Introduction
Multiple myeloma (MM) is a neoplastic disorder
characterized by expansion and accumulation of clonal plasma cells
in the bone marrow (BM), secretion of monoclonal immunoglobulin and
presence of osteolytic bone lesions (1,2).
This plasma cell dyscrasia is the second most common hematological
malignancy (after non-Hodgkins lymphoma) accounting for ~1% of all
cancer diagnoses (3). Over the
last two decades, significant progress has been made in the
management of MM because of the introduction of new therapeutic
agents, such as proteasome inhibitors (PIs) and immunomodulatory
agents (IMiDs) (4).
PIs such as bortezomib and carfilzomib have assumed
a central role for the management of MM. Bortezomib can be used at
all phases of MM treatment, from frontline to combination therapy,
or for the re-treatment of relapsed disease (5–8).
Despite these newer agents, responses to therapy are transient and
patients treated with PIs tend to develop resistance and to become
refractory to their treatment regimens (9–11).
Therefore, MM remains an incurable disease with a fatal outcome and
new approaches to enhance the response to proteasome-targeted drugs
are an unmet need.
In a previous study, we evaluated the methylation
pattern in a group of genes in MM patients and observed an
association between Deleted in colorectal cancer
(DCC) hypermethylation and poor survival (12), suggesting that this gene could play
a role in MM tumorigenesis. It is well known that DCC
encodes a cell surface receptor whose ligand is Netrin 1. This
receptor can act in cell-cell and cell-matrix interactions and it
is involved in both epithelial and neuronal-cell differentiation
(13,14). DCC has been proposed as a
tumor suppressor gene because most of the advanced colorectal
carcinomas show losses of the 18q region where this gene is located
(15,16). The loss of DCC expression is
not restricted to colorectal cancer, and it is also found in other
tumors such as stomach, pancreas, esophagus, prostate, bladder,
breast, neuroblastomas and gliomas (17,18).
However, the rarity of point mutations in DCC coding
sequences, associated to the lack of a tumor predisposition
phenotype in DCC hemizygous mice and to a correlation with
the relapse site in gastric cancer, has raised questions about its
role as a tumor suppressor (19–21).
Hence, this study was performed to gain an understanding of the
DCC biological function and its role in MM tumorigenesis, as
well as to evaluate this receptor contribution to bortezomib
treatment response.
Materials and methods
Cell line culture
The cell lines RPMI8226 and SKO007 were a kind gift
from Octavia L. Caballero (Ludwig Institute for Cancer Research,
New York branch) and the cell line U266 was provided by Anamaria
Camargo Aranha (Ludwig Institute for Cancer Research, São Paulo
branch). All cell lines were maintained in suspension with RPMI
1640 medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
supplemented with 10% fetal bovine serum (Thermo Fisher Scientific,
Inc., Waltham, MA, USA), 1% non-essential amino acids (MEM NEAA)
and 0.01 µg/ml of penicillin-streptomycin (both Gibco; Thermo
Fisher Scientific, Inc.). These cells were grown in an incubator at
37°C with 5% CO2. Routinely, cells were treated with
drug concentrations previously described in the literature. The
bortezomib treatment was conducted by keeping the cells in the
presence of 10 nM of the drug for 48 h. For demethylation study,
RPMI8226 cells were seeded on day 0 and treated with 10 µM of
5-aza-2-deoxycytidine (decitabine, DAC; Sigma-Aldrich; Merck KGaA)
for 3 days. DNA and RNA were extracted at days 0 and 3, and stored
at −80°C.
