Introduction
Pulmonary arterial hypertension (PAH) is a chronic
severe disease characterized by the obliteration of small pulmonary
arteries (Pas) and progressive development of pulmonary vascular
resistance, which eventually leads to right heart failure and
mortality (1). In PAH, the
aberrant growth, migration and phenotypic switch of pulmonary
arterial smooth muscle cells (PASMCs) serve a vital role in the
vascular remodeling and constriction of distal Pas (2). Therefore, the inhibition of PASMC
proliferation, migration and phenotypical alteration is a
prospective strategy for the treatment of PAH.
In addition to genetic, epigenetic and environmental
factors, the function of PASMCs is affected by numerous endogenous
molecules, including platelet-derived growth factor BB (PDGF-BB),
transforming growth factor-β and fibroblast growth factor (FGF)
(3,4). It has been demonstrated that PDGF-BB
may affect PASMC function via the regulation of the downstream
signals required for the progression of PAH (5). In addition, previous studies
demonstrated that the regulation of several microRNAs (miRNAs/miR)
by PDGF-BB was critical for PASMC function (6,7).
miRNAs are small endogenous non-coding RNAs that
serve as repressors of gene expression by regulating the stability
of their target mRNAs (8). It is
well-known that miRNAs are involved in the pathological processes
of PAH (9,10). Several of these miRNAs, including
miR-204 (11), miR-214 (12), let-7g (13), miR-143/145 (14), mir-130/301 (15) and miR-135a-5p (16) regulate PASMC function.
Previous studies have suggested that miR-132
inhibited vascular smooth muscle cell (VSMC) proliferation and
repressed neointimal formation (17). Furthermore, phosphatase and tensin
homolog deleted on chromosome 10 (PTEN) is a direct target of
miR-132 (18) and is involved in
the development of PAH (19).
However, the exact mechanism of this interaction remains unclear.
Therefore, the present study aimed to identify the role of miR-132
in the development of PAH in vivo and in vitro.
In the present study, it was demonstrated that
miR-132 was upregulated in the monocrotaline (MCT)-induced PAH rats
and PDGF-BB-treated PASMCs. The results additionally revealed that
miR-132 may affect the proliferation, migration and phenotypic
switching of PASMCs via the PTEN signaling pathway.
Materials and methods
Experimental animal model and
treatment
A total of 16 Sprague-Dawley male rats (~11 weeks
old, 180–200 g) were obtained from the Hunan Slack Jingda
Laboratory Animal Co., Ltd. (Changsha, China). The rats was
subjected to controlled conditions of 20–22°C, relative humidity of
50–55% and a 12-h light/dark cycle, and provided with ad
libitum access to food and water. The experiments were
performed in accordance with the National Institutes of Health
guidelines for the care and use of laboratory animals (20) and were approved by the Ethics
Committee of the Hunan University of Medicine (Huaihua, China). The
rats received a subcutaneous injection of 60 mg/kg MCT
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for the induction
of PAH. Control rats received 0.9% saline.
Hemodynamic measurements
PAH was established in the rats by the 21st day
following injection, at which point the rats were anesthetized via
an intraperitoneal injection of 60 mg/kg pentobarbital sodium. A
jugular catheter (1 mm) filled with heparinized saline was
introduced into the right external jugular vein, and subsequently
advanced through the right atrium and right ventricle (RV).
Appropriate positioning of the catheter was confirmed by the change
of the pressure waveform. Once the change in the pressure waveform
was stable, the RV systolic pressure (RVSP) was measured with a
multichannel physiological recorder monitoring equipment
(BL-420F).
Hematoxylin and eosin (H&E)
staining
The rats were immediately sacrificed by cervical
dislocation following the hemodynamic measurements. Sacrifice was
confirmed by a lack of breathing and blood pressure, following
which lung tissues were quickly harvested and fixed in 4%
paraformaldehyde at 4°C for 24 h. The paraffin-embedded lung tissue
samples were cut into 4-µm thick sections and stained with 0.1%
Mayer's hematoxylin (Sigma-Aldrich; Merck KGaA) for 5 min and 0.5%
eosin Y solution (Sigma-Aldrich; Merck KGaA) for 40 sec at room
temperature. Structural changes in the pulmonary vascular wall and
the wall thickness were observed using a Zeiss Axio Scope.A1
microscope (magnification, ×200; Zeiss GmbH, Jena, Germany).
Between six to eight vessels were analyzed per animal, and four
values for the thickness and two values for the diameter were
measured for each vessel. Percentage wall thickness (wall
thickness/vessel diameter) was calculated.
Cell culture and treatment
Human PASMCs were obtained from PromoCell GmbH
(Heidelberg, Germany) and were maintained in smooth muscle cell
growth medium (SMCGM; PromoCell GmbH) containing 5% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and 1% penicillin/streptomycin (Gibco; Thermo Fisher
Scientific, Inc.). Human PASMCs were starved in SMCGM containing a
low percentage of serum (0.2% FBS) for 24 h prior to treatment with
PDGF-BB (Sangon Biotech Co., Ltd., Shanghai, China) at 37°C. Cells
were treated with various concentrations of PDGF-BB (5, 10, 20, 40
and 80 ng/ml) for 24 h, or with 20 ng/ml PDGF-BB for various
incubation periods (2, 6, 12, 24 and 48 h). RNA was extracted from
PAMSCs following treatment, as described below. Each independent
experiment was performed at least 3 times.
miRNA transfection
The miR-132 mimics (miR10000426), the miR-132
inhibitors (miR20000426), the mimic negative control (NC;
miR1N0000001) and inhibitor NC (miR2N0000001) were purchased from
Guangzhou RiboBio Co., Ltd., (Guangzhou, China). The mimics and
inhibitors were synthesized on the basis of sequence of
hsa-miR-132-3p (5′-UAACAGUCUACAGCCAUGGUCG-3′). Human PASMCs were
seeded in 6-well culture plates at a density of 2×105
cells/well, incubated for 24 h, and transfected with miR-132 mimics
(20 nM), miR-132 inhibitor (50 nM) or NCs (20 nM for mimics; 50 nM
for inhibitors) for 6 h according to the instructions provided by
the manufacturer of the Hiperfect Transfection Reagent (Qiagen
GmbH, Hilden, Germany). The culture media was replaced and the
cells were further incubated for 42–66 h prior to subsequent
experiments.
