Introduction
Hypertension is a worldwide epidemic and global
health problem, and is one of the most important risk factors for
cardiovascular disease events, which are a major cause of morbidity
and mortality (1–3). In recent years, the frequency of
hypertension has been gradually increasing in China (4). It is reported that ~300 million
people in China will be living with hypertension by 2017 and the
morbidity rate increases gradually with age (4). A large number of clinical and
experimental investigations from numerous laboratories around the
world have demonstrated that chronic low-grade inflammation and the
adaptive immune response make important contributions to the
pathogenesis of various forms of systemic hypertension (5), other cardiovascular diseases
(6), and renal disease (1). However, there is mounting evidence,
supporting the concept that hypertensive stimuli induce activation
of T lymphocytes and infiltration of activated T lymphocytes into
target organs, including the peripheral blood vessels and kidney
(7); with Dahl-salt sensitive rats
and spontaneously hypertensive rats (SHRs) as example models
(8). When activated, effector T
lymphocytes contribute to blood pressure elevation by exacerbating
vascular remodeling and chronic kidney injury directly via the
release of pro-inflammatory cytokines, including tumor necrosis
factor-α (TNF-α) and interleukin-6 (IL-6) (8). Furthermore, to a large extent, the
severity of inflammation is determined by imbalances between
pro-inflammatory responses of effector T cells and
anti-inflammatory responses of regulatory T lymphocytes (Tregs)
(9). Although the role of T
lymphocytes in hypertension-mediated inflammation is clearly
defined by a large body of experimental data, the evidence that
inflammation and an adaptive immune response are induced by
hypertension is rather limited. The authors' laboratory has
demonstrated that gap junctional communication via connexins (Cxs)
in peripheral blood lymphocytes of SHRs (10) and hypertensive patients (11) is involved in the
hypertension-mediated inflammatory response, and that Cx expression
is positively correlated with the proliferation of T lymphocyte and
production of pro-inflammatory cytokines in the peripheral blood of
patients with hypertension and SHRs (10–13).
Data from the authors' laboratory and other groups have also
revealed that inhibition of Cx43-mediated gap junctional
communication can reduce the activation and proliferation of T
lymphocytes and production of pro-inflammatory cytokines under
various pro-inflammatory stimuli (10,11,14–16).
Therefore, Cx-based channels may be novel potential targets for the
treatment of hypertension-mediated inflammation.
Experimental and clinical studies have demonstrated
that T-cell-targeted immunosuppressive drugs (including
mycophenolate mofetil) and cytokine inhibitors reduce arterial
pressure or/and ameliorate renal inflammation in hypertensive
patients with rheumatoid arthritis and psoriasis, or in
pharmacologically-induced (angiotensin II infusion and
deoxycorticosterone acetate salt) or genetic rodent models of
hypertension (Dahl salt-sensitive rats and SHRs) (1,3,9,17,18);
however, these immunosuppressive therapies produce non-specific
inhibition of the immune system, and therefore, may interfere with
the normal generation of T lymphocytes, or result in unwanted and
unsafe side-effects in hypertensive patients and animals (1,19).
By contrast, estrogen replacement therapy using 17β-estradiol
reduces blood pressure (BP) via vasoprotective effects (20) and prevents a hypertension-mediated
pro-inflammatory response by inhibiting the production of
pro-inflammatory cytokines and stimulating the production of Tregs
and IL-10 (21,22). Studies have also provided further
evidence that increased pro-inflammatory and decreased
anti-inflammatory status caused by ovarian hormone deficiency is
associated with an increased frequency of hypertension in women and
numerous animal models of hypertension (23); therefore, estrogen can keep women
‘cardiovascularly younger’ than men of the same age (24). However, there are clear gaps in the
understanding of the precise mechanisms underlying how estrogen
regulates the adaptive immune response, BP and T lymphocyte
profiles (23). In order to
further expand the current knowledge of the cellular mechanisms of
estrogen in preventing T lymphocyte-associated hypertension, the
aim of the present study was to determine whether exogenous
estrogen (β-estradiol) treatment can prevent hypertension-mediated
inflammation and target organ damage by modulating Cx-mediated gap
junctional intracellular communication (GJIC) in peripheral blood T
lymphocytes. Identification of the cellular mechanisms underlying
the regulatory role of estrogen in BP and hypertension-mediated
inflammation may lead to specific treatment strategies that
ultimately reduces the incidence of hypertension.
Materials and methods
Experimental animals and drug
treatment
The 15-week-old male SHR and age-matched
Wistar-Kyoto rats (WKY) rats from Vital Beijing River Laboratory
Animal Technology Co., Ltd., (Beijing, China) were housed in
plastic cages in a room with a relative humidity of 45–55%,
temperature of 23±2°C and a 12-h light-dark cycle. Rats had free
access to drinking water and food throughout the experiment. Prior
to the onset of the experiment, all rats were allowed to train with
and acclimate to the tail-cuff plethysmography procedure for 1
week. After 1 week, the systolic blood pressure was measured for 3
consecutive days to record the hypertension prior to starting the
experiments. SHRs with a BP ≥150 mmHg were used in subsequent
experiments. The animal experimental procedures performed in the
present study were approved by the Institutional Animal Care and
Use Committees (permit number: A2016-047-03) of the Medical College
of Shihezi University (Shihezi, China) and conducted in strict
accordance with the recommendations of the Guide for the Care and
Use of Laboratory Animals of the US National Institutes of Health
(25).
Male SHRs (16-week-old; n=12) with an initial body
weight of 130–200 g were randomly divided into two groups: SHR
group (n=6) and SHR + β-estradiol (E) group (n=6). Male WKY rats of
the same age and body weight served as a control group (WKY group;
n=6). SHR in the β-estradiol treatment group received a single
subcutaneous injection of 20 µg/kg/day β-estradiol (cat. no. E8875;
≥98% pure; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) once
daily at the same time each day from 16–21 weeks of age and rats in
the other groups (WKY control group and the SHRs control group)
were injected subcutaneously with the same volume of normal saline.
The concentration of β-estradiol was selected based on the
effective dosage in a previous study (26). Following treatment with drug for 5
weeks, the tail arterial BP of all rats was measured.
Measurement of tail arterial BP
Tail systolic arterial pressure was recorded in
conscious and calm rats at 21 weeks old using a non-invasive
tail-cuff apparatus (Chengdu Taimeng Software Co. Ltd., Chengdu,
China) without heating, as described in the authors' previous study
(13,27). The BP of each rat was taken as the
average of at least three stable consecutive measurements following
removal of outliers and any readings associated with excess noise
or animal movement on each occasion. Data are expressed as mmHg and
calculated as the mean ± standard error of the mean (SEM).
Morphology and histological
analyses
At the end of the experiment, all rats were
euthanized by an intraperitoneal injection of 50 mg/kg
pentobarbital sodium (30 mg/l) at 21 weeks old. Kidneys and basilar
arteries (BA) were harvested and fixed in 4% paraformaldehyde at
4°C for 48 h. The animals were sacrificed by decapitation under an
overdose of pentobarbital (100 mg/kg) anesthesia at the end of the
experiments. Fixed renal and vascular tissues from the different
groups were dehydrated in graded concentrations of ethanol and
imbedded in paraffin, then 5 µm-thick sections were cut with a
microtome. Kidney and BA sections were stained with hematoxylin and
eosin as described in the authors' previous study (13). Histological observation of vascular
remodeling and renal injury was performed using light microscopy
(Olympus BX50 microscope; Olympus Corporation, Tokyo, Japan). A
total of 1 tissue section was selected from each rat and at least
10 random non-overlapping fields (at ×100 or 200 magnification)
were imaged to observe the presence of inflammatory cell
infiltration, thickness of the medial wall and endothelium injury
in BA, and tubular dilation, glomerulus deformation and fibrosis in
the kidneys.
Flow cytometric analysis of peripheral
blood mononuclear cells (PBMCs)
Following administration of β-estradiol or normal
saline for 5 weeks, rats from each group were euthanized by an
intraperitoneal injection of 50 mg/kg pentobarbital sodium and
peripheral blood (5 ml) was collected from the abdominal aorta into
a glass tube with EDTA, and PBMCs were separated and purified using
a rat mononuclear cell isolation kit (cat. no. P8630; Beijing
Solarbio Science & Technology, Beijing, China) and FACS™ Lysing
solution (cat. no. 349202; BD Biosciences; Becton, Dickinson and
Company, Franklin, Lakes, NJ, USA), according to the manufacturer's
protocol. All overdose euthanized rats with pentobarbital sodium
(100 mg/kg) were sacrificed by decapitation at the end of the
experiments. The cell survival rate of isolated PBMCs was assessed
by 0.4% Trypan blue staining for 10 min at room temperature.
Subsequently, PBMCs (>1×106 cells/ml) from the
different groups were transferred to a tube and stained in PBS
containing fluorescein isothiocyanate (FITC)-conjugated anti-rat
CD3 (dilution 1:1,000; cat. no. 201403), allophycocyanin
(APC)-conjugated anti-rat CD4 (dilution 1:400; cat. no. 201509),
phycoerythrin (PE)-conjugated anti-rat CD8 (dilution 1:400; cat.
no. 201705) and PE-conjugated anti-rat CD25 monoclonal antibodies
(dilution 1:400; cat. no. 202105) (all antibodies from Biolegend,
Inc., San Diego, CA, USA) for 30 min at 4°C in the dark.
Isotype-matched, FITC-, APC-, and PE-conjugated monoclonal
antibodies were used as negative controls. Following staining,
cells were analyzed within 24 h using a flow cytometer (FACSort; BD
Pharmingen; BD Biosciences; Becton, Dickinson and Company) and data
analysis was performed using BD CellQuest Pro software (version
2.0, system OS2; Becton, Dickinson and Company). Cluster of
differentiation CD4+ T cells, CD8+ T cells
and Treg cells were identified as double-positive stained cells
(CD3+ and CD4+ or CD8+, and
CD4+ and CD25+, respectively) and were
expressed as percentages of different T lymphocyte
subpopulation.