RNA extraction, cDNA synthesis and
RT-qPCR
RNA extraction was performed using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturers instructions. After extraction, 25 µg of total RNA
were treated with RQ1 RNase-Free DNase (Promega Corp.,
Madison, WI, USA) to eliminate the presence of genomic DNA and 2 µg
of total RNA were subjected to cDNA synthesis using the SuperScript
III First-Strand Synthesis System (Invitrogen; Thermo Fisher
Scientific, Inc.). The cDNA obtained was diluted 10-fold before
use. The DCC mRNA expression was determined by RT-qPCR using
an ABI 7500 Sequence Detection System (Applied Biosystems)
and SYBR-Green reagent (both Thermo Fisher Scientific,
Inc.). The primer sequences are available upon request. All
reactions were performed in triplicate. The 2−ΔΔCt
method was employed to evaluate the expression of DCC in the
MM cell lines (22). In a previous
study, using geNorm algorithm to assess gene expression stability
of different housekeeping genes in MM samples, we determined
GAPDH and ACTB as the most stable combination of
genes to be used to normalize the expression data in MM (23). Therefore, mean Ct values of these
two genes were used for the normalization of RT-qPCR data and the
results were illustrated in arbitrary units.
Methylation-specific PCR
Bisulfite treatment of DNA converts unmethylated
cytosine to uracil while methylated ones remain as cytosine. Sodium
bisulfite conversion of 2 µg of genomic DNA was performed as
previously described (24).
Bisulfite-modified DNA was used as a template for
fluorescence-based real-time PCR (qMSP) as described previously
(25). Primers and probes were the
same used by de Carvalho et al (12). All reactions were performed in
triplicate. Leukocyte DNA obtained from a healthy individual was
methylated in vitro using SSSI methyltransferase enzyme (New
England Biolabs Inc., Ipswich, MA, USA) to generate methylated DNA
at all CpG sites (positive control). The calculation of the
methylation level was performed using the 2−ΔΔCt
equation (22).
Western blotting
Cell lines were harvested, washed in ice-cold
phosphate-buffered saline solution and resuspended in lyses buffer
(50 mM Tris-HCl pH 7.4; 100 mM NaCl; 0.5% NP-40) containing a
protease inhibitor cocktail (Roche, Applied Science, Penzberg,
Germany). The proteins were resolved in SDS-PAGE and transferred to
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). The membranes were blocked in Tris-buffered saline solution
containing 0.05% of Tween-20 and 5% of nonfat dry milk for 1 h at
room temperature. Further, the membranes were incubated with the
appropriate primary antibodies for 2 h using the following
dilutions: 1:100 DCC (cat. no. A-20; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) and 1:1,000 α/β-Tubulin (cat. no. 2148; Cell
Signaling Technologies, Inc., Danvers, MA, USA). As secondary
antibodies, we used goat anti-mouse (cat. no. 14-13-06; Kirkegaard
& Perry Laboratories Inc., Gaithersburg, MD, USA) and
anti-rabbit IgG HRP-linked (cat. no. 7074; Cell Signaling
Technologies, Inc.), conjugated with peroxidase. Proteins were
detected using the Chemiluminescent HRP Substrate (EMD Millipore)
and visualized in an ImageQuantLass 4000 system (GE Healthcare Life
Sciences).
DCC esiRNA transfections
The U266 cells were transfected with esiRNAs (cat.
no. EHU014471; Mission esiRNA1; Sigma-Aldrich; Merck KGaA)
targeting DCC or with a pmaxGFP™ control vector
(Lonza Group, Ltd., Basel, Switzerland). Transfections were
performed by electroporation using a Nucleofector apparatus
following the manufacturers recommendations (Cell Line Nucleofector
kit C, program X-005; Lonza Group, Ltd.). Upon 24 h after
transfection, the number of cells expressing GFP was counted using
an inverted fluorescence microscope in order to estimate the
transfection success (data not shown) (Zeiss Imager M1-AX10). After
48, 72 or 96-h post-transfection, cells were collected and
subjected to downstream analysis.
Cell viability assay
Cell viability was quantified using PrestoBlue Cell
Viability Reagent (Thermo Fisher Scientific, Inc.) following the
manufacturers instructions. Briefly, MM cells (WT or freshly
transfected with esiDCC) were seeded into 96-well plates at an
initial density of 12×103 cells per well and incubated
at 37°C. After 48 h, 10 nM of bortezomib was added. Two h before
the end of the treatment (total of 96 h), 10 µl of PrestoBlue
reagent was added to each well and, at completion of 96
h-incubation, fluorescence (540 nm excitation/590 nm emissions) was
measured using a microplate reader (Molecular Devices, LLC,
Sunnyvale, CA, USA). Experiments were performed in sextuplicate.