Cell proliferation assay
PASMCs were seeded in a 96-well plate. Cell
viability was determined with an MTS CellTiter 96®
AQueous one solution cell proliferation assay (Promega
Corporation, Madison, WI, USA). The absorbance was detected by
spectrophotometry at 490 nM using a Berthold LB-942 instrument
(Berthold Technologies GmbH & Co., KG, Bad Wildbad,
Germany).
Cell cycle analysis
PASMCs were treated in groups as indicated in the
transfection protocol and fixed with 70% ethanol overnight at 4°C.
Following washing with cold PBS, 5 µl RNase A (10 mg/ml) was added
to the cells. Subsequently, the cells were stained with 1 mg/ml
propidium iodide solution (Sigma-Aldrich; Merck KGaA) for 15 min at
4°C. The cell cycle analysis was performed by flow cytometry
(CytoFLEX; Beckman Coulter, Inc., Brea, CA, USA) and the results
were analyzed using ModFit LT 3.3 software (Verity Software House,
Inc., Topsham, ME, USA).
Transwell assay
Cell migration was assessed using a Transwell assay.
The transfected cells were seeded in the top chamber containing
medium with 0.1% FBS. The lower chamber was filled with SMCGM
containing 5% FBS. PASMCs were allowed to migrate for 24 h at 37°C.
The cells were fixed in 4% paraformaldehyde for 30 min and then
stained with 0.1% crystal violet for 30 min, each at room
temperature. Finally, the cells from five random fields were
counted using a Zeiss Axio Vert.A1 microscope (magnification, ×200;
Zeiss GmbH) and the images were processed using Image-Pro Plus 6.0
software (Media Cybernetics, Inc., Rockville, MD, USA).
RNA preparation and reverse
transcription quantitative polymerase chain reaction (RT-qPCR)
analysis
The total RNA of PASMCs and lung tissues was
extracted using TRIzol® reagent (Thermo Fisher
Scientific, Inc.) as described previously (21). The concentration of the RNA samples
was quantified using NanoDrop One spectrophotometer (NanoDrop
Technologies; Thermo Fisher Scientific, Inc., Wilmington, DE, USA).
Purified RNA was reverse transcribed with PrimeScript™ RT reagent
(Takara Bio, Inc., Otsu, Japan) at 37°C for 15 min and 85°C for 5
sec. Mature miRNA was reverse transcribed with Bulge-Loop™ miRNA
qPCR primers (Guangzhou RiboBio Co., Ltd.) prior to qPCR according
to the manufacturer's protocol. The qPCR experiments were conducted
using an SYBR Premix ExTaq™ kit (Takara Biotechnology, Co., Ltd.,
Dalian, China) in a LightCycler 480 Real-Time PCR System (Roche
Applied Science, Mannheim, Germany). PCR was conducted as follows:
95°C for 30 sec, followed by 40 cycles of 95°C for 5 sec and 60°C
for 30 sec. The expression levels were quantified using the
2−ΔΔCq method (22)
with U6 and β-actin as the endogenous reference genes
for miRNA and mRNA expressions, respectively. The primer sequences
were as follows: α-smooth muscle actin (α-SMA) forward,
5′-TATCAGGGGGCACCACTATG-3′ and reverse,
5′-AGGAGCAGGAAAGTGTTTTAGA-3′; transgelin (SM22α) forward,
5′-TGGTGAACAGCCTGTACCCT-3′ and reverse, 5′-CACGGTAGTGCCCATCATTC-3′;
calponin 1 (CNN1) forward, 5′-TGCTACAGGGTCCAACATAGA-3′ and
reverse, 5′-GTTGCTCAGTGCGTCCTT-3′; β-actin forward,
5′-TGTCACCAACTGGGACGATA-3′ and reverse, 5′-ACCCTCATAGATGGGCACAG-3′.
The primers of miR-132 (MQPS0000604) and U6 (MQPS0000002) were
purchased from Guangzhou RiboBio Co., Ltd., (Guangzhou, China).