Expression of Cx40 and Cx43 on CD4+ or
CD8+ T cells was determined by flow cytometry as
previously described, with minor modifications (11,13).
Briefly, permeabilized PBMCs were incubated with anti-Cx40
monoclonal antibody (dilution 1:500; cat. no. sc-365107; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) or anti-Cx43 antibody
(dilution 1:500; cat. no. sc-13558; Santa Cruz Biotechnology, Inc.)
overnight at 4°C. Following washing, the PBMCs were incubated in
FITC-labeled secondary antibody (dilution 1:500; cat. no. 405305;
Biolegend, Inc.) and/or anti-CD4 and anti-CD8 antibodies. The
expression of Cx40/Cx43 in different T lymphocyte subpopulations
was analyzed using the two-color immunofluorescence flow cytometry
method as described previously (11,13).
Measurements of cytokines in the
serum
Cytokine levels in serum from the different groups
were measured in duplicate using commercially available ELISA assay
kits for TNF-α, IL-6 and IL-10 [TNF-α, cat. no. 70-EK382HS-96;
IL-6, cat. no. 70-EK3062/2; and IL-10, cat. no. 70-EK3102/2;
Hangzhou Multi Sciences (Lianke) Biotech Co., Ltd., Hangzhou,
China] following the manufacturer's protocol. Results are expressed
as pg/ml in each sample.
GJIC assay
The effect of β-estradiol on the GJIC between
peripheral blood lymphocytes was measured using a calcein
acetoxymethyl ester (calcein AM) transfer assay as described
previously (10,11). Briefly, PBMCs (1×106
cells/ml) isolated from WKY rats were incubated in a 6-well plate
with a 2.5 µM solution of calcein AM (cat. no. 3099; Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) or the lipophilic
dye DiIC18 (10 µM; cat. no. D282; Invitrogen; Thermo
Fisher Scientific, Inc.) for 30 min at 37°C in RPMI-1640 (cat. no.
11875085; Gibco; Thermo Fisher Scientific, Inc.) containing 10%
fetal bovine serum (FBS; cat. no. SH30084; HyClone; GE Healthcare
Life Sciences, Logan, UT, USA). Following incubation, these cells
were washed five times with PBS containing 1% bovine serum albumin
(BSA; cat. no. A8010; Beijing Solarbio Science & Technology
Co., Ltd.). The donor cells
(calcein+DiIC18−) and the
recipient cells (calcein−DiIC18+)
were cocultured at 1:10 ratio. Following seeding for 30 min,
cocultured cells were incubated in RPMI-1640 medium supplemented
with concanavalin A (Con A; 5 µg/ml) and/or β-estradiol (10 nM) for
3 h (28). Following the indicated
time of incubation, each group of cocultures were collected and
suspended in PBS containing 1% BSA, and the transfer of calcein
from donor cells to the recipient cells was analyzed by flow
cytometry as described previously (11).
Cell culture and drug treatment of
peripheral blood T lymphocytes
PBMCs isolated from WKY rats were cultured in
RPMI-1640 growth medium (cat. no. 11875085; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% (v/v) heat-inactivated FBS
(cat. no. SH30084; HyClone; GE Healthcare Life Sciences), 100 IU/ml
penicillin and 100 IU/ml streptomycin (cat. no. P0781;
Sigma-Aldrich; Merck KGaA) in an atmosphere of 5% CO2
and 95% air at 37°C for 3 h. Following 3-h incubation, the cell
viability of peripheral blood T lymphocytes was determined using
trypan blue and cells were then adjusted to a concentration of
1×106 cells/ml in medium. The cultured cells were
transferred into 6-well plates and pretreated with or without
β-estradiol (10 nM) for 24 h, and then stimulated with 5 µg/ml Con
A (cat. no. C5275; Sigma-Aldrich; Merck KGaA) for another 48 h at
37°C (humidified atmosphere, 5% CO2). Untreated controls
were cultured under the same conditions without Con A and
β-estradiol. Following drug treatment for the indicated times, all
cells and culture supernatants collected were used to analyze the
expression of Cxs (Cx40 and Cx43) and the concentration of
cytokines (TNF-α and IL-6) by immunofluorescence/immunoblotting and
ELISA, respectively.
Immunofluorescence staining
Peripheral blood lymphocytes (1×105
cells/ml) isolated from WKY rats were incubated with or without Con
A and/or β-estradiol in a 5% CO2 and 95% air atmosphere
for the indicated duration at 37°C. Subsequently, cells were washed
with PBS then fixed in PBS containing 4% paraformaldehyde for 30
min at room temperature. Lymphocytes were washed with PBS three
times and permeabilized with 0.5% Triton X-100/0.5% FBS for 10 min.
Following washing with PBS, each well was blocked with 1% BSA/PBS
for 1 h at room temperature. Following blocking, peripheral blood
lymphocytes were incubated overnight at 4°C with anti-Cx40
monoclonal antibody (dilution 1:500; cat. no. sc-365107; Santa Cruz
Biotechnology, Inc.) or anti-Cx43 antibody (dilution 1:500; cat.
no. sc-13558; Santa Cruz Biotechnology, Inc.) in 1% BSA/PBS. The
cells were washed thoroughly, and the bound primary antibody was
detected by incubating the cells with secondary FITC-conjugated
goat anti-mouse IgG (dilution 1:100; cat. no. ZF0312; OriGene
Technologies, Inc., Rockville, MD, USA) for 2 h at room temperature
in the dark. Subsequently, cells were counterstained with 10 µg/ml
DAPI (cat. no. C0065; Beijing Solarbio Science & Technology
Co., Ltd.) at 37°C for 2 min to visualize the nucleus. Fluorescence
images were captured using a laser scanning confocal microscope
(Zeiss LSM510; Carl Zeiss AG, Oberkochen, Germany) with a 63× oil
immersion objective (numerical aperture, 1.40). Adobe Photoshop
software (version 4.0; Adobe Systems, Inc., San Jose, CA, USA) was
used to adjust the contrast of the images and to compose and
overlay the images. Semiquantitative analysis of the mean
fluorescence intensities of Cx40 and Cx43 was performed using
ImageJ software (version 1.52a; National Institutes of Health,
Bethesda, MD, USA). A total of 50 peripheral blood lymphocytes from
each group in ~25 fields were evaluated in at least three
experimental repeats.
Immunoblotting
Peripheral blood lymphocytes (1×106
cells/well) from WKY rats were transferred to 6-well plates.
Following Con A (5 µg/ml) or/and β-estradiol (10 nM) treatment for
the indicated time, the expression levels of Cx40 and Cx43 in PBMCs
were determined by immunoblot analysis as previously described
(11,13). PBMCs were lysed with ice-cold lysis
buffer (25 mM bicine, 150 mM sodium chloride, pH 7.6; cat. no.
78510; Pierce; Thermo Fisher Scientific, Inc.) containing 1 mM
phenylmethylsulfonyl fluoride for 30 min at 4°C. Lysed lymphocytes
were sonicated and centrifuged at 10,000 × g for 20 min at 4°C. The
supernatant was collected and the protein concentration was
determined with a Bradford protein assay kit (cat. no. GK5021;
Generay Biotech Co., Ltd., Shanghai, China). Each lane was loaded
with an equal amount of protein (25 µg/lane) and separated by
SDS-PAGE on 10% gels. Separated proteins were transferred to a
polyvinylidene fluoride membrane as described previously (11,13).
The following antibodies were used for western blot analysis: Mouse
monoclonal anti-Cx40 antibody (1:1,000; overnight incubation at
4°C; cat. no. sc-365107; Santa Cruz Biotechnology, Inc.), mouse
monoclonal anti-Cx43 antibody (1:1,000; overnight incubation at
4°C; cat. no. sc-13558; Santa Cruz Biotechnology, Inc.), mouse
monoclonal anti-β-actin antibody (1:1,000; overnight incubation at
4°C; cat. no. TA-09; OriGene Technologies, Inc.) and horseradish
peroxidase-conjugated goat anti-mouse secondary antibody (1:10,000;
incubation for 1.5 h at room temperature; cat. no. ZB-5305; OriGene
Technologies, Inc.). Cx signals were visualized using an enhanced
chemiluminescence reagent (cat. no. RPN2109; GE Healthcare Life
Sciences) and quantified using Quantity One analysis software
(version 4.6.8, Bio-Rad Laboratories, Inc., Hercules, CA, USA).
β-actin was used as an internal standard to normalize the protein
levels of Cxs in each sample.
Statistical analysis
All experimental data are presented as the mean ±
SEM. Statistical analysis was performed using GraphPad Prism 5.0
software (GraphPad Software, Inc., La Jolla, CA, USA). Data with
more than two groups were compared using one-way analysis of
variance, followed by a post-hoc test (Tukey's multiple comparison
test). All of the experiments were performed at least three times
independently. P<0.05 was considered to indicate a statistically
significant difference.