Data was expressed as mean ± SD of the sextuplicate assays.
Cell death assay
Briefly, 1×105 MM cells were seeded in
each well of a 12-well dish with 1 ml of RPMI-1640 medium. A total
of 24 h after seeding, bortezomib was added. After 48 h of
incubation with bortezomib, cells were harvested by centrifugation
(250 × g; 5 min), washed with PBS and resuspended in 100 µl
of binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl and 2.5 mM
CaCl2). Next, 0.5 µl of Annexin V-conjugated with FITC
(cat. no. 556419; BD Biosciences, Franklin Lakes, NJ, USA) and 0.5
µl of Propidium Iodide (PI; cat. no. P4170; Sigma-Aldrich; Merck
KGaA) were added to the cells and incubated for 60 min at room
temperature in the dark. Flow cytometer was performed using
standard procedures on Accuri C6 (BD Biosciences). MM cells marked
only with 0.5 µl of Annexin V or with 0.5 µl of PI were used to
calibrate the flow cytometer according to Annexin V and PI
labeling. This assay was performed in triplicate. For this
analysis, the percentage of cell death represents the sum of MM
cells staining Annexin V+/PI- and Annexin V+/PI+.
Statistical analysis
The statistical significance of DCC
transcripts or methylation levels was calculated using the Welch t
test or one-way ANOVA, as appropriated. Comparisons of the values
obtained in cell viability and cell death assays were performed
using one-way ANOVA. All multiple paired comparisons were conducted