Western blot analysis
Western blot analysis of total protein content was
performed as previously described (23). Briefly, protein was extracted from
PASMCs and lung tissues in ice-cold radioimmunoprecipitation assay
lysis buffer (P0013B, Beyotime Institute of Biotechnology,
Shanghai, China) and quantified using Bicinchoninic Acid Protein
Assay Reagent (Pierce; Thermo Fisher Scientific, Inc.). Protein
lysate (40 µg/lane) was separated by 10% SDS-PAGE and blotted onto
polyvinylidene difluoride membranes. The membranes were blocked
with 5% non-fat milk for 1 h at room temperature and incubated with
the following primary antibodies at 4°C overnight: Rabbit
anti-α-SMA monoclonal antibody (dilution, 1:1,000; A2547,
Sigma-Aldrich; Merck KGaA); anti-SM22α polyclonal antibody
(dilution, 1:1,000; HPA001925, Sigma-Aldrich; Merck KGaA);
anti-PTEN polyclonal antibody (dilution, 1:1,000; D261095, Sangon
Biotech Co., Ltd.); anti-p-PTEN polyclonal antibody (dilution,
1:1,000; D155023, Sangon Biotech Co., Ltd.); anti-α-tubulin
monoclonal antibody (dilution, 1:5,000; A01410, GenScript,
Piscataway, NJ, USA) and anti-β-actin monoclonal antibody
(dilution, 1:5,000; A00702, GenScript). Following washing,
membranes were incubated for 2 h at room temperature with
horseradish peroxide-conjugated goat-anti-mouse (D110087) or
goat-anti-rabbit (D110058) immunoglobulin G secondary antibodies
(dilution, 1:6,000; Sangon Biotech Co., Ltd.). Finally, the protein
expression levels were detected with an enhanced chemiluminescent
substrate (cat. no. 32209, Thermo Fisher Scientific, Inc.) and
recorded with ChemiDoc XRS+ system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The results were analyzed using ImageJ 1.52
software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
The data are presented as the mean ± standard
deviation of at least three independent experiments. Data analysis
was conducted using GraphPad Prism 5.0 software (GraphPad Software,
Inc., La Jolla, CA, USA). Comparisons between two groups were
performed by a double-sided Student's t-test. A one-way analysis of
variance test followed by a Student-Newman-Keuls post-hoc test was
performed for multiple comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
miR-132 expression levels in
MCT-induced PAH rats
To investigate whether miR-132 serves a role in PAH,
the miR-132 expression levels in MCT-induced PAH rats were
initially detected by RT-qPCR assays. The results indicated that
the expression levels of miR-132 were upregulated in the rat lung
tissues and PAs following 3 weeks of MCT exposure (Fig. 1A). The rats demonstrated increased
RVSP and RV/(left ventricle + septum) ratios compared with that of
the untreated group after 3 weeks of MCT exposure (Fig. 1B and C). Fig. 1D revealed that the medial thickness
of the pulmonary artery was increased in the MCT-induced rats.
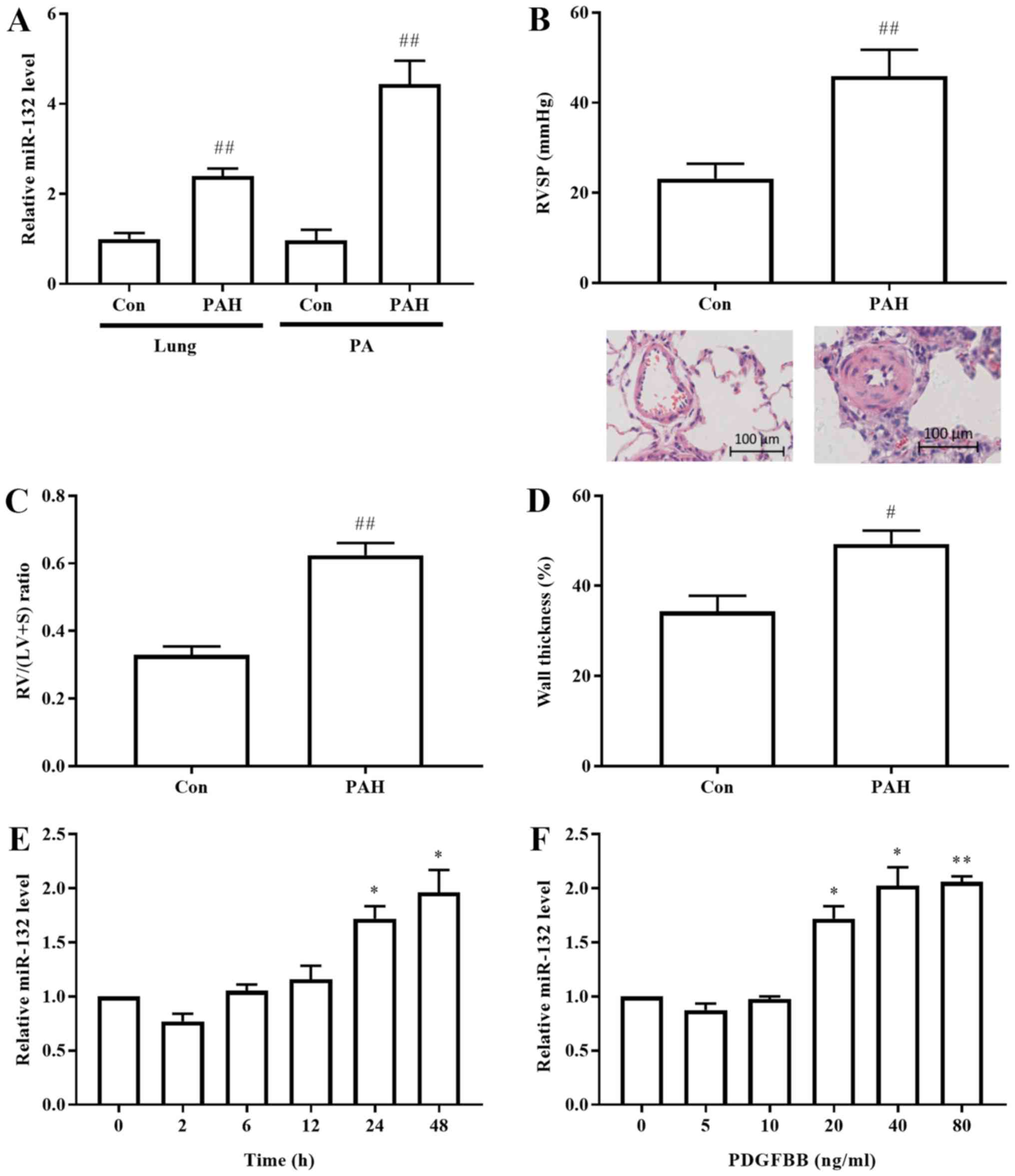 | Figure 1.miR-132 expression is increased in
MCT-induced PAH rats and PDGF-BB-treated PASMCs. (A) The levels of
miR-132 were measured by reverse transcription polymerase chain
reaction in lung tissues and PA from rats. (B) RVSP and (C) RV/(LV
+ S) ratios in the control and PAH rats were compared. (D) The
degree of pulmonary arterial wall thickening in the rats was
observed by hematoxylin and eosin staining. (E) PASMCs were treated
with 20 ng/ml PDGF-BB for different incubation periods. (F) PASMCs
were treated with various concentrations of PDGF-BB for 24 h and
the miR-132 levels were assessed. Data are presented as the mean ±
standard deviation (n=6). Scale bar, 100 µm. #P<0.05
and ##P<0.01 vs. control. *P<0.05 and **P<0.01
vs. control without PDGF-BB treatment. miR, microRNA; con, control;
PDGF-BB, platelet-derived growth factor BB; PAH, pulmonary arterial
hypertension; PA, pulmonary artery; RVSP, right ventricular
systolic pressure; RV, right ventricle/ventricular; LV, left
ventricle/ventricular; S, septum. |
miR-132 expression levels in
proliferating human PASMCs
Subsequently, the miR-132 expression levels in
PASMCs that were stimulated by PDGF-BB were determined. As
demonstrated in Fig. 1E, the
miR-132 levels increased gradually following treatment of PASMCs
with 20 ng/ml PDGF-BB from 2–48 h, indicating a time-dependent
response. In addition, PDGF-BB resulted in a rapid increase of
miR-132 expression levels in a dose-dependent manner (Fig. 1F).
