Introduction
Abdominal aortic aneurysm (AAA) involves chronic
transmural inflammation and structural deterioration of the tissue
architecture, leading to a progressively enlarged abdominal aorta.
AAAs usually remain asymptomatic until a rupture occurs, which is
associated with high morbidity and mortality rates in the adult
human population (1,2), particularly in male patients over the
age of 65 years (3). Using an AAA
mouse model, it has been previously demonstrated that statins exert
beneficial effects on AAA progression by inhibiting endoplasmic
reticulum (ER) stress and C/EBP homologous protein (Chop)
expression (4). Another study
demonstrated that the selective inhibition of c-Jun N-terminal
kinase (JNK), also known as stress-activated protein kinase, not
only prevented the development of AAA, but also caused the
regression of established AAA (5).
Angiotensin-converting enzyme inhibitors and angiotensin II
receptor blockers (ARBs) may also reduce AAA mortality (6), but to the best of our knowledge,
there are no pharmacological methods that are able to efficiently
decrease the expansion or prevent the rupture of an AAA in humans
(7).
Activator protein 1 (AP-1) is a dimeric
transcription factor that controls gene expression, cell
proliferation, differentiation and apoptosis (8,9).
AP-1 consists of Jun (c-Jun, JunB and JunD) and Fos (c-Fos, FosB,
Fra1 and Fra2) family members. AP-1 proteins regulate target gene
expression by binding to specific DNA sequences (10,11).
Jun proteins are able to homo- and heterodimerize, whereas Fos
proteins are unable to homodimerize, but heterodimerize with Jun
proteins for DNA binding. Jun-Fos heterodimers bind preferentially
to a heptamer consensus sequence known as the TPA response element
(12). Activated JNK upregulates
c-Jun and facilitates c-Jun phosphorylation at ser63 and ser73.
Additionally, activated c-Jun is involved in the formation of AP-1
(13). Angiotensin II (Ang II), a
small peptide and principal component of the renin-angiotensin
system (14), may also activate
AP-1 by stimulating reactive oxygen species generation and
activating the JNK pathway (14,15).
C/EBP homologous protein (Chop), also termed
GADD153, is a transcription factor and a member of the basic
leucine zipper domain family (16), encoded by DNA damage-inducible
transcript 3 protein (Ddit3). Chop may be activated at multiple
levels during ER stress (17) and
is commonly used as an ER stress indicator. Chop overexpression
promotes apoptosis in several cell lines (18,19).
It has previously been demonstrated that Chop is overexpressed in
an Ang II-induced AAA mouse model (4). Another study revealed that Chop may
not only act as a regulator of C/EBP target genes, but may also be
tethered to AP-1 and thereby activate AP-1 target genes (20).
In the present study, the molecular mechanisms
underlying AAA development were evaluated with a focus on the roles
of c-Jun/AP-1 and Chop, and it was determined that c-Jun/AP-1 is
overexpressed in an Ang II-induced AAA model and Ang II-treated
mouse aortic smooth muscle cell line (MOVAS cells). Chop was
synchronously overexpressed; the regulation of Chop expression and
apoptosis by c-Jun/AP-1 in AAA was then investigated. c-Jun/AP-1
may have had an essential role as a transcriptional regulator of
Ddit3, resulting in Chop overexpression and the acceleration of AAA
development.
Materials and methods
Animals and AAA models
All animals were housed at the animal care facility
of Tongji Medical College, (Wuhan, China) under specific
pathogen-free conditions and were fed a normal diet. Mice were
housed in temperature-controlled cages (20±1°C, 55±5% humidity)
with a 12 h light-dark cycle and given free access to water and
food. All animal experiments were performed in accordance with the
Animal Research Reporting of in vivo Experiments and the
National Institutes of Health guidelines for animal welfare
(21), and the study was approved
by the Animal Research Committee of Tongji Medical College.
A total of 30 10-week-old apolipoprotein E-deficient
(ApoE−/−; C57BL/6 background) male mice (~20 g) were
purchased from Beijing Huafukang Biotechnology Co., Ltd. (Beijing,
China). Mice were randomly divided into 2 groups (n=15 per group)
and implanted with osmotic pumps (Model 2004; Durect Corporation,
Cupertino, CA, USA) containing either Ang II (1,000 ng/kg/min;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) or saline (sham) for
4 weeks (22). Mice were
anesthetized with isoflurane using an anesthesia machine prior to
surgery. After 4 weeks, animal aortic tissues and blood samples
were obtained following sacrifice, and samples were stored at −80°C
for western blotting and immunofluorescence.
Histological analysis
Aortas were fixed in 4% paraformaldehyde dissolved
in PBS at room temperature for 24 h and embedded in paraffin for
histological analyses. Serial cross-sections (3 µm) were produced
from the aorta. For the morphometric analysis, the sections were
stained with hematoxylin and eosin at room temperature. Briefly,
the sections were incubated with hematoxylin for 5–10 min following
rehydration, washed with 1% ethanol hydrochloride for 5 sec, and
stained with eosin for 3 min.
Immunofluorescence staining
The aorta specimens were prepared as described
above. The arterial sections were blocked in PBS with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) at room temperature for 1 h, and incubated overnight at
4°C with primary antibodies against c-Jun/Ap-1 (cat. no. 711202;
dilution, 1:200; Invitrogen; Thermo Fisher Scientific, Inc.) and
α-smooth muscle actin (α-SMA; cat. no. BM0002; dilution, 1:50;
Boster Biological Technology, Pleasanton, CA, USA). Antibodies were
replaced with secondary antibodies labelled with Alexa
Fluor® 568-conjugated goat anti-rabbit Immunoglobulin G
(IgG; cat. no. A-11034; dilution, 1:250; Invitrogen; Thermo Fisher
Scientific, Inc.) and Alexa Fluor® 488-conjugated goat
anti-mouse IgG (cat. no. A32727; dilution, 1:250; Invitrogen;
Thermo Fisher Scientific, Inc.). Cell nuclei were stained with DAPI
at room temperature for 20 min. Images were obtained using an
E2000U confocal microscope (magnification, ×200 and ×400; Nikon
Corporation, Tokyo, Japan) and were merged using Image Pro Plus
(version 6.0; Media Cybernetics, Rockville, MD, USA).
Cell culture and treatment
MOVAS cells were purchased from the American Type
Culture Collection (Manassas, VA, USA) and were cultured in
Dulbecco's Modified Eagle's medium (DMEM) containing 10% FBS (cat.
no. 10099-141; Gibco; Thermo Fisher Scientific, Inc.) in a
humidified atmosphere at 37°C with 5% CO2. MOVAS cells
were treated with various concentrations of Ang II (1, 10, 20, 50
and 100 nM) for 24 h. c-Jun small interfering RNA (siRNA), designed
and synthesized by Guangzhou RiboBio Co., Ltd. (Guangzhou, China),
was transfected at a final concentration of 50 nM using
Lipofectamine® 2000 (cat. no. 11668019; Invitrogen;
Thermo Fisher Scientific, Inc.) for 24 or 48 h prior to subsequent
experimentation, following the manufacturer's protocol. The
c-Jun-si1 target sequence was 5′-GCCAACTCATGCTAACGCA-3′, the
c-Jun-si2 target sequence was 5′-CAGCTTCCTGCCTTTGTAA-3′, and the
c-Jun-si3 target sequence was 5′-GCGCATGAGGAACCGCATT-3′.