Results
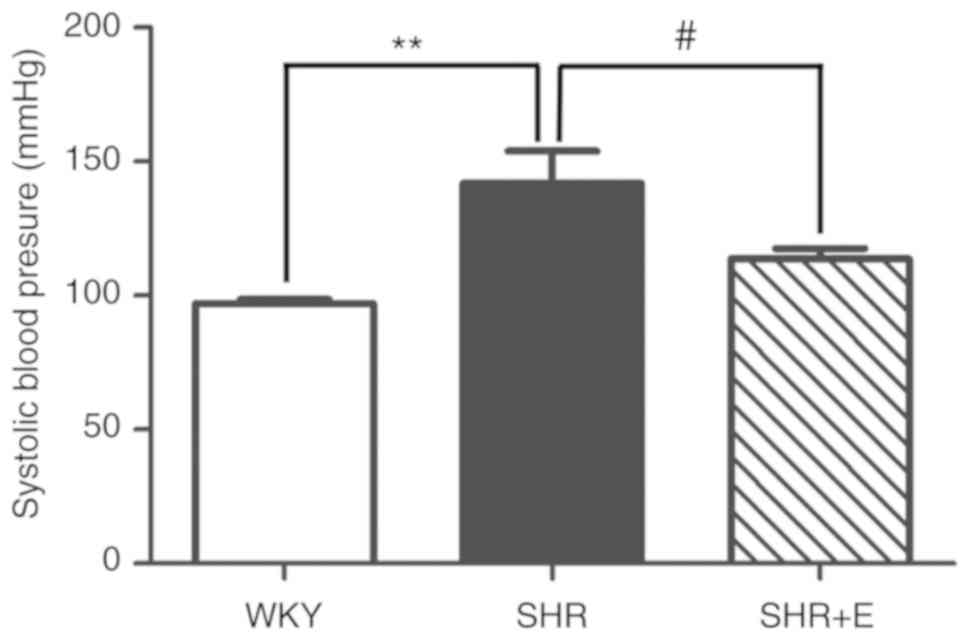
β-estradiol reduces systolic arterial
pressure in SHRs
Tail systolic arterial pressure was determined using
tail-cuff plethysmography in all rats at 21 weeks. Compared with
the WKY rats, tail arterial BP was significantly increased in the
SHR group (97.03±1.63 vs. 144.91±12.1 mmHg, respectively;
P<0.01; Fig. 1). However,
compared with the SHR group, β-estradiol significantly ameliorated
the increase in tail arterial pressure in the SHR + β-estradiol (E)
group (144.91±12.1 vs. 110.0±5.9 mmHg, respectively; P<0.05;
Fig. 1). There was no difference
in tail arterial pressure between the SHR+E group and WKY group
(110.0±5.9 vs. 97.03±1.63 mmHg, respectively; P>0.05; Fig. 1).
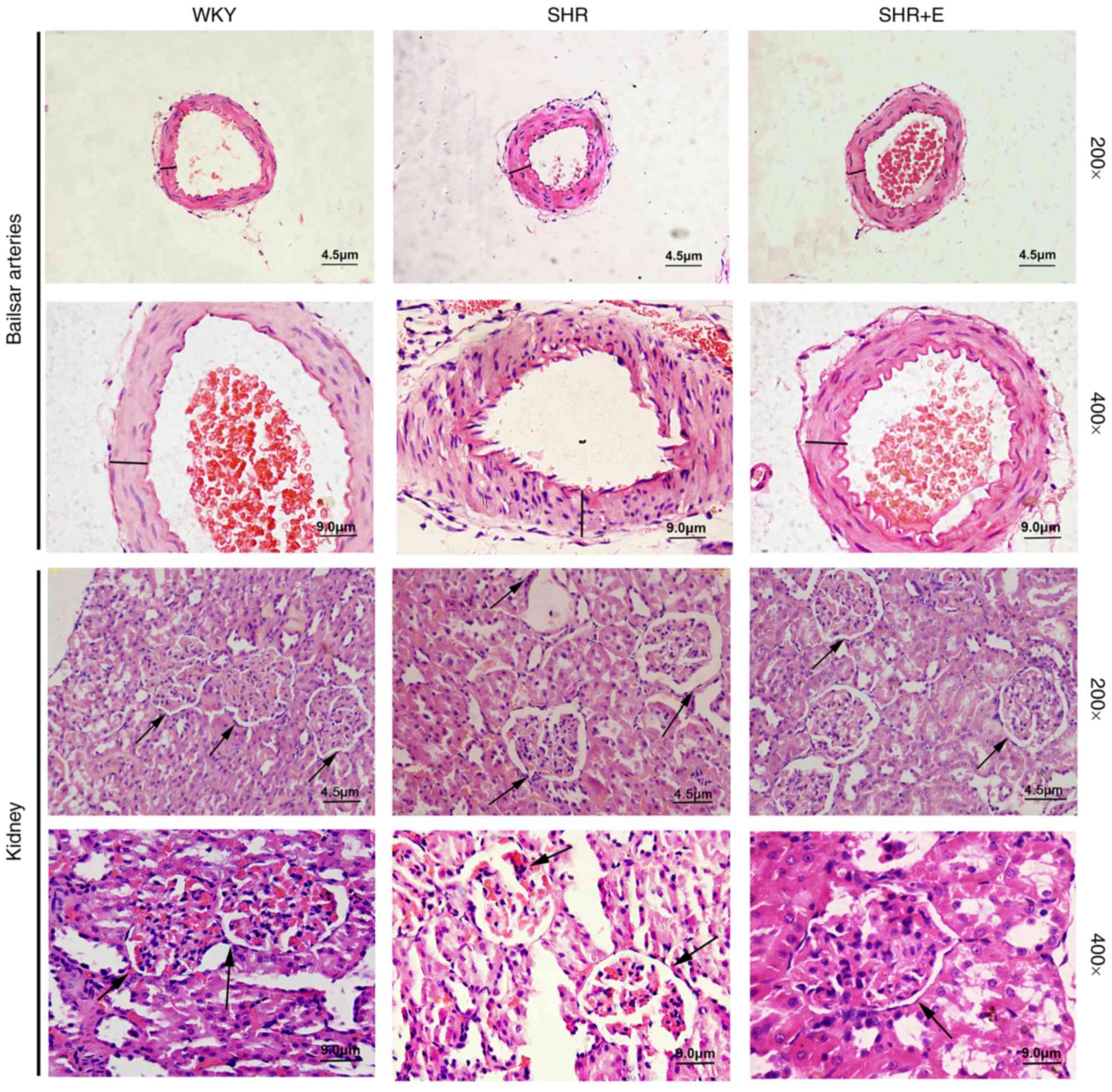
β-estradiol prevents vascular
remodeling and renal damage in SHRs
Hematoxylin and eosin staining results revealed that
cerebral arteries of SHRs exhibited increased medial wall thickness
and severe endothelium injury with increased infiltration of
inflammatory cells compared with WKYs (Fig. 2). Furthermore, hypertension also
resulted in marked renal damage, demonstrated by enlarged renal
tubules, atrophy of glomerular and tubular epithelial cells,
interstitial expansion and accumulation of inflammatory cells in
SHRs (Fig. 2). By contrast,
administration of β-estradiol ameliorated hypertension-induced
structural changes and inflammation in cerebral arteries and
kidneys compared with the SHR group (Fig. 2).
β-estradiol suppresses
hypertension-mediated inflammation by reversing the imbalance of T
lymphocyte subsets and inhibiting pro-inflammatory cytokines
production in SHR
To investigate whether estrogen ameliorates
hypertension-mediated inflammation, the effect of β-estradiol on
the percentage of lymphocyte subpopulations in peripheral blood of
SHR was evaluated using flow cytometry. In the SHR group without
β-estradiol supplement, there was a significant increase in the
percentage of CD4+/CD8+ T cell subset ratios
compared with WKY rats (WKY vs. SHR, 1.69±0.13 vs. 2.28±0.11,
respectively; P<0.01; Fig. 3B);
whereas there were no statistically significant differences among
CD3+ (WKY vs. SHR, 48.12±3.12 vs. 52.53±1.68%;
P>0.05; Fig. 3B) and
CD4+ (WKY vs. SHR, 64.76±1.23 vs. 68.66±1.44%;
P>0.05; Fig. 3B) populations in
the SHR group compared with the WKY group. Compared with the WKY
group, the percentage of CD4+CD25+ T cells
obtained from the peripheral blood of SHR was significantly
decreased (WKY vs. SHR, 7.83±0.46 vs. 5.26±0.31%; P<0.01;
Fig. 3B). However, β-estradiol
treatment resulted in a significant decrease in the percentage of
CD3+ T cells (SHR vs. SHR + E, 52.53±1.68 vs.
43.96±1.26%; P<0.05; Fig. 3B)
and CD4+/CD8+ T cell subset ratios (SHR vs.
SHR + E, 2.28±0.11 vs. 1.78±0.14; P<0.05; Fig. 3B) compared with SHR. In particular,
β-estradiol induced a significant increase in the percentage of
CD4+CD25+ T lymphocytes in SHRs compared with
SHR controls (SHR vs. SHR + E, 5.26±0.31 vs. 6.62±0.29%; P<0.05;
Fig. 3B), which may be associated
with increased Treg production stimulated by β-estradiol.