Effects of miR-132 on human PASMC
proliferation
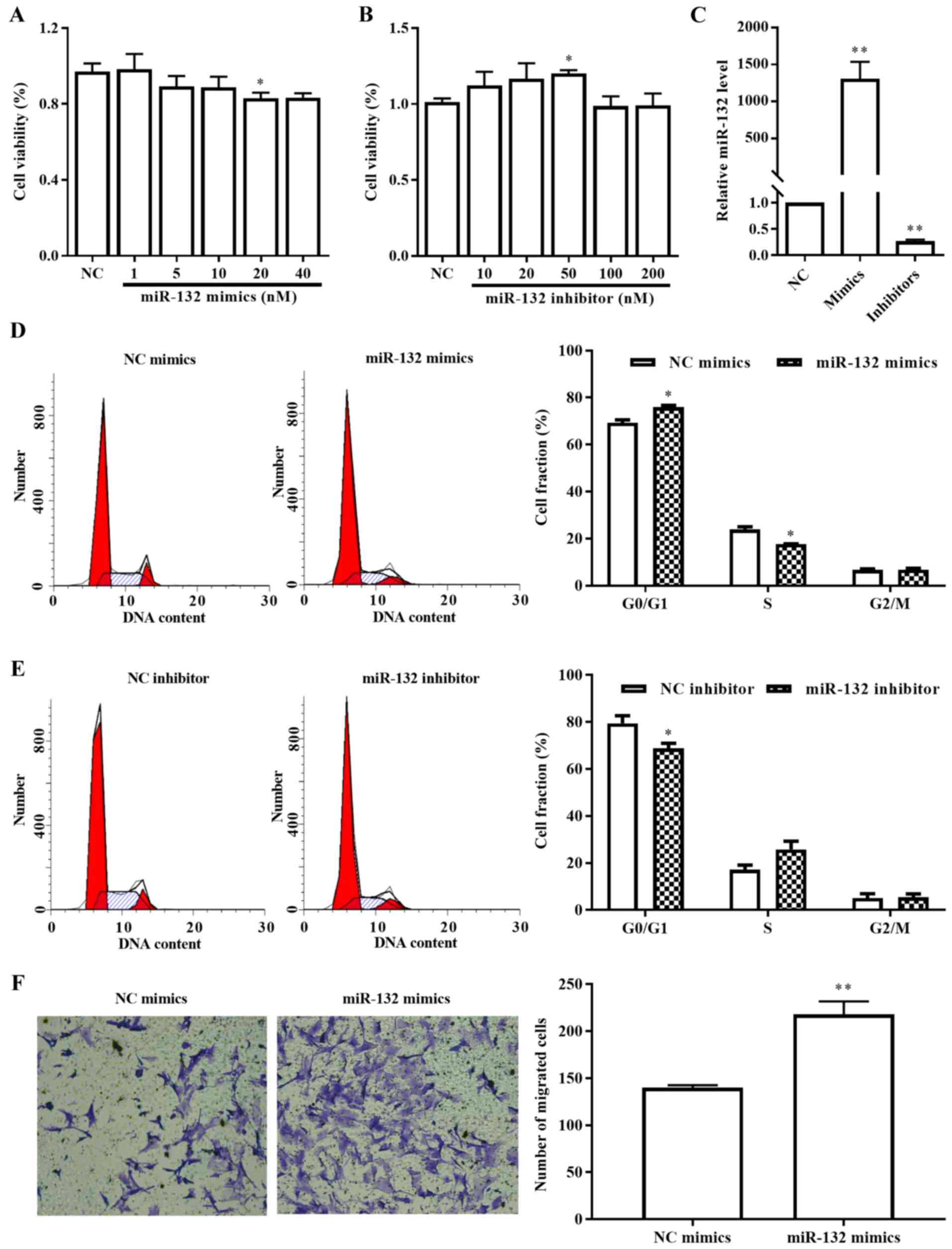
To explore the role of miR-132 in PAH, its effects
on the proliferation and cell cycle of PASMCs were detected using
cell viability and flow cytometry assays. The results indicated
that 20 nM miR-132 mimics significantly inhibited cell
proliferation (Fig. 2A), while 50
nM miR-132 inhibitor promoted cell proliferation (Fig. 2B). Upregulation and downregulation
of miR-132 following transfection of PASMCs with mimics and
inhibitors, respectively, was demonstrated by RT-qPCR analysis
(Fig. 2C). Furthermore, the
results of the cell cycle assay indicated that the percentages of
the cells that were present in the G2/M and S phases were decreased
following miR-132 mimics (20 nM) treatment (Fig. 2D), indicating that the
anti-proliferative effect of miR-132 was attributed to cell cycle
arrest. In contrast to the miR-132 mimics, the miR-132 inhibitor
(50 nM) decreased the percentage of the cells at the
G0/G1-phase (Fig. 2E).