Immunocytochemistry and confocal
imaging
MOVAS cells (4×105 cells/well) were
plated in 6-well glass slide chambers (Iwaki Glass, Tokyo, Japan)
and treated with (or without) Ang II (20 nM) for 36 h. Cells were
washed with PBS twice and fixed in 4% paraformaldehyde for 15 min
at room temperature. Following washing with PBS, cells were blocked
in fresh bovine serum albumin (BSA; cat. no. AR1006; Boster
Biological Technology) buffer (0.5% Triton X-100, 2% BSA, and 0.1%
Tween 20 dissolved in PBS) at room temperature for 1 h, and
incubated with the primary antibodies anti-c-Jun/AP-1, anti-Chop
(cat. no. GB11024; dilution, 1:100; Servicebio, Inc., Boston, MA,
USA) and anti-α-SMA, overnight at 4°C. Cells were treated with
secondary antibodies (goat anti-mouse IgG) and images were obtained
as aforementioned.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) assay
Apoptotic cells characterized by DNA fragmentation
in aortic tissues and MOVAS cells were determined by a TUNEL assay
using an In Situ Cell Death Detection kit (cat. no. 11684817910;
Roche Applied Science, Penzberg, Germany). First, aortic tissues
were fixed as aforementioned, dehydrated, embedded, sectioned (5-µm
thickness) and incubated with 0.9% NaCl for 10 min at room
temperature. Sections were washed twice with PBS, and then
incubated with biotinylated nucleotides and terminal
deoxynucleotidyl transferase at 37°C for 1 h, followed by
incubation with 50 µl TUNEL reaction mixture for 1 h at 37°C in a
dark and humidified room. Sections were then stained with DAPI (5
µg/ml) at room temperature for 20 min. Following four washes in
PBS, samples were mounted using anti-fade mounting medium (cat. no.
IH0252; Beijing Leagene Biotech Co., Ltd., Beijing, China) and
analyzed under a E2000U confocal microscope (4–7 fields were
randomly selected; magnifications, ×200 and 400). The staining
protocol of MOVAS cells was similar. Light green indicated normal
DNA (TUNEL-) and bright green indicated damaged DNA (TUNEL+).
Cell Counting Kit-8 (CCK-8) assay
MOVAS cells were seeded in 96-well plates (5,000
cells/well), and cell proliferation was monitored every 24 h (24,
48 and 72 h) following treatment with Ang II using the CCK-8 assay
(cat. no. CK04; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). Following the manufacturer's protocol, the absorbance was
read at 450 nm.
Monolayer wound healing assay
For the monolayer wound healing assay, MOVAS cells
(6×105 cells/well) were seeded in 6-well plates, and a
scratch was introduced on the cell layer after 24 h. A total of
three separate wounds were scratched using a 200 µl pipette tip,
and floating cells and cell debris were washed away with PBS. Fresh
medium with 10% FBS was added, along with 20 nM Ang II for the
experimental group, with continued cultured at 37°C. The wound was
imaged at 0 and 24 h, under a transmission microscope
(magnification, ×100), and the gap distances were measured.
Western blotting assay
Mouse aortas were used for protein extraction with
radioimmunoprecipitation assay lysis buffer (cat. no. P0013B;
Beyotime Institute of Biotechnology, Haimen, China), following
centrifugation at 12,000 × g for 20 min at 4°C. Protein
concentrations were determined using the bicinchoninic acid assay
kit (cat. no. WLA004a; Wanleibio, Co., Ltd., Shanghai, China),
according to the manufacturer's protocol. For western blotting,
cell lysates (~35 µg per lane) were resolved via 10% (wt/vol)
SDS-PAGE and transferred to a polyvinylidene difluoride membrane
(Merck KGaA). The membranes were blocked with 5% non-fat dry milk
in TBS-Tween 20 for 1.5 h at room temperature, and incubated
overnight at 4°C with primary antibodies against c-Jun/Ap-1 (cat.
no. 711202; dilution, 1:200; Invitrogen; Thermo Fisher Scientific,
Inc.), Chop (cat. no. mAb 2895; dilution 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA), GAPDH (cat. no. A00227-1;
dilution 1:100; Boster Biological Technology), and β-actin (cat.
no. BM0627; dilution 1:100; Boster Biological Technology). The
membranes were washed four times, and incubated with the
horseradish peroxidase-conjugated secondary antibody (cat. no.
111-035-003; dilution 1:8,000; Jackson Immuno Research
Laboratories, Inc., West Grove, PA, USA) in blocking buffer for 1 h
at room temperature. The bands were visualized using an enhanced
chemiluminescence kit (Pierce; Thermo Fisher Scientific, Inc.).
ImageJ 1.42q (National Institutes of Health, Bethesda, MD, USA) was
used for densitometry analyses.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
TRIzol® (cat. no. 10296028; Invitrogen;
Thermo Fisher Scientific, Inc.) was used to extract total RNA from
MOVAS cells following treatment. RNA samples were
reverse-transcribed into cDNA with different primers using the
First Strand Synthesis kit (RR036A; Takara Biotechnology Co., Ltd.,
Dalian, China), following the manufacturer's protocol. The RT
reaction was conducted at 37°C for 15 min and then at 85°C for 5
sec. cDNA was then stored at −20°C until use. qPCR was performed
using SYBR® Premix Ex Taq (RR820A; Takara Biotechnology
Co., Ltd.) and an ABI PRISM 7900HT Sequence Detection System
(Applied Biosystems; Thermo Fisher Scientific, Inc.). qPCR was
performed as follows: 95°C for 30 sec, and then 40 cycles of 95°C
for 5 sec and 60°C for 30 sec. Relative gene expression was
determined using the 2−∆∆Cq method (23). GAPDH was used as an internal
reference. The following primers were used: mouse c-Jun, forward
5′-GGGAGCATTTGGAGAGTCCC-3′, reverse 5′-TTTGCAAAAGTTCGCTCCCG-3′;
mouse Ddit3 forward 5′-CTGCCTTTCACCTTGGAGAC-3′, reverse
5′-CGTTTCCTGGGGATGAGATA-3′; mouse GAPDH, forward
5′-GGGAAATTCAACGGCACAGT-3′ and reverse 5′-AGATGGTGATGGGCTTCCC-3′.
All PCRs were performed in triplicate. All results were analyzed
using SDS Software (version 2.4; Applied Biosystems; Thermo Fisher
Scientific, Inc.).
Prediction of c-Jun/Ap-1 binding sites
in the mouse Ddit3 promoter
The mouse Ddit3 promoter region sequence (−1.09 kb
to 0 kb) was downloaded from the UCSC Genome Browser (http://genome.ucsc.edu/). Potential transcription
factors and binding sites were predicted using the Promoter 2.0
Prediction Server (http://www.cbs.dtu.dk/services/Promoter/) and JASPAR
database (http://jaspar.genereg.net/).
Chromatin immunoprecipitation (ChIP)
assay
A ChIP kit was purchased from Beyotime Institute of
Biotechnology (cat. no. P2078), and ChIP assay was conducted
following the manufacturer's protocol. MOVAS cells were
cross-linked (40% methanol solution, 37°C for 15 min) and then
sonicated (0°C, 50% power with 4 cycles of 5 sec on, 5 sec off)
when they reached 90% confluency in 10 cm cell culture dishes,
followed by IP with a polyclonal anti-c-Jun/Ap-1 antibody (cat. no.