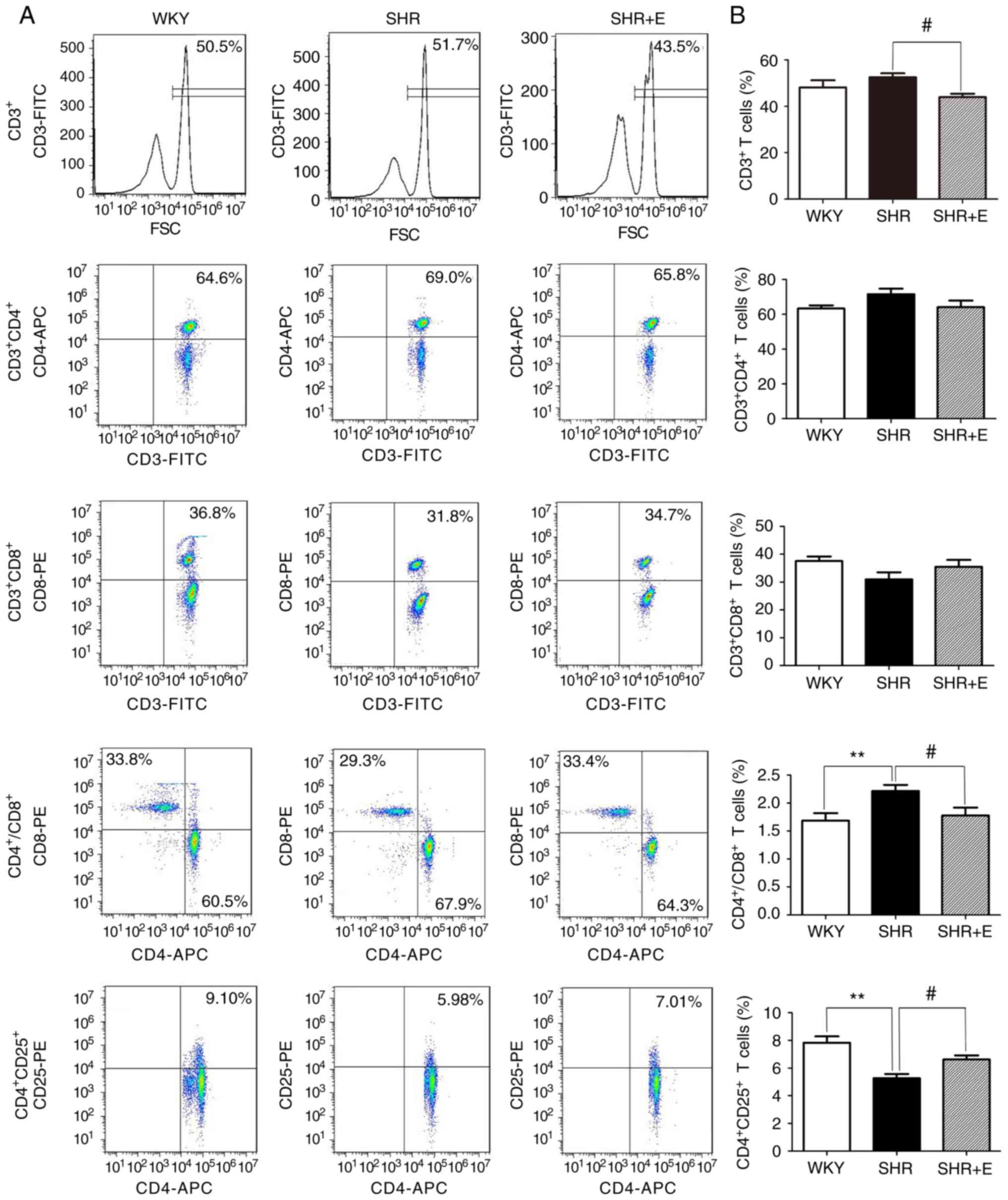 | Figure 3.β-estradiol supplement reverses the
changes in the percentages of various T lymphocyte subpopulations
in peripheral circulation of SHR. The percentage of
CD3+, CD3+CD4+,
CD3+CD8+, CD4+ CD25+ T
cells was analyzed by flow cytometry. (A) Flow cytometry dot plots
represent the percentages of circulating T lymphocytes subtypes in
the peripheral blood of SHR and WKY rats. Decrease in the
CD3+ T cell population and
CD4+/CD8+ T cell subset ratios, and increased
percentage of CD4+CD25+ T cells were observed
following β-estradiol treatment in SHR. (B) Bar graphs represent
the percentage of various T cell subpopulations and
CD4+/CD8+ T cell subset ratios. All numerical
data are displayed as the mean ± standard error (n=6/group).
**P<0.01 vs. WKY group and SHR group without any drug treatment.
#P<0.05 SHR group without any drug treatment vs. SHR
group given β-estradiol. SHR, spontaneously hypertensive rat; WKY,
Wistar-Kyoto rats; CD, cluster of differentiation; SHR + E, SHR +
β-estradiol; FITC, fluorescein isothiocyanate; PE, phycoerythrin;
APC, allophycocyanin; FSC, forward scatter. |
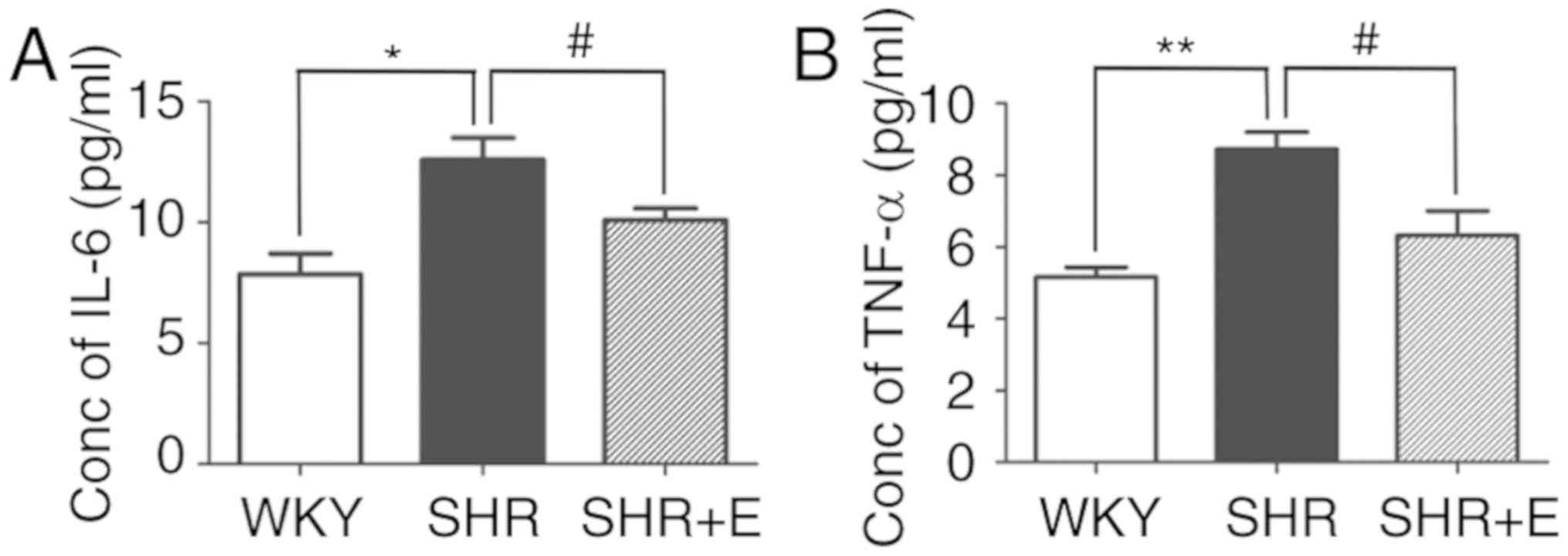
The influence of β-estradiol on TNF-α and IL-6
secretion was evaluated in serum isolated from SHRs. The analysis
revealed that serum from SHRs that received β-estradiol exhibited a
significantly lower level of plasma IL-6 (SHR vs. SHR + E,
12.37±0.67 vs. 10.11±0.42 pg/ml; P<0.05; Fig. 4A) and TNF-α (SHR vs. SHR + E,
8.94±0.40 vs. 6.17±0.96 pg/ml; P<0.05; Fig. 4B) compared with the SHR control
group.
Effect of β-estradiol on the
expression of Cxs in peripheral blood lymphocyte subsets of
SHRs
To determine whether β-estradiol affects the
expression of Cxs, conventional flow cytometry was used to evaluate
total protein expression of Cx40 and Cx43 in peripheral blood
lymphocyte subsets from SHRs. Consistent with the authors' previous
study (12), flow cytometric
analysis revealed that the percentages of
CD8+Cx40+ (WKY vs. SHR, 28.14±1.55 vs.
34.80±1.22%; P<0.01), CD4+Cx43+ (WKY vs.
SHR, 44.75±3.02 vs. 53.52±0.97%; P<0.05) and
CD8+Cx43+ (WKY vs. SHR, 28.09±1.80 vs.
38.82±1.44%; P<0.01) double-positive peripheral blood T
lymphocytes were significantly increased in SHRs compared with
WKYs; whereas, there were no statistically significant differences
in the percentages of Cx40 and CD4 double-positive T lymphocytes
between the SHR and WKY groups (Fig.
5A and B). β-estradiol supplementation exhibited an inhibitory
effect on the abundance of Cx40 and Cx43 in CD4+ or
CD8+ cells. β-estradiol significantly decreased the
amount of CD8+Cx40+ (SHR vs. SHR + E,
34.80±1.22 vs. 30.53±1.24%; P<0.05),
CD4+Cx43+ (SHR vs. SHR+E, 53.52±0.97 vs.
49.65±0.87%; P<0.05; Fig. 5A and
B) and CD8+Cx43+ (SHR vs. SHR + E,
38.82±1.44 vs. 33.3±1.89%; P<0.05) double-positive peripheral
blood T lymphocytes compared with the SHR controls (Fig. 5A and B).