by means of the Bonferronis post-test method to maintain the 5%
significance level.
Results
Methylation status of DCC in MM
cells
First of all, to better understand the regulation of
DCC expression in MM cells, we evaluated the presence of
DCC transcripts and protein in three different MM lines
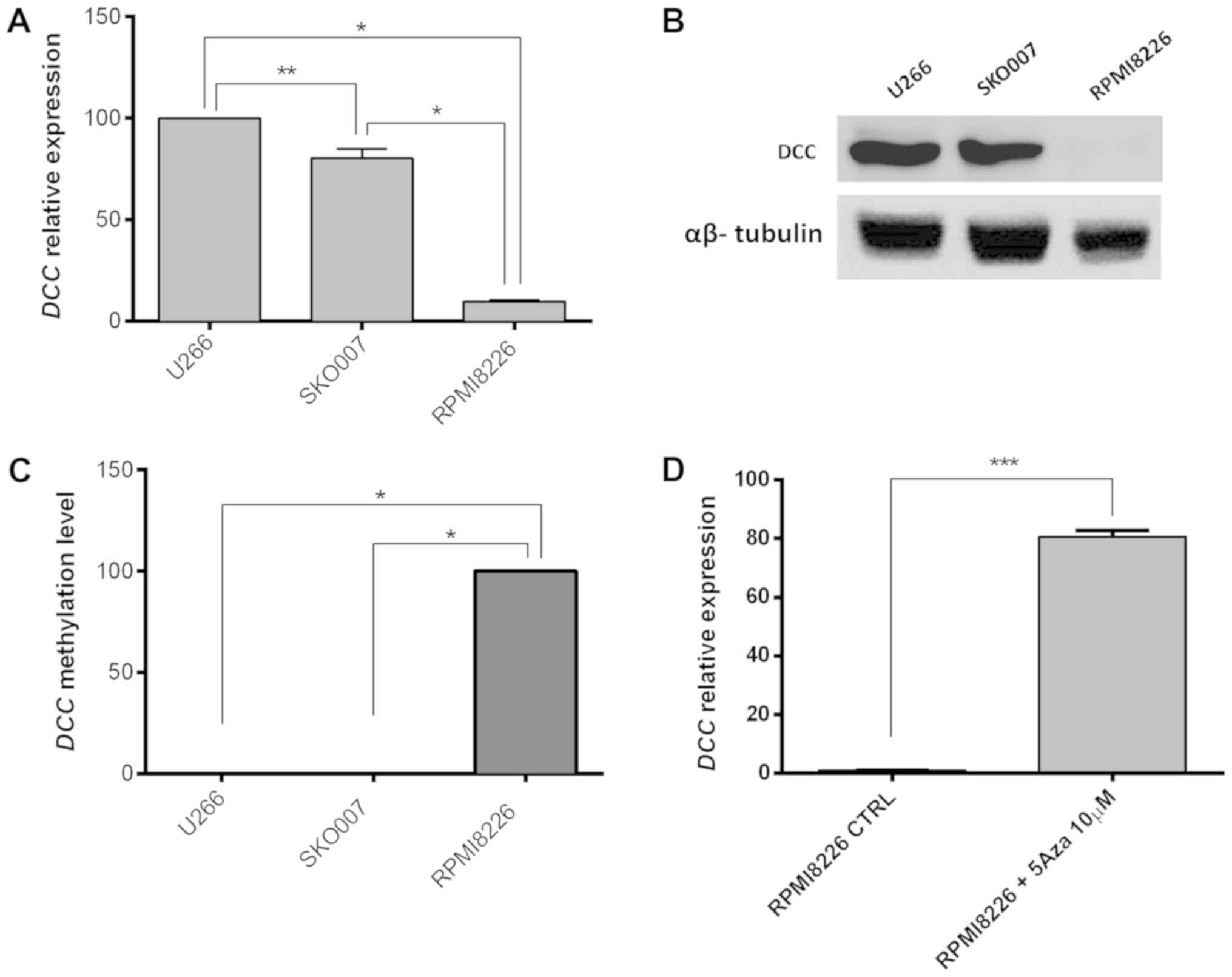
(RPMI8226, SKOO07 and U266). This analysis showed high DCC
mRNA expression in SKO007 and U266, while RPMI8226 presented a
lower transcript level of this gene (100, 80.19 e 9.66%,
respectively; Fig. 1A) and, as
expected, the DCC polypeptides were observed only in U266 and
SKO007 cells (Fig. 1B). In order
to correlate the status of DCC methylation with the
transcript and protein expression observed, the methylation pattern
of the promoter region of this gene was evaluated in MM cell lines.
According to this analysis, the DCC promoter region was
highly methylated in the RPMI8226 cell line, which presents
DCC low expression, whereas no methylation could be detected
in the DCC high expression cell lines SKO007 and U266
(Fig. 1C; P<0.0001).
To confirm that DCC low expression in
RPMI8226 was associated with the hypermethylation of its promoter
region, this MM cell line was treated with DAC (a recognized DNA
demethylating agent). Remarkably, an 82-times increment could be
observed in the DCC expression in RPMI8226 cells treated
with this demethylating agent (Fig.
1D; P=0.0002).