711202; Invitrogen; Thermo Fisher Scientific, Inc.). Normal IgG
(cat. no. ab172730; Abcam, Cambridge, UK) was used as a negative
control. The supernatant was used as an input control. Precipitated
DNA was amplified by PCR using Ddit3-specific primers (forward,
5′-CTGAGTGGCGGATGTAAGGG-3′; reverse, 5′-GGTCCAGGAGCCTACCAATC-3′).
PCR products (74 bp) were analyzed by 2% agarose gel (cat. no.
5261; Takara Biotechnology Co., Ltd.) electrophoresis and
visualized using GoldView™ (cat. no. G8140; Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China) under an
ultraviolet light.
Statistical analyses
All results are presented as the mean ± standard
error of the mean from three independent experiments. Statistical
differences were evaluated by one-way analysis of variance followed
by a Tukey's comparison test. Statistical significance was
evaluated using GraphPad Prism (version 5.0; GraphPad Software
Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
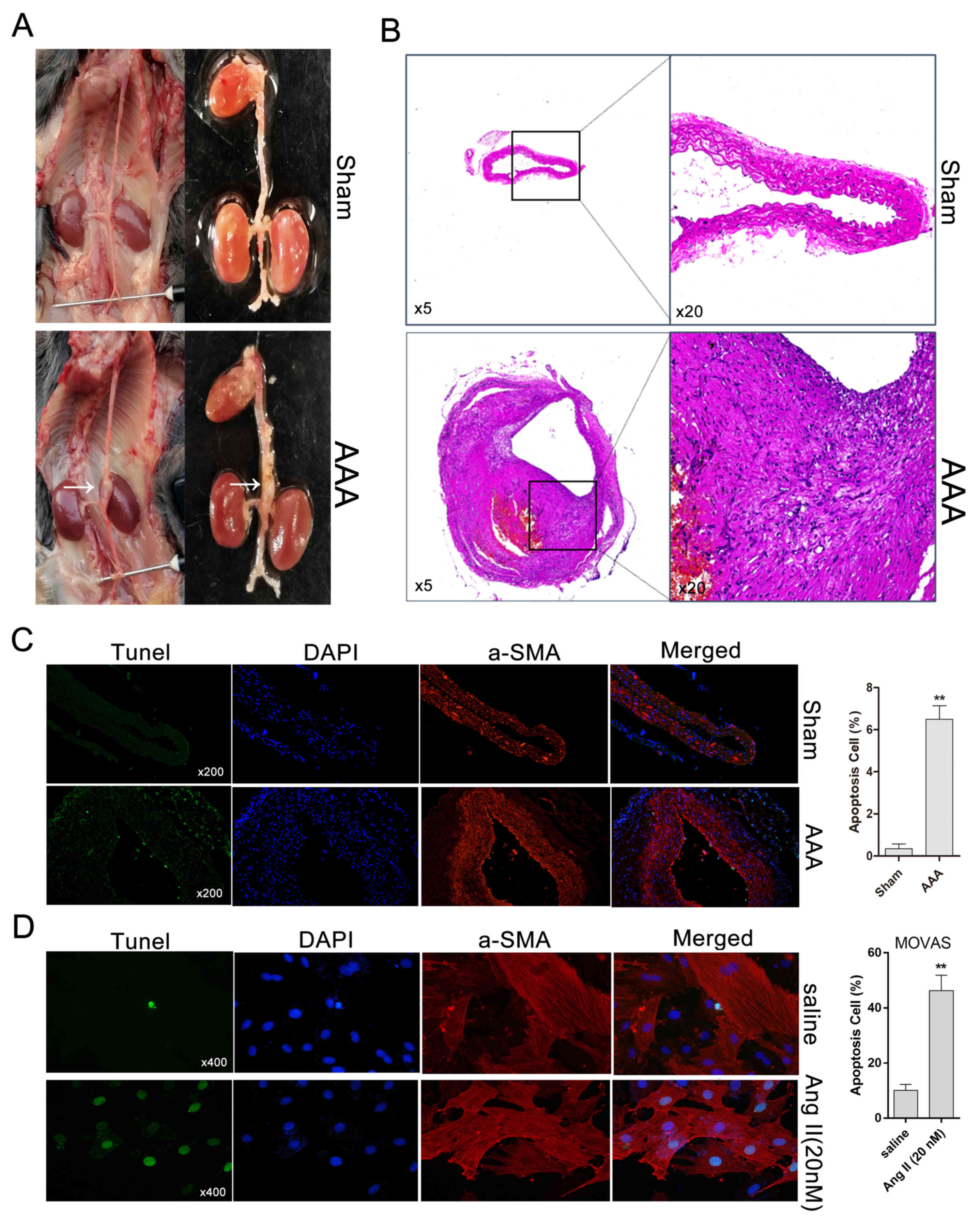
Abdominal aortic aneurysm was induced
by Ang II in ApoE−/− mice
After 4 weeks of Ang II treatment, AAA developed in
ApoE−/− mice but not in saline-infused
ApoE−/− mice, which exhibited normal aortas (Fig. 1A). Histologically, compared with
control aortas, Ang II-induced AAA was characterized by expansion
and thickening of the aortic wall, and the cells of the aortic wall
exhibited a disordered arrangement (Fig. 1B). Subsequently, apoptosis in Ang
II-induced AAA and Ang II-treated MOVAS cells was evaluated by
TUNEL staining. Compared with the sham group (saline-treated), the
Ang II-treated AAA group exhibited an increase in the percentage of
TUNEL+ cells (Fig. 1C). The
percentage of apoptotic MOVAS cells with DNA fragmentation under
Ang II treatment was significantly higher compared with that for
saline-treated cells (Fig.
1D).
