C, Hönscheid P, et al:
Neuropilin-2 and its ligand VEGF-C predict treatment response after
transurethral resection and radiochemotherapy in bladder cancer
patients. Int J Cancer. 136:443–451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rinderknecht M, Villa A, Ballmer-Hofer K,
Neri D and Detmar M: Phage-derived fully human monoclonal antibody
fragments to human vascular endothelial growth factor-c block its
interaction with VEGF receptor-2 and 3. PLoS One. 5:e119412010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Eruslanov E, Stoffs T, Kim WJ, Daurkin I,
Gilbert SM, Su LM, Vieweg J, Daaka Y and Kusmartsev S: Expansion of
CCR8(+) inflammatory myeloid cells in cancer patients with
urothelial and renal carcinomas. Clin Cancer Res. 19:1670–1680.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li D, Duell EJ, Yu K, Risch HA, Olson SH,
Kooperberg C, Wolpin BM, Jiao L, Dong X, Wheeler B, et al: Pathway
analysis of genome-wide association study data highlights
pancreatic development genes as susceptibility factors for
pancreatic cancer. Carcinogenesis. 33:1384–1390. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Berenguer J, Lagerweij T, Zhao XW, Dusoswa
S, van der Stoop P, Westerman B, de Gooijer MC, Zoetemelk M, Zomer
A, Crommentuijn MHW, et al: Glycosylated extracellular vesicles
released by glioblastoma cells are decorated by CCL18 allowing for
cellular uptake via chemokine receptor CCR8. J Extracell Vesicles.
7:14466602018. View Article : Google Scholar : PubMed/NCBI
|