MM cells response to bortezomib
treatment
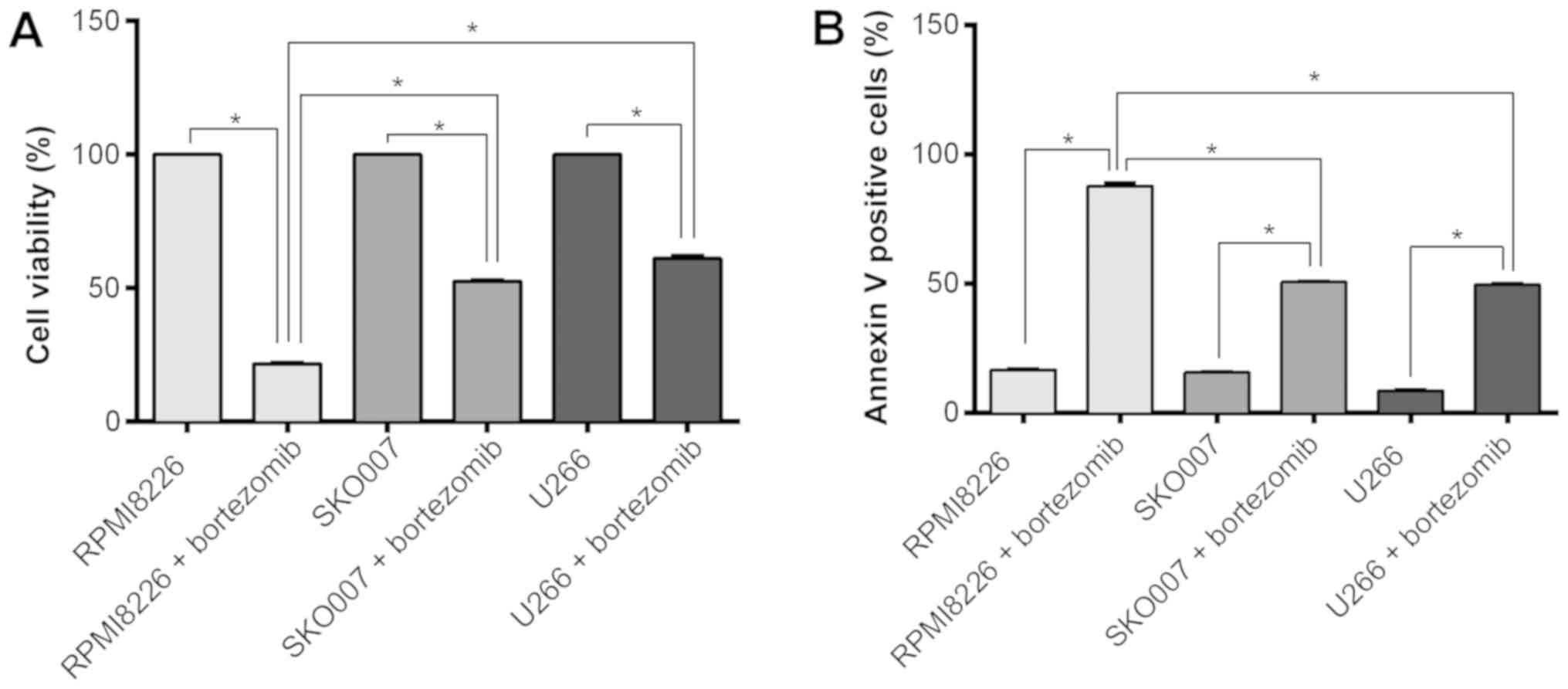
To evaluate the association between bortezomib
resistance in MM cells and the DCC expression, it was
determined the bortezomib effect in SKO007 and U266 (high
expression of DCC), as well as in RPMI8226 (low expression
of DCC). This assay showed a significant lower number of
viable cells in RPMI8226 submitted to bortezomib treatment in
comparison to the other two MM cell lines (P<0.0001; Fig. 2A). In accordance with this, the
total number of dead cells after bortezomib treatment is higher in
the RPMI8226 in comparison to SKO007 or U266 (P<0.0001; Fig. 2B). These results demonstrated that
the effect of the bortezomib on RPMI8226 (the cell line with the
lowest expression level of DCC) is stronger in comparison to
the other two MM cell lines, which presented higher DCC
expression.