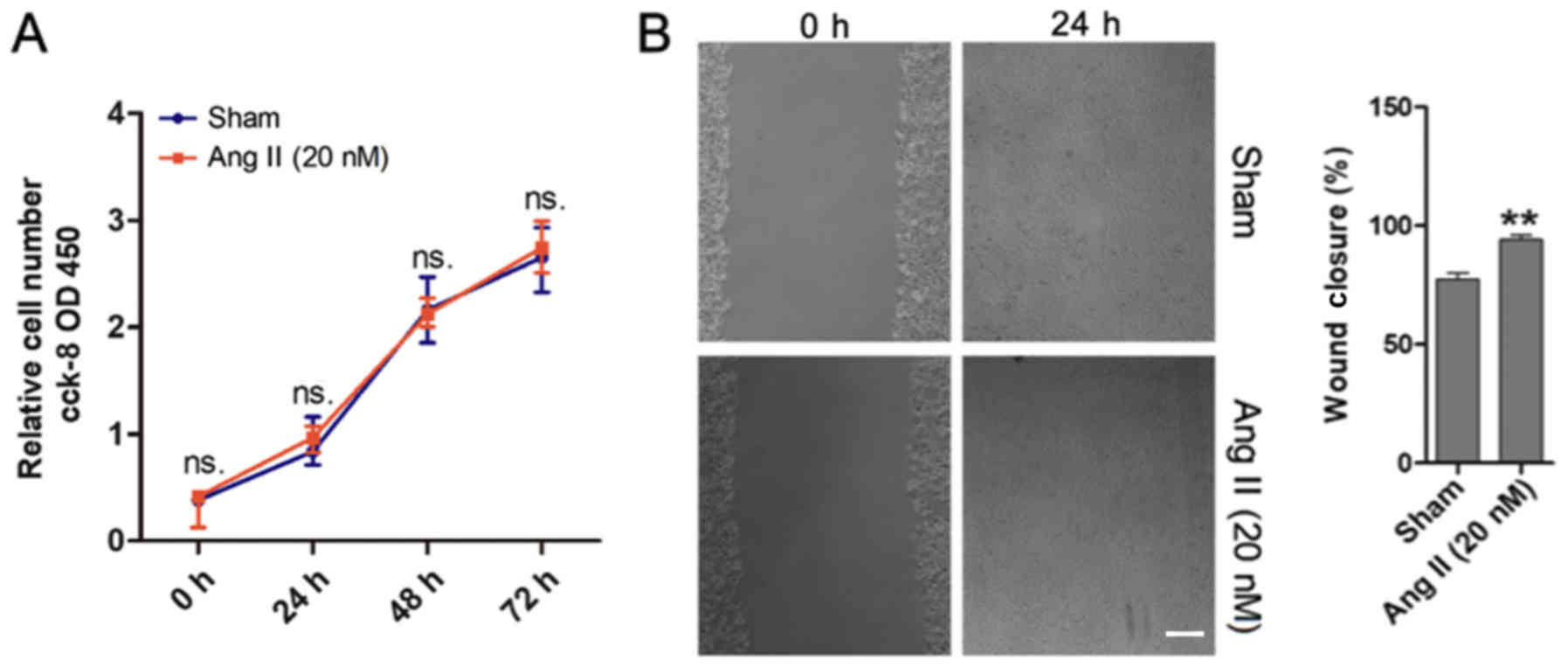
Ang II affects MOVAS cell migration in
vitro
To investigate the role of Ang II in the cell
proliferation and migration of MOVAS cells, CCK-8 and monolayer
wound healing assays were performed. The results demonstrated that
Ang II did not affect MOVAS cell proliferation (Fig. 2A). However, the monolayer wound
healing assay revealed that Ang II promoted MOVAS cell migration
(Fig. 2B), and this was in
agreement with the results from Greene et al (24).
c-Jun and Chop were simultaneously
upregulated in vivo and in vitro in response to Ang II
To investigate the expression of c-Jun during Ang
II-induced AAA formation, immunofluorescence was used. c-Jun levels
were observed to be higher in Ang II-induced AAA compared with
normal aortas (Fig. 3A). Western
blotting also demonstrated that Chop was markedly upregulated
(Fig. 3B), consistent with the
results of a previous study (4).
To examine c-Jun and Chop expression levels in MOVAS cells treated
with Ang II, cells were co-stained with α-SMA and either c-Jun or
Chop. c-Jun and Chop expression were increased following treatment
with Ang II. Western blotting revealed that c-Jun and Chop protein
levels increased in response to Ang II in a dose-dependent manner
following treatment with 0–50 nM Ang II; however, following
treatment with 100 nM Ang II, the expression levels of c-Jun and
Chop markedly decreased compared with 20 nM Ang II (Fig. 3C). These results were confirmed by
immunofluorescence, which demonstrated that c-Jun was elevated and
was primarily localized in the nuclei of MOVAS cells (Fig. 3D). Furthermore, Chop was also
observed to be localized in the nuclei and cytoplasm of MOVAS cells
(Fig. 3E).
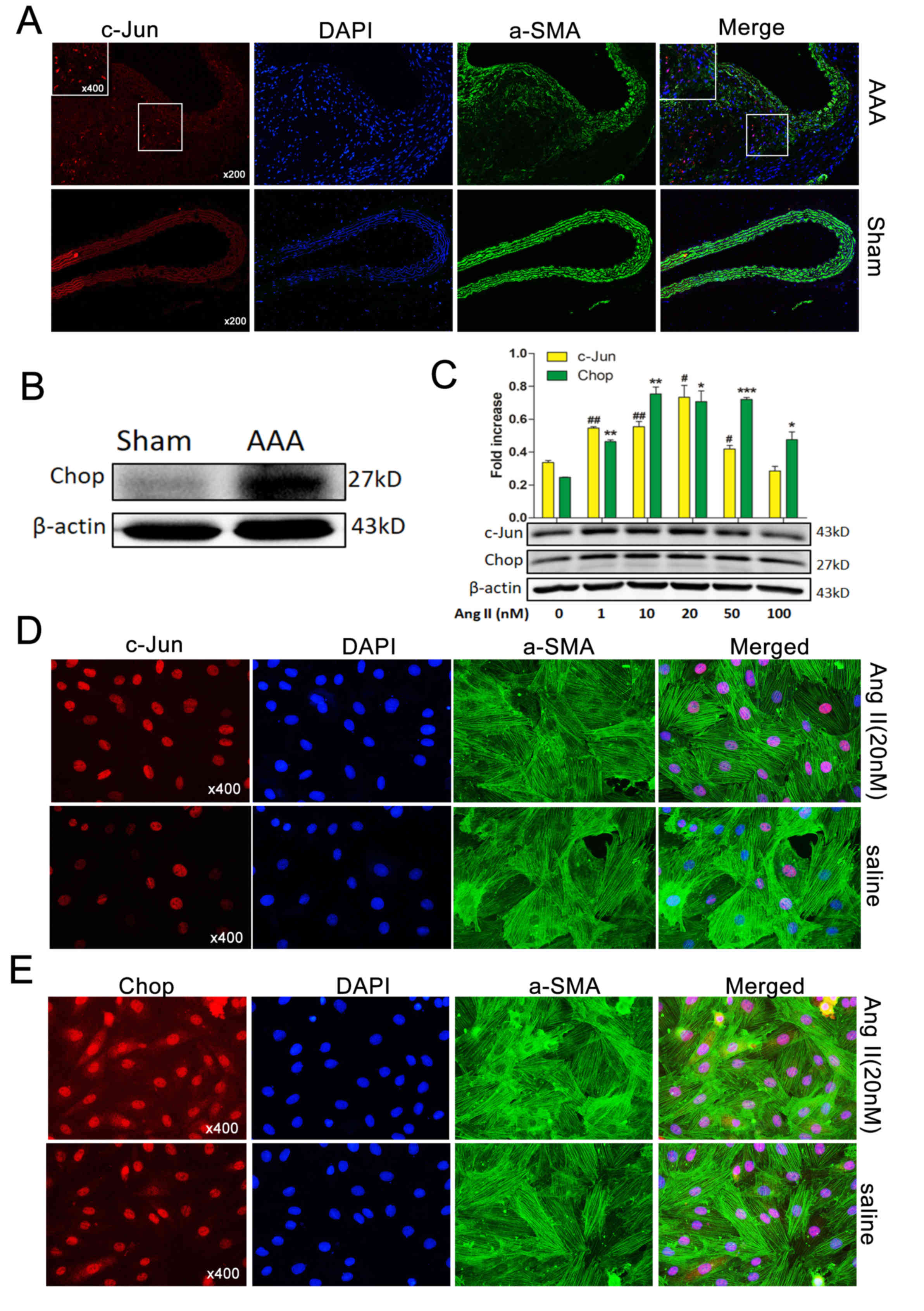 | Figure 3.Ap-1 and Chop are induced by Ang II
in vivo and in vitro. (A) Immunofluorescence staining
with c-Jun/Ap-1 (red) expression and localization in the aortas of
Ang II-induced AAA mice and normal aortas. α-SMA, green; DAPI,
blue. Magnifications, ×200 and 400. (B) Western blotting exhibiting
Chop expression in normal aortas (control) and Ang II-induced AAA
in ApoE−/− mice. β-Actin was used as an internal
control. (C) Western blotting demonstrated that c-Jun/Ap-1 and Chop
protein levels in MOVAS cells were induced in a dose dependent
manner by varying concentrations (0, 1, 10, 20 and 50 nM) of Ang II
for 36 h. Immunofluorescence staining demonstrating (D) c-Jun/Ap-1