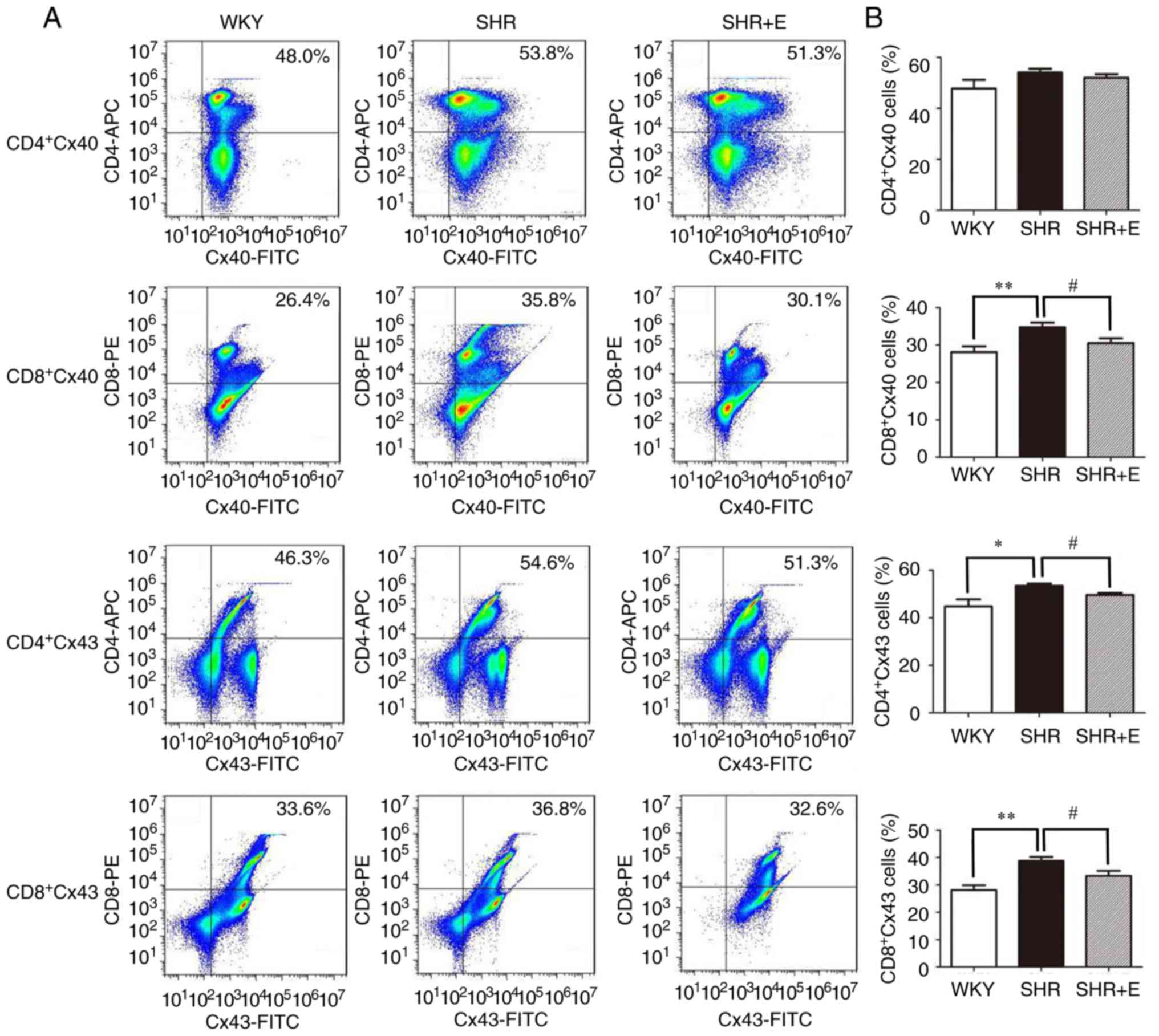 | Figure 5.β-estradiol supplement reduces the
percentages of Cx40- and Cx43-expressing CD4+ and
CD8+ T cells in SHR. (A) Representative dot plot and (B)
collated data presenting percentages of Cx40- and Cx43-positive
CD4+ and CD8+ T cells in each group. Data are
presented as the mean ± standard error of three independent
experiments (n=6/group). *P<0.05 and **P<0.01, WKY group vs.
SHR group without any drug treatment; #P<0.05, SHR
group without any drug treatment vs. SHR group with β-estradiol
treatment. Cx, connexin; SHR, spontaneously hypertensive rat; WKY,
Wistar-Kyoto rats; SHR + E, SHR + β-estradiol; CD, cluster of
differentiation; FITC, fluorescein isothiocyanate; PE,
phycoerythrin; APC, allophycocyanin. |
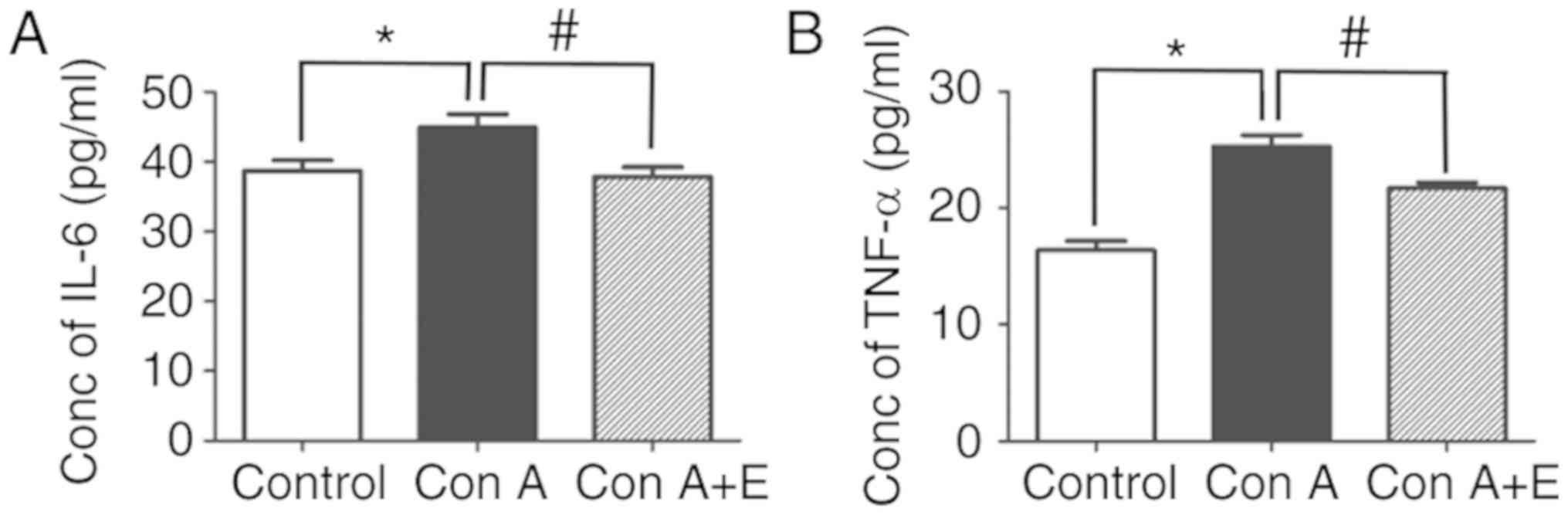
Pretreatment with β-estradiol inhibits
the release of pro-inflammatory cytokines in T-cell
mitogen-stimulated peripheral blood lymphocytes
To further assess whether β-estradiol has inhibitory
effects on the release of pro-inflammatory cytokines, the levels of
pro-inflammatory cytokines released by peripheral blood T
lymphocytes were determined using the culture supernatant of
peripheral blood lymphocytes in vitro. IL-6 (Control vs. Con
A, 38.68±1.53 vs. 44.92±1.90 pg/ml; P<0.05; Fig. 6A) and TNF-α (Control vs. Con A,
16.39±0.80 vs. 25.28±0.99 pg/ml, P<0.05; Fig. 6B) concentrations were significantly
enhanced by Con A compared with the untreated controls. However,
pre-treatment with β-estradiol (10 nM) significantly attenuated the
secretion of IL-6 (Con A vs. Con A + E, 44.92±1.90 vs. 37.87±1.36
pg/ml; P<0.05; Fig. 6A) and
TNF-α (Con A vs. Con A + E, 25.28±0.99 vs. 21.72±0.49 pg/ml;
P<0.05; Fig. 6B) in peripheral
blood T lymphocytes. Overall, the data demonstrated that
β-estradiol treatment has potent inhibitory effects on T
lymphocyte-induced inflammatory responses.
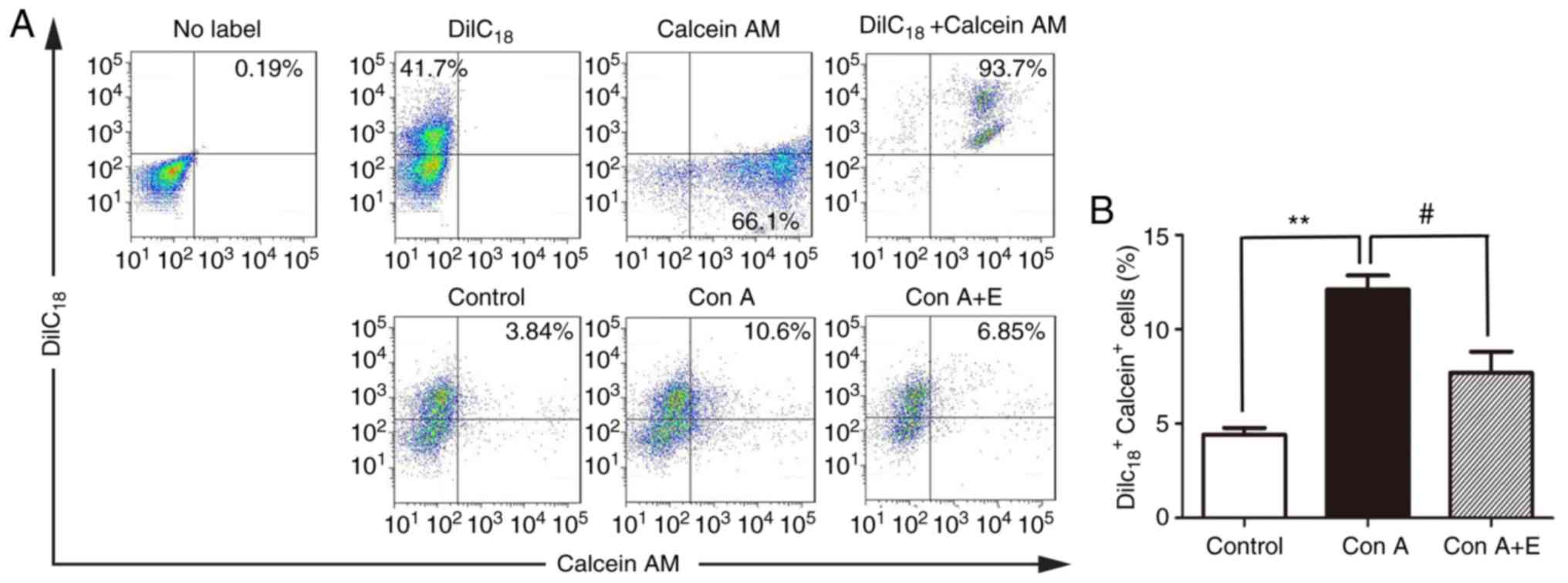
β-estradiol suppresses the function of
gap junctions between peripheral blood lymphocytes in the presence
of T cell mitogenic stimuli
To confirm whether Cxs-based gap junctions are
involved in inhibitory effects of β-estradiol on
hypertension-mediated inflammation, a functional in vitro
assay based on the transfer of a gap junction permeant dye was used
to evaluate the functional alteration of Cx-mediated gap junctions
in peripheral blood lymphocytes in the presence of β-estradiol
or/and Con A. Dye transfer between lymphocytes was evaluated by
flow cytometry. Following Con A treatment, there was a significant
increase in calcein AM dye transfer between cocultured cells, as
demonstrated by an increased percentage of
DiIC18-calcein double-positive cells compared with the
unstimulated control (Control vs. Con A, 4.41±0.37 vs. 12.1±0.76%,
P<0.01; Fig. 7A and B).