Effects of miR-132 on human PASMC
migration
In addition to cell proliferation, the migration of
PASMCs has been considered to be a contributor in the development
of PAH. The results of the Transwell assay indicated that the
overexpression of miR-132 significantly increased the migration of
human PASMCs (Fig. 2F).
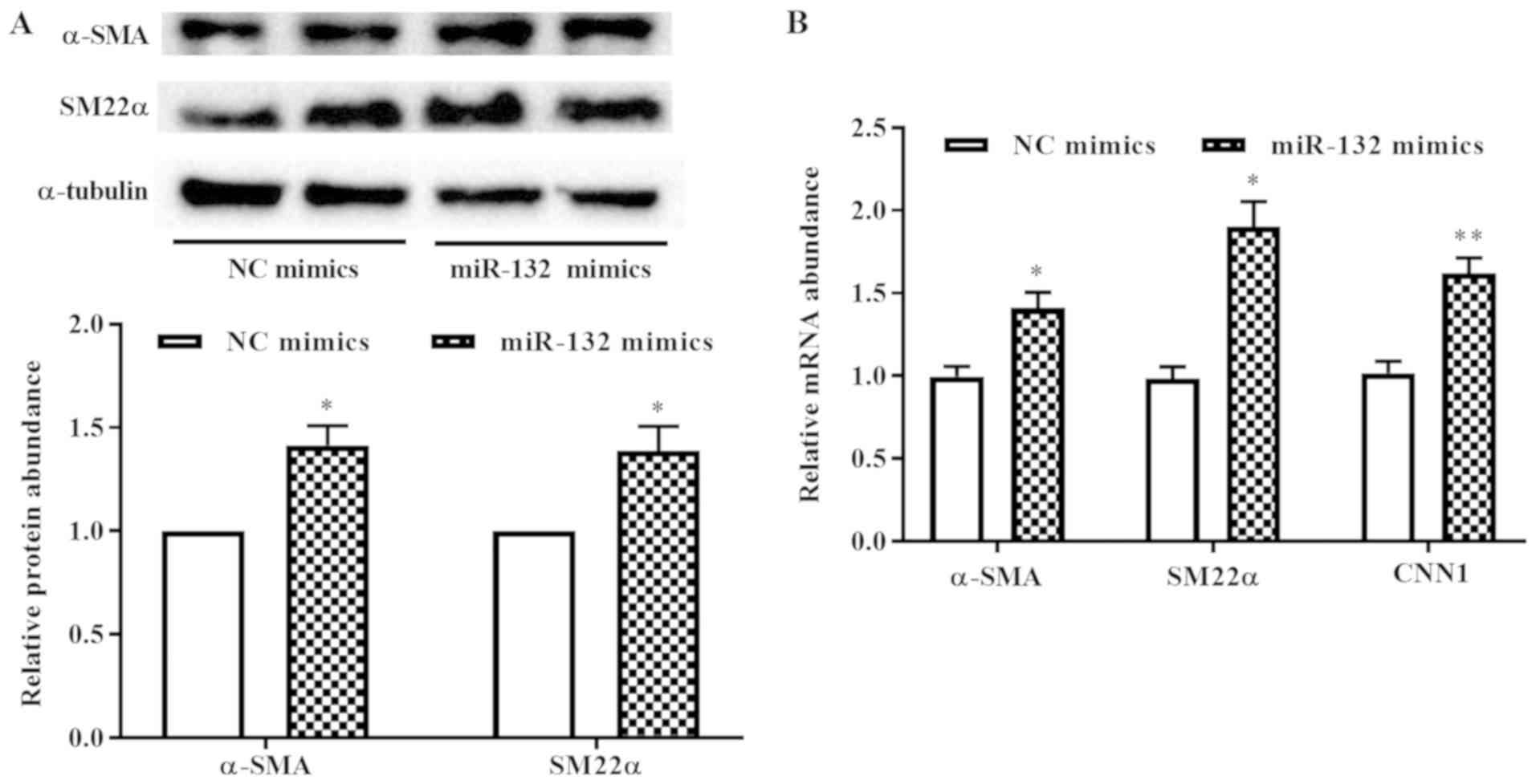
Effects of miR-132 on the human PASMC
phenotype
To additionally validate the role of miR-132 in PAH,
the phenotypic switch of PASMCs was investigated by determining the
expression of mRNAs and proteins of smooth muscle cell (SMC)
phenotypic markers following overexpression of miR-132.
Overexpression of miR-132 in human PASMCs increased the levels of
α-SMA, SM22α and CNN1 compared with those in the NC samples
(Fig. 3A and B), indicating that
miR-132 may be essential for maintaining the PASMC contractile
phenotype.