(red) and (E) Chop (red) expression and localization in MOVAS cells
induced (or not) by Ang II (20 nM) for 36 h. α-SMA (green), DAPI
(blue). Magnification, ×400. Results are presented as the mean ±
standard error of the mean; n=3. #P<0.05, *P<0.05,
##P<0.01, **P<0.01 and ***P<0.001 vs. Control
(0 nM). Ap-1, activator protein 1; Chop, C/EBP homologous protein;
Ang II, angiotensin II; α-SMA, α-smooth muscle actin AAA, abdominal
aortic aneurysm; ApoE−/−, apolipoprotein
E-deficient. |
Chop expression was suppressed by
siRNA-mediated c-Jun silencing
Based on the aforementioned results, it was
hypothesized that there may be underlying connections between
c-Jun/Ap-1 and Ddit3. AP-1 is a transcription factor consisting of
a dimer of c-Jun and c-Fos. However, whether Ddit3 is activated by
Ap-1 is unknown. Following this line of thought, c-Jun was
downregulated using c-Jun-targeted siRNAs and the expression of
Chop was assessed. RT-qPCR revealed that c-Jun transcript levels
were reduced by >60% at 24 h post-transfection with c-Jun-si2
and c-Jun-si3 [P<0.001 vs. negative control (NC)-si], but
c-Jun-si1 had no effect (P>0.05 vs. NC-si). GAPDH was used as an
internal control (Fig. 4A).
Western blotting revealed that the protein levels of c-Jun were
also decreased following transfection with c-Jun-si2 and c-Jun-si3
for 48 h in MOVAS cells (P<0.05 vs. NC-si) but not with
c-Jun-si1 (P>0.05 vs. NC-si), also using GAPDH as an internal
control (Fig. 4B). MOVAS cells
were transfected with effective siRNAs (c-Jun-si2 and c-Jun-si3)
for 24 h, and the transfected cells were incubated with Ang II (20
nM) for 24 h prior to mRNA or protein assessments. RT-qPCR
subsequently demonstrated that the mRNA expression levels of Ddit3
were significantly reduced (P<0.05 vs. NC-si) (Fig. 4C), and western blotting revealed
that c-Jun and Chop protein levels were also reduced following
transfection with c-Jun-si2 and c-Jun-si3 (P<0.05 vs. NC-si).
β-actin was used as an internal control (Fig. 4D). These results indicated that
c-Jun/AP-1 mediates Chop expression in MOVAS cells.
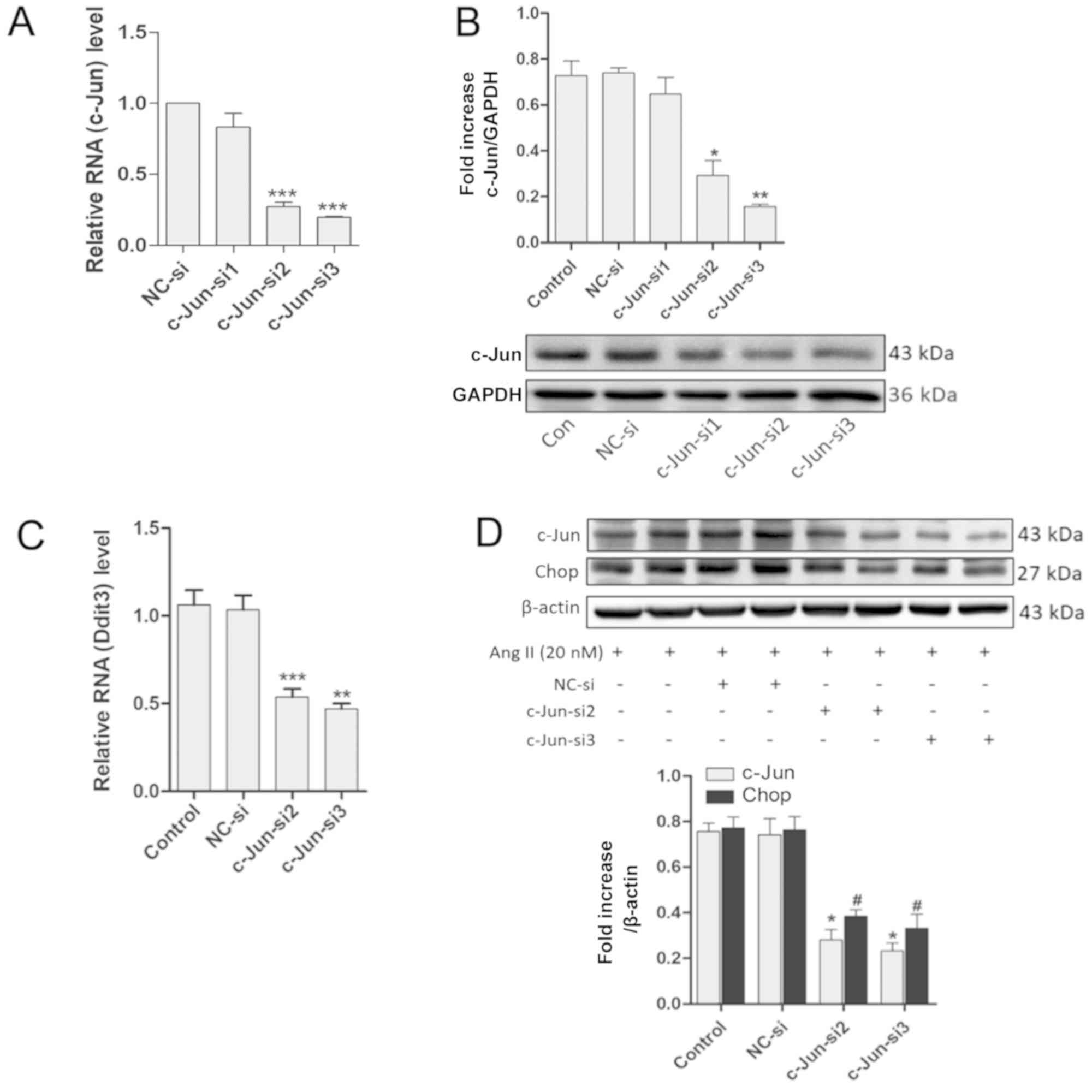 | Figure 4.Downregulation of c-Jun/Ap-1 by
c-Jun-siRNA suppresses Chop expression. (A) mRNA levels of c-Jun
were assessed by RT-qPCR in MOVAS cells transfected with siRNAs for
24 h; GAPDH was used as an internal control (n≥3). (B) Western
blotting demonstrated the Ap-1 levels in MOVAS cells transfected
with siRNAs for 48 h; GAPDH was used as an internal control (n=3).