DCC silencing decreases cell viability
and promotes cell death
To put some light in the functional role of DCC in
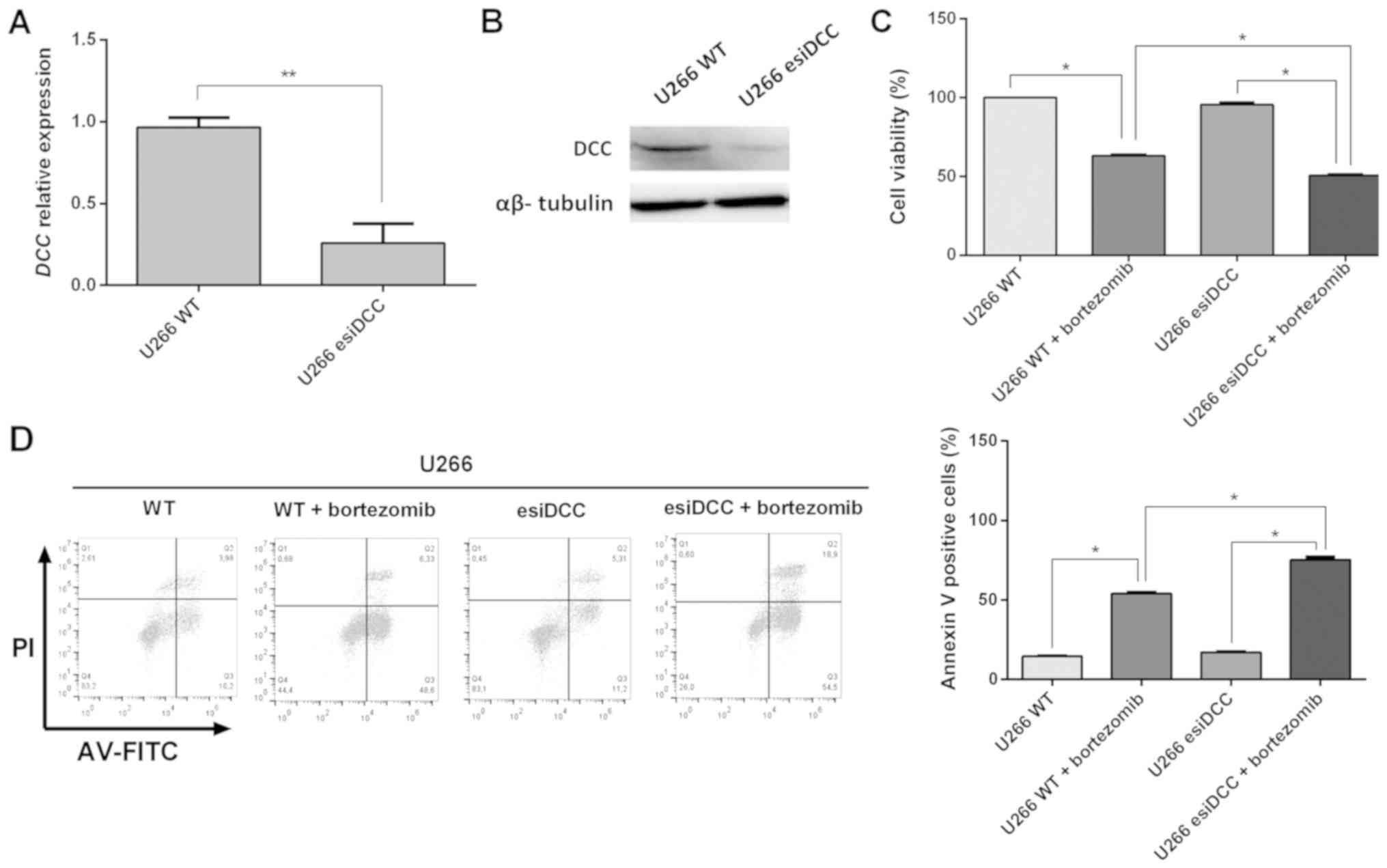
MM tumorigenesis, we conducted a transient DCC knockdown in
the U266 line. The DCC silencing was confirmed by
quantitative PCR and western blot analysis. As shown in Fig. 3, when compared to control cells,
DCC-silenced cells exhibited a dramatic reduction in
DCC transcript and polypeptide levels.
No significant difference in cell viability of
U266-WT and U266-esiDCC cells was observed in the absence of
bortezomib. However, DCC-silenced cells exhibited a
significant decrease in the cell viability in the presence of this
PI (36.9 and 49.9%, respectively; Fig.