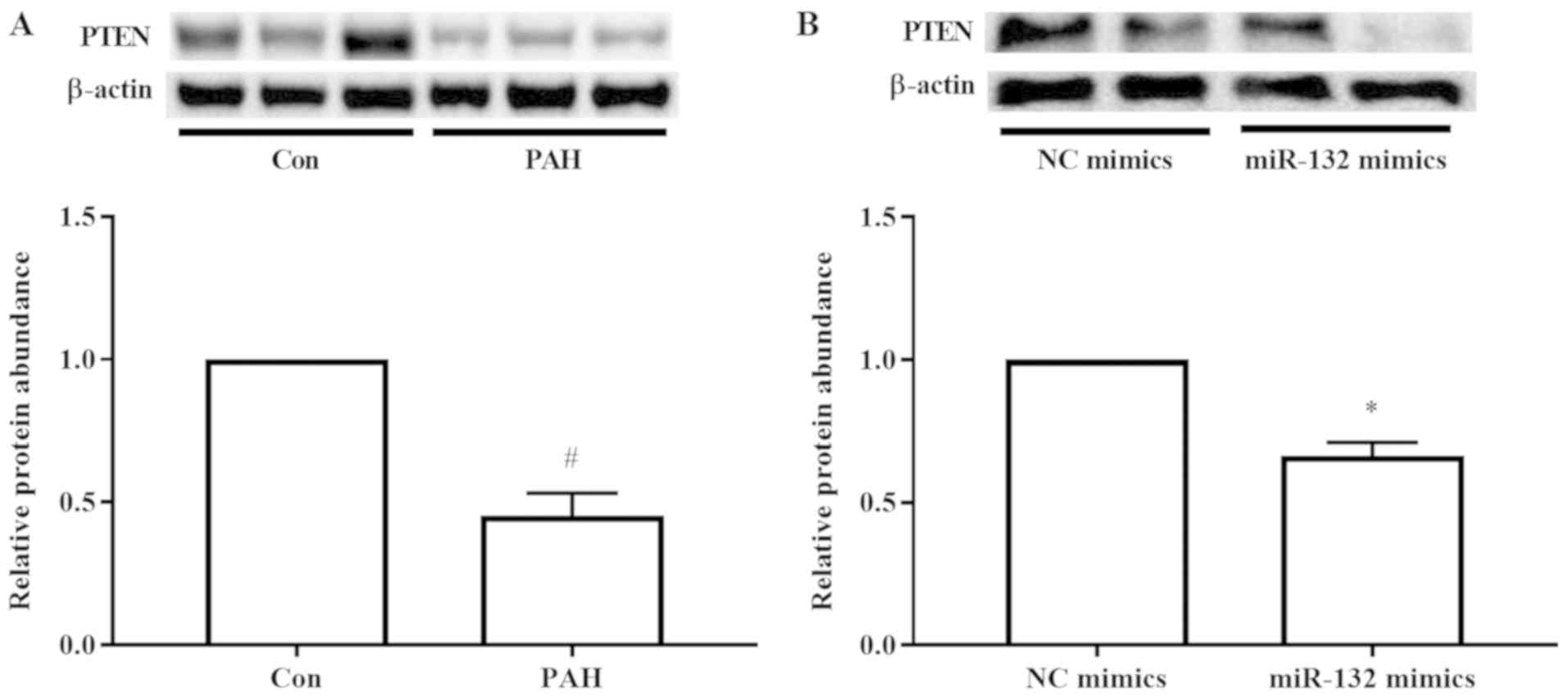
PTEN is a direct target of
miR-132
It has been demonstrated that PTEN is a direct
target of miR-132 in vascular smooth muscle cells (VSMCs) (18). To examine the interaction between
miR-132 and PTEN in PAH, the expression levels of PTEN were
detected in vivo and in vitro. The results indicated
that PTEN expression levels were decreased in MCT-induced PAH
(Fig. 4A). Furthermore, it was
identified that transfection of human PASMCs with miR-132 mimics
downregulated the expression levels of PTEN significantly (Fig. 4B).
Discussion
PAH is a fatal disease without effective treatment
and is primarily characterized by the excessive proliferation of
PASMCs. Numerous studies have revealed that miRNAs serve a
fundamental role in the pathogenesis of PAH by affecting the
functions of PASMCs and pulmonary artery endothelial cells
(24,25). In the present study, the role of
miR-132 in PAH in vivo and in PASMCs in vitro was
examined.
miR-132 is a highly conserved miRNA involved in the
development of the central nervous and cardiovascular systems.
Several studies have suggested that miR-132 expression is
upregulated in certain cardiovascular diseases, including cardiac
hypertrophy, hypertension and atherosclerosis (17,26,27).
These studies demonstrated that miR-132 affected the proliferation
and migration of cardiomyocytes, endothelial cells and VSMCs.
Similarly, in the present study, miR-132 was highly expressed in
PAH and its expression levels were markedly increased following
PDGF-BB treatment, which indicated that miR-132 contributed to the
development of PAH.
miR-132 has been demonstrated to regulate the
function of vessel cells by different mechanisms of action. For
example, miR-132 promoted the proliferation of endothelial cells
via p120RasGAP (28) and inhibited
the proliferation and migration of VSMCs (17) by targeting leucine-rich repeat
flightless interacting protein 1. In contrast to these results, the
present study revealed that miR-132 exerted multiple functions in
PASMCs and served a complex role in the development of PAH, namely
as an anti-proliferative factor in PASMCs and an aid to the
maintenance of the contractile phenotype, while facilitating cell
migration.
The major structural change observed during the
development of PAH is the remodeling of small PAs, and typical
vessel lesions that consist of neointima formation, medial
thickening, adventitial fibrosis and angioproliferative plexiform
lesions (29,30). These processes are accompanied with
proliferative and anti-apoptotic phenotypes of pulmonary
endothelial cells, smooth muscle cells and fibroblasts (29,30).
The data from the present study suggest that the pro-migrative
effect of miR-132 contributed to the formation of plexiform
lesions. However, its anti-proliferative and differentiation
effects that were noted in vivo appeared to be contradictory
to the results from the in vitro studies. The first
possibility is that the proliferation of PASMCs is induced by other
key factors including hypoxia, FGF and PDGF-BB. A second possible
explanation is that the differentiation effect of miR-132 may
facilitate the formation of SMCs and SMC-like cells. Taken
collectively, these results provide novel evidence that miR-132 may
be involved in the development of PAH and that it is detrimental to
the development of vessel lesions. However, additional studies are
required to ensure that the inhibition of miR-132 may prevent and
reverse the progression of PAH.