MOVAS cells were transfected with siRNAs for 24 h and the
transfected cells were treated with Ang II (20 nM) for 24 h. (C)
The mRNA expression level of Ddit3 were detected by RT-qPCR; GAPDH
was used as an internal control (n≥3). (D) Protein levels were
detected by western blotting with anti-c-Jun, anti-Chop, and
anti-β-actin (internal control) antibodies (n=3). All results are
presented as the mean ± standard error of the mean.
#P<0.05; *P<0.05, **P<0.01, ***P<0.001 vs.
NC-si. Ap-1, activator protein 1; Chop, C/EBP homologous protein;
RT-qPCR, reverse transcription quantitative-polymerase chain
reaction; siRNA, small interfering RNA; Ang II, angiotensin II;
NC-si, negative control siRNA; Ddit3, DNA damage-inducible
transcript 3 protein. |
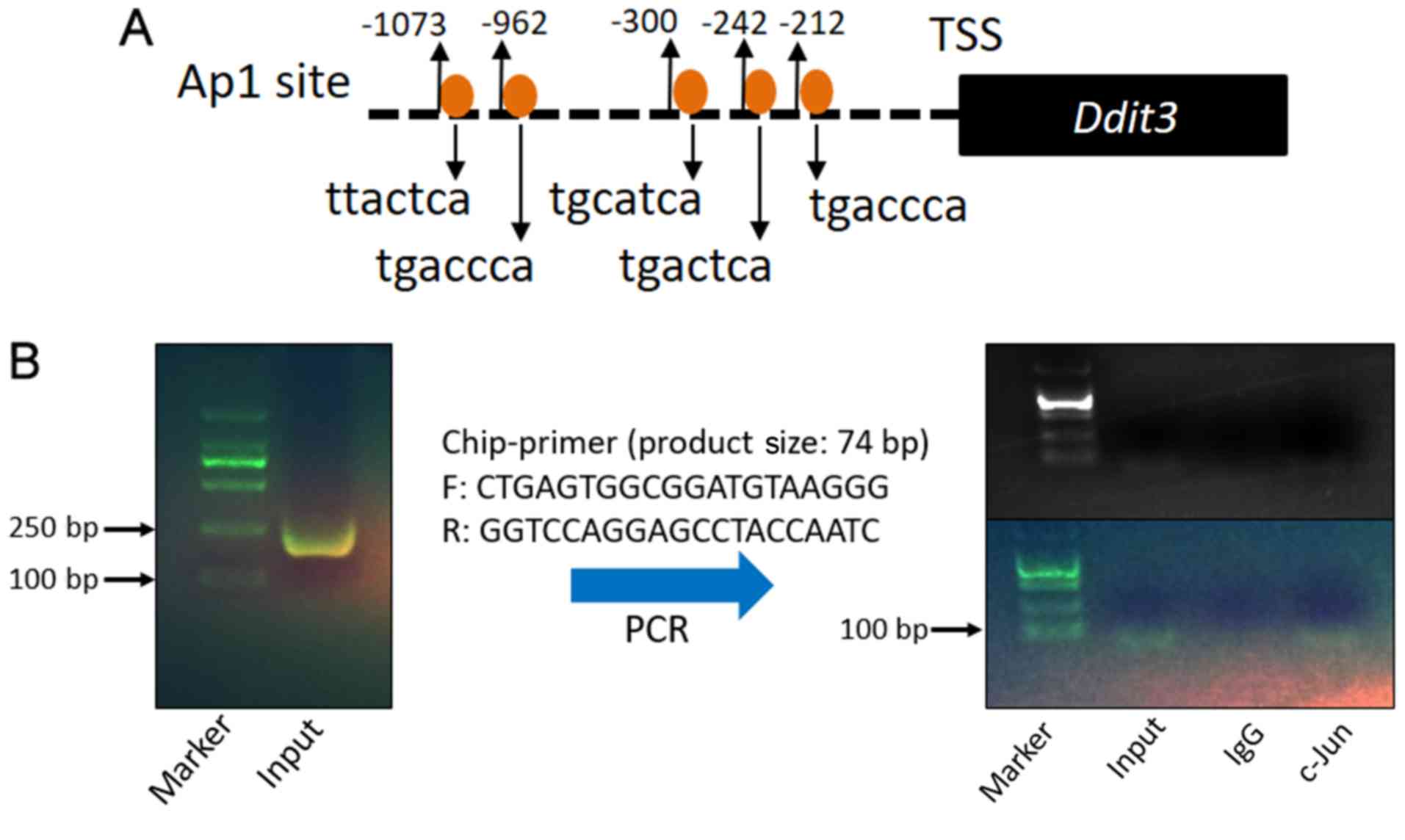
c-Jun/Ap-1 binds to the mouse promoter
region in MOVAS cells
To further evaluate the aforementioned hypothesis,
the promoter sequence of mouse Ddit3 (encoding Chop) was evaluated
using online databases (−1.09 kb to 0 kb). The sequence analysis
suggested five potential c-Jun/Ap-1 binding sites in the Ddit3
promoter region (Fig. 5A). ChIP
assays were performed to further test whether AP-1 binds to the
Ddit3 promoter in MOVAS cells. DNA was crushed into fragments
(100–250 bp) following sonication and amplified by ChIP-PCR using
Ddit3 promoter-specific primers. The DNA fragments pulled down by
anti-c-Jun/AP-1 primary antibodies were amplified by Ddit3
promoter-specific primers (Fig.
5B), indicating that c-Jun/Ap-1 binds directly to the Ddit3
promoter and may regulate its expression.
Discussion
Previous evidence suggests that Ang II serves
important roles in cardiovascular homeostasis by directly
regulating ER signaling and upregulating Chop expression (4), the latter being involved in a number
of diseases, including atherosclerosis (25), hypertension (26) and AAA (4,27).
Furthermore, Chop is implicated in the programmed cell death
pathway during ER stress (18,19,28),
and apoptosis in vascular smooth muscle cells (VSMCs) is associated
with AAA formation, and may contribute to arterial wall thinning,
expansion and eventual rupture (4,27).
Previous studies have indicated that Ang II may promote ER stress
signaling via eukaryotic translation initiation factor 2-α kinase 3
and the phospho-eukaryotic translation initiation factor
2α-mediated upregulation of cyclic AMP-dependent transcription
factor ATF4 (29,30), a transcription factor of Ddit3, and
may lead to the expression of Chop. Consistent with these
observations, Ang II was also demonstrated to stimulate
intracellular reactive oxygen species generation (14,31),
and to upregulate c-Jun/AP-1 expression by activating the
mitogen-activated protein kinase-JNK pathway (32). Therefore, it was hypothesized that
c-Jun/AP-1 may cause Chop overexpression and may indirectly induce
apoptosis, thus accelerating the development of AAA.