Treatment with β-estradiol significantly suppressed the Con
A-induced increase in dye transfer between lymphocytes (Con A vs.
Con A + E, 12.1±0.76 vs. 7.69±1.13%, P<0.05; Fig. 7A and B).
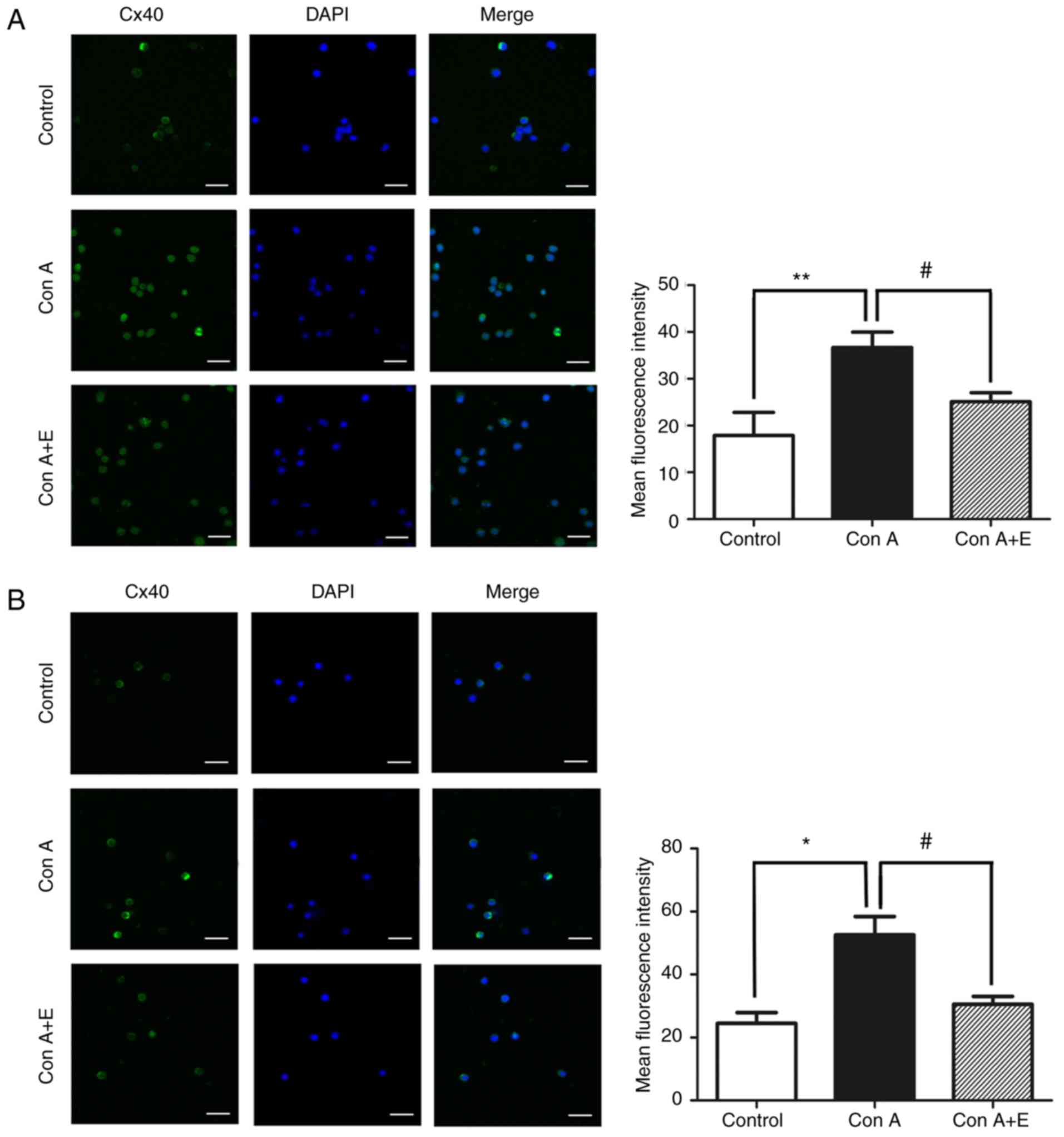
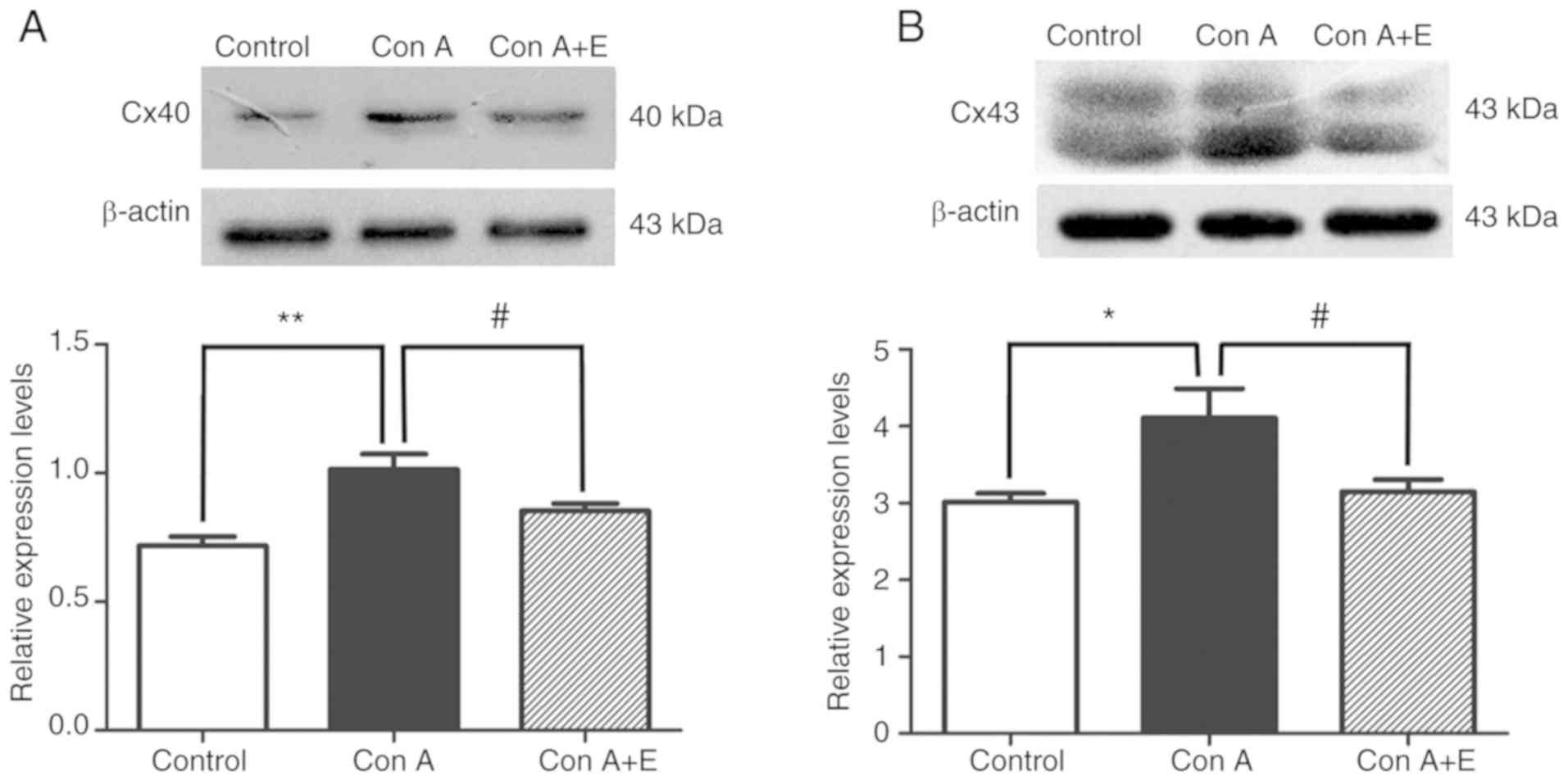
Pretreatment with β-estradiol inhibits
the expression of Cx40 and Cx43 in Con A stimulated peripheral
blood lymphocytes
The effect of β-estradiol on the expression of Cx40
and Cx43 in Con A-stimulated peripheral blood lymphocytes was
assessed using confocal microscopy and immunoblotting. The protein
levels of Cx40 and Cx43 were significantly enhanced in Con
A-stimulated peripheral blood lymphocytes, as demonstrated by
immunofluorescence (P<0.01 for Cx40; P<0.05 for Cx43;
Fig. 8A and B) and western blot
analysis (P<0.01 for Cx40; P<0.05 for Cx43; Fig. 9A and B); by contrast, pretreatment
with β-estradiol significantly reduced the Con A-induced increase
Cx40 and Cx43 expression in peripheral blood lymphocytes as
demonstrated by immunofluorescence (P<0.05 for Cx40 and Cx43;
Fig. 8A and B) and western blot
analysis (P<0.05 for Cx40 and Cx43; Fig. 9A and B).
Discussion
In the present study, the effects of β-estradiol on
the hypertension-mediated adaptive immune response were
investigated in male SHRs. The primary findings of the present
study were that β-estradiol significantly prevented inflammation
and target organ damage (kidneys and arteries) in male SHRs.
Furthermore, while hypertension appears to promote imbalances in
the adaptive immune response through increased expression of Cxs
(Cx40 and Cx43), which have previously been demonstrated to be
pro-inflammatory (10–16), β-estradiol appeared to alleviate
hypertension-mediated imbalances of the adaptive immune response
and T cell mitogenic stimuli-induced adaptive immune response, at
least in part, by suppressing the expression of Cxs or the function
of Cx gap junctions. β-estradiol inhibited the release of
pro-inflammatory cytokines induced by spontaneous hypertension or T
cell activation. Therefore, the findings of this study support the
dual anti-hypertensive and anti-inflammatory role of estrogen and
extends the understanding of the cellular mechanisms underlying the
regulatory role of estrogen in BP and hypertension-mediated
inflammation.