The heterogeneous functions of miR-132 in SMCs are
similar with those noted for the Krüppel-like factor 4 (KLF4),
which is an additional key molecule involved in the regulation of
SMC phenotypic switching and in the development of vascular
diseases including PAH (31).
Increased KLF4 levels in SMCs were demonstrated in injured vessels
in vivo (32,33), and in response to PDGF-BB in
vitro (32). KLF4 decreased
SMC growth arrest by cellular tumor antigen p53-independent
activation of cyclin-dependent kinase inhibitor 1, whereas it
concomitantly induced SMC dedifferentiation via suppression of the
expression of SMC-specific differentiation markers (32). This promoted SMC migration via
binding to the collagen VIII alpha 1 promoter (32).
The present study explored the molecular mechanisms
of miR-132 with regard to the regulation of PASMCs, with a focus on
the miR-132-targeted gene PTEN (18), which is also downregulated in
MCT-induced PAH (34). PTEN is
well-known as a critical regulator of cell proliferation, apoptosis
and migration (35). It has been
demonstrated that inhibition of PTEN may attenuate PASMC
proliferation and increase their migration through activation of
RAC alpha serine/threonine-protein kinase (AKT) (35–37).
Therefore, the present study aimed to investigate whether miR-132
may regulate PASMC function via the PTEN-AKT pathway. The data
indicated that the expression of PTEN was decreased in the lung
tissues of PAH rats and in miR-132-overexpressing PASMCs; however,
the levels of phosphorylated AKT protein were also reduced (data
not shown). PTEN is a suppressor of AKT phosphorylation and
interacts with numerous downstream molecules (35). Thus, the targeting of PTEN by
miR-132 may regulate the proliferation, migration and phenotype of
PASMCs via the activation and/or suppression of other signaling
pathways. The exact mechanism will be additionally explored in
future studies.
In summary, the present study suggests that miR-132
served an important and pleiotropic role in modulating PASMC
proliferation, migration and phenotypic switching by targeting
PTEN. These data led to the identification of miR-132 as a
potential target for the treatment of pulmonary vascular remodeling
diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific
Research Fund of Hunan Provincial Education Department (grant no.
17B188).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZZZ and WHW conceived and designed the study. QP,
YHS and JXL performed the experiments. ZZZ wrote the paper. WW
reviewed and edited the manuscript. All authors read and approved
the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Hunan University of Medicine.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lau EMT, Giannoulatou E, Celermajer DS and
Humbert M: Epidemiology and treatment of pulmonary arterial
hypertension. Nat Rev Cardiol. 14:603–614. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen KH, Dasgupta A, Lin J, Potus F,
Bonnet S, Iremonger J, Fu J, Mewburn J, Wu D, Dunham-Snary K, et
al: Epigenetic dysregulation of the Drp1 binding partners MiD49 and
MiD51 increases mitotic mitochondrial fission and promotes
pulmonary arterial hypertension: Mechanistic and therapeutic
implications. Circulation. 138:287–304. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maron BA and Leopold JA: Emerging concepts
in the molecular basis of pulmonary arterial hypertension: Part II:
Neurohormonal signaling contributes to the pulmonary vascular and
right ventricular pathophenotype of pulmonary arterial
hypertension. Circulation. 131:2079–2091. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ten Freyhaus H, Berghausen EM, Janssen W,
Leuchs M, Zierden M, Murmann K, Klinke A, Vantler M, Caglayan E,
Kramer T, et al: Genetic ablation of PDGF-Dependent signaling
pathways abolishes vascular remodeling and experimental pulmonary
hypertension. Arterioscler Thromb Vasc Biol. 35:1236–1245. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tallquist M and Kazlauskas A: PDGF
signaling in cells and mice. Cytokine Growth Factor Rev.
15:205–213. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sysol JR, Natarajan V and Machado RF: PDGF
induces SphK1 expression via Egr-1 to promote pulmonary artery
smooth muscle cell proliferation. Am J Physiol Cell Physiol.
310:C983–C992. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen J, Cui X, Li L, Qu J, Raj JU and Gou
D: MiR-339 inhibits proliferation of pulmonary artery smooth muscle
cell by targeting FGF signaling. Physiol Rep. 5:e134412017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Picao-Osorio J, Johnston J, Landgraf M,
Berni J and Alonso CR: MicroRNA-encoded behavior in drosophila.
Science. 350:815–820. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiao T, Xie L, Huang M and Shen J:
Differential expression of microRNA in the lungs of rats with
pulmonary arterial hypertension. Mol Med Rep. 15:591–596. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thenappan T, Ormiston ML, Ryan JJ and
Archer SL: Pulmonary arterial hypertension: Pathogenesis and
clinical management. BMJ. 360:j54922018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Courboulin A, Paulin R, Giguère NJ,
Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher
S, Côté J, et al: Role for miR-204 in human pulmonary arterial
hypertension. J Exp Med. 208:535–548. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sahoo S, Meijles DN, Al Ghouleh I, Tandon
M, Cifuentes-Pagano E, Sembrat J, Rojas M, Goncharova E and Pagano
PJ: MEF2C-MYOCD and Leiomodin1 suppression by miRNA-214 promotes
smooth muscle cell phenotype switching in pulmonary arterial
hypertension. PLoS One. 11:e01537802016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang WF, Xiong YW, Zhu TT, Xiong AZ, Bao
HH and Cheng XS: MicroRNA let-7g inhibited hypoxia-induced
proliferation of PASMCs via G0/G1 cell cycle arrest by targeting
c-myc. Life Sci. 170:9–15. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deng L, Baker AH and Bradshaw AC: MicroRNA
delivery strategies to the lung in a model of pulmonary
hypertension. Methods Mol Biol. 1521:325–338. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bertero T, Cottrill K, Krauszman A, Lu Y,
Annis S, Hale A, Bhat B, Waxman AB, Chau BN, Kuebler WM and Chan
SY: The microRNA-130/301 family controls vasoconstriction in
pulmonary hypertension. J Biol Chem. 290:2069–2085. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu HM, Jia Y, Zhang YX, Yan J, Liao N, Li
XH and Tang Y: Dysregulation of miR-135a-5p promotes the
development of rat pulmonary arterial hypertension in vivo and in
vitro. Acta Pharmacol Sin. 2018.