A number of AAA induction methods in animal models
has been developed, and three approaches are frequently used by
researchers, including the porcine pancreatic elastase model
(33), the calcium chloride model
(34) and the Ang II model
(35). Notably, Ang II induced AAA
in ApoE−/− mice is the most widely used AAA animal model
among the three methods, and it has a number of features similar to
the human AAA disease, including progressive abdominal aorta
dilation, diffuse inflammation, VSMC apoptosis and extracellular
matrix deformation (36). In the
present study this latter model was adopted, and ApoE−/−
mice were treated with Ang II (1,000 ng/kg/min) for 4 weeks
(24). Based on the adverse
effects encountered in a previous study (4), 30 ApoE−/− mice were
randomly and equally assigned to Ang II or saline treatment groups,
which were delivered via osmotic minipumps. In vivo,
apoptosis was induced in the Ang II-induced AAA model, and the
TUNEL-positive signal seemed to be localized in the adventitia,
since AAA involves structural deterioration of the tissue
architecture, leading to a progressively enlarged abdominal aorta.
Concurrently, the tunica media became loose, and resident VSMCs and
infiltrating macrophages have been demonstrated to release matrix
metalloproteinases (37), which
increase VSMC migration from the tunica media to the adventitia,
which may explain the large number of VSMCs observed in the
adventitia following AAA. Furthermore, the migration ability and
apoptosis of MOVAS cells treated with Ang II were increased. Thus,
combining in vivo and in vitro data, it may be that
increased VSMC apoptosis and migration lead to AAA (38,39).
c-Jun/AP-1 and Chop were also observed to be
upregulated in Ang II-induced AAA and Ang II-treated MOVAS cells.
Additionally, the present results demonstrated that the increase in
c-Jun and Chop expression levels in response to Ang II was
dose-dependent, with c-Jun and Chop expression levels decreasing at
higher doses (100 nM). A possible interpretation is that Ang II may
induce ER stress, intracellular reactive oxygen species generation
and inflammation, leading to cell apoptosis. A previous study
demonstrated that VSMC cell apoptosis was directly dependent on Ang
II concentration, and that 50 nM Ang II may induce 25–30% cell
apoptosis; 100 nM Ang II led to cell death without activation of an
apoptotic pathway (4). In
addition, the present study indicated that Chop was suppressed when
c-Jun was silenced. A previous study revealed that Ang II may
generate a consistent and substantial amount of reactive oxygen
species, leading to a long-lasting activation of AP-1 (14). Activated AP-1 directly controls
gene expression and indirectly regulates cell functions, including
cell proliferation, migration, differentiation and apoptosis
(8,9). The present results revealed that AP-1
may be involved in the expression of Chop, and may therefore affect
cell apoptosis and aortic tissue remodeling. Furthermore, a ChIP
assay demonstrated that c-Jun/Ap-1 may bind to a consensus sequence
of the mouse Ddit3 promoter region, suggesting that c-Jun/AP-1 may
be a transcriptional regulator of Ddit3. In addition, a previous
study indicated that the selective inhibition of JNK not only
prevented the development of AAA, but also caused its regression in
Ang II-induced AAA models by ameliorating the extracellular matrix
metabolism and enhancing aortic tissue repair (5). Unfortunately, the link between this
amelioration and Chop expression was not examined.
The present work had certain limitations. Firstly,
the focus of the study was to determine whether c-Jun/AP-1 may
mediate Chop expression. In the in vitro assays the impact
of ATF4 signaling on ER stress was not assessed; siRNA-mediated
downregulation of ATF4 would have been useful as a negative control
for the evaluation of Chop expression and for controlling the
impact of ER stress. Secondly, to further evaluate the hypothesis
that c-Jun/AP-1 binds to the proximal promoter of the Ddit3 gene in
MOVAS cells increasing Chop expression, the upregulation of c-Jun,
in addition to its downregulation, could have been evaluated with
respect to Chop expression. Ang II-treated and c-Jun-overexpressing
cells could have been analyzed with respect to c-Jun binding to the
Ddit3 promoter. Thirdly, in order to further prove that Ap-1 is a
Ddit3 transcription factor, more group comparisons of ChIP assays
are required. c-Jun knockdown and/or overexpression may confirm
whether Ap-1 is a transcription factor of Ddit3, and whether
treatment with Ang II promotes Ap-1 binding to the Ddit3 promoter
in MOVAS cells. Thus, additional studies are required to determine
the precise role of c-Jun/AP-1 in AAA, and to confirm whether
c-Jun/AP-1 causes Chop overexpression, thereby accelerating AAA
development.
In conclusion, the present study demonstrated that
c-Jun/AP-1 was overexpressed in an Ang II-induced AAA model and in
Ang II-treated MOVAS cells, and that it mediated the expression of
Chop. Therefore, c-Jun/AP-1 may be a novel target for AAA
therapy.
Acknowledgements
Not applicable.
Funding
This study was funded by research grants from The
National Natural Science Foundation of China (grant no. 81370417)
and Hubei Natural Science Foundation Project (grant no.
2018CFB465).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
JY and MZ were involved in the conception and
supervision of the project. JY and DS were involved in the design
of the study. DS performed the experiments. DS, YL and JQ analyzed
the results. DS and SM performed and analyzed the CCK-8 and
migration assays. DS, YL and JQ prepared the paper. All the authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were performed in accordance
with the Animal Research Reporting of in vivo Experiments
and the National Institutes of Health guidelines for animal
welfare, and the study was approved by the Animal Research
Committee of Tongji Medical College (Wuhan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AAA
|
abdominal aortic aneurysm
|
|
Ang II
|
angiotensin II
|
|
Ap-1
|
activator protein-1
|
|
Chop
|
C/EBP homologous protein
|
|
ApoE−/−
|
apolipoprotein E-deficient
|
|
ChIP
|
chromatin immunoprecipitation
|
References
|
1
|
Wassef M, Upchurch GR Jr, Kuivaniemi H,
Thompson RW and Tilson MD III: Challenges and opportunities in
abdominal aortic aneurysm research. J Vasc Surg. 45:192–198. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lu H, Rateri DL, Bruemmer D, Cassis LA and
Daugherty A: Novel mechanisms of abdominal aortic aneurysms. Curr
Atheroscler Rep. 14:402–412. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Svensjö S, Björck M and Wanhainen A:
Editor's choice: Five-year outcomes in men screened for abdominal
aortic aneurysm at 65 years of age: A population-based cohort
study. Eur J Vasc Endovasc Surg. 47:37–44. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li Y, Lu G, Sun D, Zuo H, Wang DW and Yan
J: Inhibition of endoplasmic reticulum stress signaling pathway: A
new mechanism of statins to suppress the development of abdominal
aortic aneurysm. PLoS One. 12:e01748212017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoshimura K, Aoki H, Ikeda Y, Fujii K,
Akiyama N, Furutani A, Hoshii Y, Tanaka N, Ricci R, Ishihara T, et
al: Regression of abdominal aortic aneurysm by inhibition of c-Jun
N-terminal kinase. Nat Med. 11:1330–1338. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kristensen KE, Torp-Pedersen C, Gislason
GH, Egfjord M, Rasmussen HB and Hansen PR: Angiotensin-converting
enzyme inhibitors and angiotensin II receptor blockers in patients
with abdominal aortic aneurysms: Nation-wide cohort study.