It is well established that hypertension and
hypertension-induced organ damage are associated with excessive
inflammatory responses in experimental models of hypertension and
hypertensive patients (17,29).
Male hypertensive animal models (angiotensin II,
deoxycorticosterone acetate-salt and SHR) and hypertensive patients
exhibit a marked increase in the number of circulating T
lymphocytes, vascular and renal infiltration of CD4+ and
CD8+ T cells, and the levels of pro-inflammatory
cytokines, including IL-6 and TNF-α, compared with normotensive
controls (3,30,31).
The present data also demonstrated that hypertension-mediated
inflammation and hypertension-induced vascular and renal
infiltration of inflammatory cells is associated with alterations
in the number of effector T lymphocytes (CD4+ and
CD8+ T cells) and Tregs, and the production of
pro-inflammatory cytokines in the peripheral blood of SHRs.
Although it is clear that T lymphocytes have a key role in
hypertension development, the precise mechanism leading to
activation and proliferation of T lymphocytes in hypertension
remains poorly defined. Notably, other data from the authors' group
and the current findings demonstrate the importance of Cx-mediated
gap junctions between peripheral blood T lymphocytes in
hypertension-mediated pro-inflammatory responses (10–13);
increased Cx43 expression and enhanced Cx43-mediated gap junctional
communication, in particular, is important for pro-inflammatory
responses driven by T lymphocytes and the production of T
cell-induced cytokines under different pro-inflammatory stimuli
(10,11). In the present study, Cx40 and Cx43
expression was increased in peripheral blood lymphocytes from SHRs,
and there was a strong association between the levels of
pro-inflammatory cytokines (IL-6 and TNF-α) in the serum/culture
supernatant and the expression levels of Cx40 or Cx43 in peripheral
blood T lymphocytes from SHR/Con A-stimulated cells. Furthermore,
the present results also demonstrated that pro-inflammatory stimuli
(Con A) enhanced gap junctional communication between peripheral
blood lymphocytes from WKY rats. The data further demonstrated that
gap junctions are involved in the pathological mechanisms of
hypertension-mediated inflammation and interventions targeted to
Cx43-mediated gap junctions in effector T lymphocytes may represent
a novel therapeutic strategy for the treatment of hypertension.
While T cell-targeted immunosuppressant agents and
inhibition of pro-inflammatory cytokines prevent or ameliorate BP
elevation and hypertension-mediated inflammation in experimental
models of hypertension and in hypertensive patients (1,3,9,17,18,21),
these immunosuppressive drugs may result in unwanted side effects
in patients and animals with hypertension (21). In females, estrogen has been
revealed to have a central role in preventing the development of
hypertension and suppressing inflammation-mediated diseases
(32,33), which has been well established in
studies of women with ovarian hormone deficiency and male animal
models of hypertension (20–24,33,34).
Estrogen has also been reported to provide protective effects
against hypertension-induced vascular remodeling and renal damage
by reducing infiltration of leukocytes or effector T lymphocytes
(total T cells, CD4+ or Th17 cells) into organs (brain,
heart, kidney and vasculature), and enhancing the number of
anti-inflammatory Tregs in renal tissues (21,31,35)
in female hypertension models (SHR and female Rag-1−/−
mice infused with angiotensin II following adoptive transfer of
CD3+ T cells from wild-type male mice). Similarly,
estrogen replacement (17β-estradiol) was also demonstrated to
reduce leukocyte/macrophage infiltration in cardiac and vascular
tissues from ovariectomized hypertensive rats, aldosterone
salt-treated rats and ovariectomized rats subjected to balloon
injury of the right carotid artery (26,36).
In the current study it was also observed that β-estradiol
administration to male SHRs reduced BP and prevented vascular
remodeling (arterial wall thickening), and inhibited leukocyte
infiltration in the vascular wall and kidneys. The results
demonstrated that estrogen has a protective role in the regulation
of BP and target organ damage. Another important finding from the
present study was that long term treatment with β-estradiol
significantly attenuated the imbalance between effector and
regulatory T cell subsets in SHRs, potentially by reducing serum
levels of TNF-α and IL-6, and the CD4/CD8 ratio, and enhancing the
number of Tregs; whereas β-estradiol exhibited no effect on the
proportion of CD4+ and CD8+ T cells in the
peripheral blood of SHRs. The increasing percentage of Tregs in
β-estradiol-treated SHRs may be associated with
β-estradiol-stimulated conversion of CD4+ T cells into
Tregs (21) and decreased
pro-inflammatory cytokines levels imply that β-estradiol attenuates
the hypertension-mediated pro-inflammatory response in the
circulation.
Furthermore, a previous study indicated that the
menopause or estrogen deficiency in monocytes cultured ex
vivo is associated with increased levels of pro-inflammatory
cytokines produced by monocytes (IL-1, IL-6 and TNF-α) (22); while higher physiological or
supraphysiological levels of estrogen attenuate aberrant expression
or production of TH1-driven pro-inflammatory cytokines (IL-6,
TNF-α, interferon-γ and monocyte chemoattractant protein-1) in
estrogen-deficient animals (32),
and in various inflammatory and cardiovascular diseases, including
stroke (37), balloon injury of
carotid arteries (36) and
hypertension (34). To further
determine the anti-inflammatory role of estrogen against
inflammatory stimuli-induced secretion of pro-inflammatory
cytokines, the effect of β-estradiol pretreatment on IL-6 and TNF-α
secretion in PBMCs following exposure to T cell mitogenic stimulus
(Con A) was investigated to determine whether β-estradiol
suppresses secretion of the two cytokines. Pretreatment with
β-estradiol significantly reduced Con A-stimulated secretion of
IL-6 and TNF-α in peripheral blood lymphocytes from WKY rats, which
supports the findings from the SHR serum. These results also
confirmed an association between estrogen signaling and the
production of pro-inflammatory cytokines.
Several other studies have reported that estrogen
inhibits inflammatory infiltration via an estrogen
receptor-dependent mechanism (28,33,38–40).
Subsequently the mechanisms by which estrogen suppresses
hypertension-mediated inflammation and secretion of
pro-inflammatory cytokines, and whether this effect is associated
with the expression of Cxs and the function of Cx-mediated gap
junctions between peripheral blood lymphocytes was determined.
β-estradiol treatment markedly suppressed the expression of Cxs in
peripheral blood lymphocytes from SHRs and in Con A-stimulated
peripheral blood lymphocytes in vitro, and reduced
Cx-mediated gap junctional communication. The role of Cx gap
junctions in hypertension-mediated inflammatory response was
preliminarily demonstrated in the authors' previous studies
(10–13), whereby blockade of Cx43-mediated
gap junctions between T lymphocytes led to reduced expression and
production of various pro-inflammatory cytokines by peripheral
blood lymphocytes in SHRs and hypertensive patients (10,11).
Therefore, on the basis of these observations, it is logical to
hypothesize that β-estradiol supplementation could affect the
peripheral blood lymphocyte subsets in SHRs and suppress production
of pro-inflammatory cytokines by inhibiting the expression and
function of Cxs in peripheral blood lymphocytes.
The current study has certain limitations. The
levels of estrogen in peripheral blood/culture supernatant were not
detected, although estrogen effectively ameliorated BP elevation,
hypertension-mediated inflammation and hypertension-induced organ
damage in SHRs, and reduced Con A-induced secretion of
pro-inflammatory cytokines in vitro. Additionally, although
estrogen receptors α and β are expressed on the majority of immune
cells (41) and estrogen receptor
α signaling in T lymphocytes is required for the protective effect
of estrogen via estradiol-mediated inhibition of Th1 and Th17 cell
differentiation (33), whether
estrogen receptors α and β are expressed in peripheral blood
lymphocytes from different experimental hypertensive animals and
hypertensive patients is unknown, and their expression profile in
hypertensive models following estrogen treatment has not been
established. Consequently, future studies are required to determine
whether estrogen inhibits hypertension-mediated inflammation in
different hypertensive models and hypertensive patients via
estrogen receptor-dependent or independent signaling pathways.
Finally, the detailed anti-inflammatory mechanism of estrogen via
regulation of Cxs-based channel functions (gap junction channels or
hemi-channels) during hypertension or pro-inflammatory
stimuli-mediated inflammatory responses was not investigated,
although Cx-mediated hemi-channels are known to be activated in
inflammatory/pathological conditions. Further studies will also be
required to determine the role of gap junctions or hemi-channels in
the protective effects of estrogen in hypertension-mediated
inflammation; this may involve using gap junction- and
hemi-channel-specific mimetic peptides (42) in combination with
lentivirus-mediated RNA interference knockdown of Cx43.
In conclusion, the results of the present study
indicate that β-estradiol inhibits BP elevation and target organ
damage in male SHRs. In addition, the results suggest that
β-estradiol contributes to the inhibition of hypertension- or
pro-inflammatory stimuli-mediated inflammatory responses and
production of pro-inflammatory cytokines. These effects may be
mediated, at least in part, by suppressing the function of Cx-based
gap junctions and Cx protein expression (Cx40 and Cx43). These
results provide novel insight into the molecular mechanisms by
which estrogen can reduce hypertension. The present results and
previous studies indicate that Cxs and Cx-mediated gap junctions
are critical contributing factors in hypertension-mediated
inflammation and may be useful as targets for the treatment of
hypertension and other cardiovascular diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81660271 to
KM; grant no. 81460098 to XL; grant no. 81600325 to LZ; and grant
no. 81560081 to JS) and the International Cooperation Project of
Shihezi University (grant no. GJHZ201603 to KM).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KTM, XZL and YYZ conceived and designed the
experiments. XN performed the experiments. LZ, XM, LYS, LL and JQS
analyzed the data. LZ wrote the manuscript. All authors read and
approved the final manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study does not report on studies
involving human participants, human data or human tissue. The
protocol of the study was approved by Institutional Animal Care and
Use Committees (permit no. A2016-047-03) of the Medical College of
Shihezi University, and all animal handling and experimental
procedures were performed in accordance with guidelines for the
Care and Use of Laboratory Animals published by the US National
Institutes of Health (Public Health Service Policy on Humane Care
and Use of Animals, DHEW Publication no. 96-01, PHS Policy revised
in 2002).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Abais-Battad JM, Dasinger JH, Fehrenbach
DJ and Mattson DL: Novel adaptive and innate immunity targets in
hypertension. Pharmacol Res. 120:109–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Agita A and Alsagaff MT: Inflammation,
immunity, and hypertension. Acta Med Indones. 49:158–165.