|
|
17
|
Choe N, Kwon JS, Kim JR, Eom GH, Kim Y,
Nam KI, Ahn Y, Kee HJ and Kook H: The microRNA miR-132 targets
Lrrfip1 to block vascular smooth muscle cell proliferation and
neointimal hyperplasia. Atherosclerosis. 229:348–355. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jin W, Reddy MA, Chen Z, Putta S, Lanting
L, Kato M, Park JT, Chandra M, Wang C, Tangirala RK and Natarajan
R: Small RNA sequencing reveals microRNAs that modulate angiotensin
II effects in vascular smooth muscle cells. J Biol Chem.
287:15672–15683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lai YJ, Hsu HH, Chang GJ, Lin SH, Chen WJ,
Huang CC and Pang JS: Prostaglandin E1 attenuates pulmonary artery
remodeling by activating phosphorylation of CREB and the PTEN
signaling pathway. Sci Rep. 7:99742017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
National Research Council, . Guide for the
care and use of laboratory animals. 8th. Washington (DC): National
Academies Press (US); 2011, PubMed/NCBI
|
|
21
|
Wu WH, Hu CP, Chen XP, Zhang WF, Li XW,
Xiong XM and Li YJ: MicroRNA-130a mediates proliferation of
vascular smooth muscle cells in hypertension. Am J Hypertens.
24:1087–1093. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zeng Z, Huang Q, Shu Z, Liu P, Chen S, Pan
X, Zang L and Zhou S: Effects of short-chain acyl-CoA dehydrogenase
on cardiomyocyte apoptosis. J Cell Mol Med. 20:1381–1391. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bienertova-Vasku J, Novak J and Vasku A:
MicroRNAs in pulmonary arterial hypertension: Pathogenesis,
diagnosis and treatment. J Am Soc Hypertens. 9:221–234. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leopold JA and Maron BA: Molecular
mechanisms of pulmonary vascular remodeling in pulmonary arterial
hypertension. Int J Mol Sci. 17:E7612016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ucar A, Gupta SK, Fiedler J, Erikci E,
Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann
A, et al: The miRNA-212/132 family regulates both cardiac
hypertrophy and cardiomyocyte autophagy. Nat Commun. 3:10782012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eskildsen TV, Jeppesen PL, Schneider M,
Nossent AY, Sandberg MB, Hansen PB, Jensen CH, Hansen ML, Marcussen
N, Rasmussen LM, et al: Angiotensin II regulates microRNA-132/-212
in hypertensive rats and humans. Int J Mol Sci. 14:11190–11207.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Anand S, Majeti BK, Acevedo LM, Murphy EA,
Mukthavaram R, Scheppke L, Huang M, Shields DJ, Lindquist JN,
Lapinski PE, et al: MicroRNA-132-mediated loss of p120RasGAP
activates the endothelium to facilitate pathological angiogenesis.
Nat Med. 16:909–914. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ghataorhe P, Rhodes CJ, Harbaum L, Attard
M, Wharton J and Wilkins MR: Pulmonary arterial
hypertension-progress in understanding the disease and prioritizing
strategies for drug development. J Intern Med. 282:129–141. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lythgoe MP, Rhodes CJ, Ghataorhe P, Attard
M, Wharton J and Wilkins MR: Why drugs fail in clinical trials in
pulmonary arterial hypertension, and strategies to succeed in the
future. Pharmacol Ther. 164:195–203. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yoshida T and Hayashi M: Role of
kruppel-like factor 4 and its binding proteins in vascular disease.
J Atheroscler Thromb. 21:402–413. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Alexander MR and Owens GK: Epigenetic
control of smooth muscle cell differentiation and phenotypic
switching in vascular development and disease. Annu Rev Physiol.
74:13–40. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Y, Sinha S, McDonald OG, Shang Y,
Hoofnagle MH and Owens GK: Kruppel-like factor 4 abrogates
myocardin-induced activation of smooth muscle gene expression. J
Biol Chem. 280:9719–9727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ravi Y, Selvendiran K, Meduru S, Citro L,
Naidu S, Khan M, Rivera BK, Sai-Sudhakar CB and Kuppusamy P:
Dysregulation of PTEN in cardiopulmonary vascular remodeling
induced by pulmonary hypertension. Cell Biochem Biophys.
67:363–372. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee YR, Chen M and Pandolfi PP: The
functions and regulation of the PTEN tumour suppressor: New modes
and prospects. Nat Rev Mol Cell Biol. 19:547–562. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu Y, Cao Y, Sun S, Zhu J, Gao S, Pang J,
Zhu D and Sun Z: Transforming growth factor-beta1 upregulation
triggers pulmonary artery smooth muscle cell proliferation and
apoptosis imbalance in rats with hypoxic pulmonary hypertension via
the PTEN/AKT pathways. Int J Biochem Cell Biol. 77:141–154. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu B, Gong Y, Yan G, Wang D, Qiao Y, Wang
Q, Liu B, Hou J, Li R and Tang C: Down-regulation of lncRNA MEG3
promotes hypoxia-induced human pulmonary artery smooth muscle cell
proliferation and migration via repressing PTEN by sponging miR-21.
Biochem Biophys Res Commun. 495:2125–2132. 2018. View Article : Google Scholar : PubMed/NCBI
|