Arterioscler Thromb Vasc Biol. 35:733–740. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kent KC: Clinical practice. Abdominal
aortic aneurysms. N Engl J Med. 371:2101–2108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shaulian E and Karin M: AP-1 as a
regulator of cell life and death. Nat Cell Biol. 4:E131–E136. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wisdom R, Johnson RS and Moore C: c-Jun
regulates cell cycle progression and apoptosis by distinct
mechanisms. EMBO J. 18:188–197. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shaulian E: AP-1-The Jun proteins:
Oncogenes or tumor suppressors in disguise? Cell Signal.
22:894–899. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hess J, Angel P and Schorpp-Kistner M:
AP-1 subunits: Quarrel and harmony among siblings. J Cell Sci.
117:5965–5973. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chinenov Y and Kerppola TK: Close
encounters of many kinds: Fos-Jun interactions that mediate
transcription regulatory specificity. Oncogene. 20:2438–2452. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Plotnikov A, Zehorai E, Procaccia S and
Seger R: The MAPK cascades: Signaling components, nuclear roles and
mechanisms of nuclear translocation. Biochim Biophys Acta.
1813:1619–1633. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu S, Gao J, Ohlemeyer C, Roos D, Niessen
H, Köttgen E and Gessner R: Activation of AP-1 through reactive
oxygen species by angiotensin II in rat cardiomyocytes. Free Radic
Biol Med. 39:1601–1610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Frank GD, Eguchi S, Yamakawa T, Tanaka S,
Inagami T and Motley ED: Involvement of reactive oxygen species in
the activation of tyrosine kinase and extracellular
signal-regulated kinase by angiotensin II. Endocrinology.
141:3120–3126. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Friedman AD: GADD153/CHOP, a DNA
damage-inducible protein, reduced CAAT/enhancer binding protein
activities and increased apoptosis in 32D c13 myeloid cells. Cancer
Res. 56:3250–3256. 1996.PubMed/NCBI
|
|
17
|
Hotamisligil GS: Endoplasmic reticulum
stress and atherosclerosis. Nat Med. 16:396–399. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim I, Xu W and Reed JC: Cell death and
endoplasmic reticulum stress: Disease relevance and therapeutic
opportunities. Nat Rev Drug Discov. 7:1013–1030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ubeda M, Vallejo M and Habener JF: CHOP
enhancement of gene transcription by interactions with Jun/Fos AP-1
complex proteins. Mol Cell Biol. 19:7589–7599. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guide for the Care and Use of Laboratory
Animals, . NIH Publication No. pp. 85–23. National Institutes of
Health; Bethesda, MD: 1996
|
|
22
|
Daugherty A, Manning MW and Cassis LA:
Angiotensin II promotes atherosclerotic lesions and aneurysms in
apolipoprotein E-deficient mice. J Clin Invest. 105:1605–1612.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Greene EL, Lu G, Zhang D and Egan BM:
Signaling events mediating the additive effects of oleic acid and
angiotensin II on vascular smooth muscle cell migration.
Hypertension. 37:308–312. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cominacini L, Garbin U, Mozzini C,
Stranieri C, Pasini A, Solani E, Tinelli IA and Pasini AF: The
atherosclerotic plaque vulnerability: Focus on the oxidative and
endoplasmic reticulum stress in orchestrating the macrophage
apoptosis in the formation of the necrotic core. Curr Med Chem.
22:1565–1572. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun Y, Zhang T, Li L and Wang J: Induction
of apoptosis by hypertension via endoplasmic reticulum stress.
Kidney Blood Press Res. 40:41–51. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qin Y, Wang Y, Liu O, Jia L, Fang W, Du J
and Wei Y: Tauroursodeoxycholic acid attenuates angiotensin ii
induced abdominal aortic aneurysm formation in apolipoprotein
E-deficient mice by inhibiting endoplasmic reticulum stress. Eur J
Vasc Endovasc Surg. 53:337–345. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zinszner H, Kuroda M, Wang X, Batchvarova
N, Lightfoot RT, Remotti H, Stevens JL and Ron D: CHOP is
implicated in programmed cell death in response to impaired
function of the endoplasmic reticulum. Genes Dev. 12:982–995. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Harding HP, Novoa I, Zhang Y, Zeng H, Wek
R, Schapira M and Ron D: Regulated translation initiation controls
stress-induced gene expression in mammalian cells. Mol Cell.
6:1099–1108. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vattem KM and Wek RC: Reinitiation
involving upstream ORFs regulates ATF4 mRNA translation in
mammalian cells. Proc Natl Acad Sci USA. 101:11269–11274. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dai DF, Johnson SC, Villarin JJ, Chin MT,
Nieves-Cintrón M, Chen T, Marcinek DJ, Dorn GW II, Kang YJ, Prolla
TA, et al: Mitochondrial oxidative stress mediates angiotensin
II-induced cardiac hypertrophy and Galphaq overexpression-induced
heart failure. Circ Res. 108:837–846. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chiu CZ, Wang BW and Shyu KG: Angiotensin
II and the JNK pathway mediate urotensin II expression in response
to hypoxia in rat cardiomyocytes. J Endocrinol. 220:233–246. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pyo R, Lee JK, Shipley JM, Curci JA, Mao
D, Ziporin SJ, Ennis TL, Shapiro SD, Senior RM and Thompson RW:
Targeted gene disruption of matrix metalloproteinase-9 (gelatinase
B) suppresses development of experimental abdominal aortic
aneurysms. J Clin Invest. 105:1641–1649. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bi Y, Zhong H, Xu K, Zhang Z, Qi X, Xia Y
and Ren L: Development of a novel rabbit model of abdominal aortic
aneurysm via a combination of periaortic calcium chloride and
elastase incubation. PLoS One. 8:e684762013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ghoshal S and Loftin CD: Cyclooxygenase-2
inhibition attenuates abdominal aortic aneurysm progression in
hyperlipidemic mice. PLoS One. 7:e443692012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Trachet B, Fraga-Silva RA, Piersigilli A,
Tedgui A, Sordet-Dessimoz J, Astolfo A, Van der Donckt C, Modregger
P, Stampanoni MF, Segers P and Stergiopulos N: Dissecting abdominal
aortic aneurysm in Ang II-infused mice: Suprarenal branch ruptures
and apparent luminal dilatation. Cardiovasc Res. 105:213–222. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Browatzki M, Larsen D, Pfeiffer CA, Gehrke
SG, Schmidt J, Kranzhofer A, Katus HA and Kranzhofer R: Angiotensin
II stimulates matrix metalloproteinase secretion in human vascular
smooth muscle cells via nuclear factor-kappaB and activator protein
1 in a redox-sensitive manner. J Vasc Res. 42:415–423. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schubl S, Tsai S, Ryer EJ, Wang C, Hu J,
Kent KC and Liu B: Upregulation of protein kinase cdelta in
vascular smooth muscle cells promotes inflammation in abdominal
aortic aneurysm. J Surg Res. 153:181–187. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cai Z, Zhao G, Yan J, Liu W, Feng W, Ma B,
Yang L, Wang JA, Tu L and Wang DW: CYP2J2 overexpression increases
EETs and protects against angiotensin II-induced abdominal aortic
aneurysm in mice. J Lipid Res. 54:1448–1456. 2013. View Article : Google Scholar : PubMed/NCBI
|