2017.PubMed/NCBI
|
|
3
|
Bomfim GF, Rodrigues FL and Carneiro FS:
Are the innate and adaptive immune systems setting hypertension on
fire? Pharmacol Res. 117:377–393. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Committee of Cardio-Cerebro-Vascular
Diseases of Gerontological Society of China and Chinese College of
Cardiovascular Physicians of Chinese Medical Doctor Association:
Chinese expert consensus on the diagnosis and treatment of
hypertension in the elderly (2017). Zhonghua Nei Ke Za Zhi.
56:885–893. 2017.(In Chinese). PubMed/NCBI
|
|
5
|
Jafri S and Ormiston ML: Immune regulation
of systemic hypertension, pulmonary arterial hypertension, and
preeclampsia: Shared disease mechanisms and translational
opportunities. Am J Physiol Regul Integr Comp Physiol.
313:R693–R705. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schiffrin EL: Immune mechanisms in
hypertension and vascular injury. Clin Sci (Lond). 126:267–274.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marvar PJ, Vinh A, Thabet S, Lob HE, Geem
D, Ressler KJ and Harrison DG: T lymphocytes and vascular
inflammation contribute to stress-dependent hypertension. Biol
Psychiatry. 71:774–782. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schiffrin EL: The immune system: Role in
hypertension. Can J Cardiol. 29:543–548. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rodriguez-Iturbe B, Pons H and Johnson RJ:
Role of the immune system in hypertension. Physiol Rev.
97:1127–1164. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ni X, Li XZ, Fan ZR, Wang A, Zhang HC,
Zhang L, Li L, Si JQ and Ma KT: Increased expression and
functionality of the gap junction in peripheral blood lymphocytes
is associated with hypertension-mediated inflammation in
spontaneously hypertensive rats. Cell Mol Biol Lett. 23:402018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ni X, Wang A, Zhang L, Shan LY, Zhang HC,
Li L, Si JQ, Luo J, Li XZ and Ma KT: Up-regulation of gap junction
in peripheral blood T lymphocytes contributes to the inflammatory
response in essential hypertension. PLoS One. 12:e01847732017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang HC, Zhang ZS, Zhang L, Wang A, Zhu
H, Li L, Si JQ, Li XZ and Ma KT: Connexin 43 in splenic lymphocytes
is involved in the regulation of CD4+CD25+ T
lymphocyte proliferation and cytokine production in hypertensive
inflammation. Int J Mol Med. 41:13–24. 2018.PubMed/NCBI
|
|
13
|
Ni X, Zhang L, Peng M, Shen TW, Yu XS,
Shan LY, Li L, Si JQ, Li XZ and Ma KT: Hydrogen sulfide attenuates
hypertensive inflammation via regulating connexin expression in
spontaneously hypertensive rats. Med Sci Monit. 24:1205–1218. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Elgueta R, Tobar JA, Shoji KF, De Calisto
J, Kalergis AM, Bono MR, Rosemblatt M and Sáez JC: Gap junctions at
the dendritic cell-T cell interface are key elements for
antigen-dependent T cell activation. J Immunol. 183:277–284. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mendoza-Naranjo A, Bouma G, Pereda C,
Ramírez M, Webb KF, Tittarelli A, López MN, Kalergis AM, Thrasher
AJ, Becker DL and Salazar-Onfray F: Functional gap junctions
accumulate at the immunological synapse and contribute to T cell
activation. J Immunol. 187:3121–3132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oviedo-Orta E, Gasque P and Evans WH:
Immunoglobulin and cytokine expression in mixed lymphocyte cultures
is reduced by disruption of gap junction intercellular
communication. FASEB J. 15:768–774. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Idris-Khodja N, Mian MO, Paradis P and
Schiffrin EL: Dual opposing roles of adaptive immunity in
hypertension. Eur Heart J. 35:1238–1244. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mattson DL, James L, Berdan EA and Meister
CJ: Immune suppression attenuates hypertension and renal disease in
the Dahl salt-sensitive rat. Hypertension. 48:149–156. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wenzel UO, Bode M, Kurts C and Ehmke H:
Salt, inflammation, IL-17 and hypertension. Br J Pharmacol. doi 10:
1111/bph.14359. 2018. View Article : Google Scholar
|
|
20
|
Xing D, Nozell S, Chen YF, Hage F and
Oparil S: Estrogen and mechanisms of vascular protection.
Arterioscler Thromb Vasc Biol. 29:289–295. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tipton AJ and Sullivan JC: Sex differences
in T cells in hypertension. Clin Ther. 36:1882–1900. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Camilleri G, Borg M, Brincat S,
Schembri-Wismayer P, Brincat M and Calleja-Agius J: The role of
cytokines in cardiovascular disease in menopause. Climacteric.
15:524–530. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sandberg K, Ji H, Einstein G, Au A and Hay
M: Is immune system-related hypertension associated with ovarian
hormone deficiency? Exp Physiol. 101:368–374. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pollow DP, Uhrlaub J, Romero-Aleshire M,
Sandberg K, Nikolich-Zugich J, Brooks HL and Hay M: Sex differences
in T-lymphocyte tissue infiltration and development of angiotensin
II hypertension. Hypertension. 64:384–390. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chiu CZ, Wang BW, Chung TH and Shyu KG:
Angiotensin II and the ERK pathway mediate the induction of
myocardin by hypoxia in cultured rat neonatal cardiomyocytes. Clin
Sci (Lond). 119:273–282. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mori T, Kai H, Kajimoto H, Koga M, Kudo H,
Takayama N, Yasuoka S, Anegawa T, Kai M and Imaizumi T: Enhanced
cardiac inflammation and fibrosis in ovariectomized hypertensive
rats: A possible mechanism of diastolic dysfunction in
postmenopausal women. Hypertens Res. 34:496–502. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kubota Y, Umegaki K, Kagota S, Tanaka N,
Nakamura K, Kunitomo M and Shinozuka K: Evaluation of blood
pressure measured by tail-cuff methods (without heating) in
spontaneously hypertensive rats. Biol Pharm Bull. 29:1756–1758.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Adori M, Kiss E, Barad Z, Barabás K,
Kiszely E, Schneider A, Kövesdi D, Sziksz E, Abrahám IM, Matkó J
and Sármay G: Estrogen augments the T cell-dependent but not the
T-independent immune response. Cell Mol Life Sci. 67:1661–1674.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
McMaster WG, Kirabo A, Madhur MS and
Harrison DG: Inflammation, immunity, and hypertensive end-organ
damage. Circ Res. 116:1022–1033. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Singh MV, Chapleau MW, Harwani SC and
Abboud FM: The immune system and hypertension. Immunol Res.
59:243–253. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sandberg K, Ji H and Hay M: Sex-specific
immune modulation of primary hypertension. Cell Immunol.
294:95–101. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Salem ML: Estrogen, a double-edged sword:
Modulation of TH1- and TH2-mediated inflammations by differential
regulation of TH1/TH2 cytokine production. Curr Drug Targets
Inflamm Allergy. 3:97–104. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lélu K, Laffont S, Delpy L, Paulet PE,
Périnat T, Tschanz SA, Pelletier L, Engelhardt B and Guéry JC:
Estrogen receptor α signaling in T lymphocytes is required for
estradiol-mediated inhibition of Th1 and Th17 cell differentiation
and protection against experimental autoimmune encephalomyelitis. J
Immunol. 187:2386–2393. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Novella S, Heras M, Hermenegildo C and
Dantas AP: Effects of estrogen on vascular inflammation: A matter
of timing. Arterioscler Thromb Vasc Biol. 32:2035–2042. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tipton AJ, Baban B and Sullivan JC: Female
spontaneously hypertensive rats have a compensatory increase in
renal regulatory T cells in response to elevations in blood
pressure. Hypertension. 64:557–564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Miller AP, Feng W, Xing D, Weathington NM,
Blalock JE, Chen YF and Oparil S: Estrogen modulates inflammatory
mediator expression and neutrophil chemotaxis in injured arteries.
Circulation. 110:1664–1669. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ritzel RM, Capozzi LA and McCullough LD:
Sex, stroke, and inflammation: The potential for estrogen-mediated
immunoprotection in stroke. Horm Behav. 63:238–253. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xing D, Oparil S, Yu H, Gong K, Feng W,
Black J, Chen YF and Nozell S: Estrogen modulates NFκB signaling by
enhancing IκBα levels and blocking p65 binding at the promoters of
inflammatory genes via estrogen receptor-β. PLoS One. 7:e368902012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Monteiro R, Teixeira D and Calhau C:
Estrogen signaling in metabolic inflammation. Mediators Inflamm.
2014:6159172014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Özdemir Kumral ZN, Kolgazi M, Üstünova S,
Kasımay Çakır Ö, Çevik ÖD, Şener G and Yeğen BÇ: Estrogen receptor
agonists alleviate cardiac and renal oxidative injury in rats with
renovascular hypertension. Clin Exp Hypertens. 38:500–509. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bhatia A, Sekhon HK and Kaur G: Sex
hormones and immune dimorphism. ScientificWorldJournal.
2014:1591502014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Willebrords J, Crespo Yanguas S, Maes M,
Decrock E, Wang N, Leybaert L, Kwak BR, Green CR, Cogliati B and
Vinken M: Connexins and their channels in inflammation. Crit Rev
Biochem Mol Biol. 51:413–439. 2016. View Article : Google Scholar : PubMed/NCBI
|