3C). As shown in Fig. 3D, the
DCC-silenced U266 cells exhibited a significant increase in
the number of dead cells upon bortezomib treatment in comparison to
U266 WT cells (75.3 vs. 54.1%, respectively). Interestingly, these
results suggest that the blockage of DCC activity increases
bortezomib-induced cell death in MM cells, suggesting that DCC
absence could be able to sensitize MM cells to bortezomib
treatment.
Discussion
Over the last decade, the introduction of novel
agents such as immunomodulatory drugs (thalidomide and
lenalidomide) and PIs has changed the treatment landscape of MM and
prolonged overall survival of patients with this plasma cell
disease (26). Nevertheless, MM is
still regarded as an incurable disease for most patients.
Therefore, a major challenge is to enlarge the molecular aspects
knowledge of this hematological malignancy and the consequent
development of rational combination therapies that could
efficiently destroy MM cells.
Previous data showing frequent hypermethylation of
DCC in MM patients and its association with poor prognosis
(12) raised the question of what
would be the DCC contribution to MM tumorigenesis. It is well
accepted that hypermethylation leads to the promoter obstruction,
which hinders gene transcription, and subsequently causes gene
silencing (27). Thus, first and
foremost, we verified if the aberrant methylation of the DCC
promoter translates into gene expression inhibition in MM cells.
Our results fully support this correlation, since the absence of
aberrant methylation in promoter region was associated to the
presence of mRNA and DCC protein, as observed in SKO007 and U266
cell lines, while, on the other hand, the DCC
hypermethylation (as observed in RPMI8226) correlates with the lack
of DCC transcription and the scarcity of DCC
polypeptides.
DCC was firstly described in colorectal
carcinomas as a potential tumor suppressor gene located on
chromosome 18q (15). This
chromosome region is often subjected to the loss of heterozygosity,
and the absence of DCC in colorectal cancer has been
correlated with poor prognosis, tumor progression and increased
risk of metastasis (16). It was
also reported that DCC might induce apoptosis in colorectal tumors
in the absence of netrin 1, while the presence of this ligand is
able to block DCC-induced cell death (28). Moreover, the DCC
overexpression in ovarian cancer cells suggests an important role
in suppressing cell viability and inducing apoptosis through
regulation of the levels of β-catenin (29,30).
On the other hand, the DCC role as a tumor
suppressor remains controversial. The chromosome 18q region, where
DCC is located, also harbors other genes known as tumor
suppressors, such as SMAD2 and SMAD4. Furthermore,
the lack of predisposition to intestinal tumors in mice carrying a
Dcc mutation was interpreted as an evidence that DCC
gene may not has a significant role in suppressing colorectal
tumors (19,31). Beside this, Bamias et al
(21) showed an association
between the loss of DCC and a better prognosis in resected gastric
cancer. These studies indicate that DCC does not act as a
tumor suppressor gene in all situations and could play different
roles in different cancer tissues. Therefore, in this context, it
seems that a major question is to understand when the presence/loss
of DCC offers selective advantages for cancer cells. To the best of
our knowledge, this is the first study to demonstrate that the
DCC absence increases MM cells sensitivity to bortezomib
treatment. The lack of DCC induces a reduction in the cell
viability rate and increases the number of dead cells in response
to bortezomib treatment, which suggest that this cell surface
receptor may not have a tumor suppressor activity in MM.
Bortezomib is a reversible inhibitor that primarily
targets the β5-subunit (PSMB5) subunit/chymotrypsin-like activity
of the 26S proteasome and also, to a somewhat lesser extent, the
caspase-like activity harbored by the β1 (PSMB6) proteasome
subunit. At higher concentrations, bortezomib inhibits trypsin-like
proteolytic activity facilitated by β2 (PSMB7) proteasome subunits
(32,33). Due to this proteasome inhibition,
there is an accumulation of misfolded proteins, resulting in
endoplasmic reticulum stress to cause unfolded protein response and
cell apoptosis (34). Other
effects of this drug in MM include inhibition of angiogenesis and
DNA repair system, as well as decrease of osteoclast activity
(35). In 2003, the United States
Food and Drug Administration (FDA) approved bortezomib for use in
relapsed/refractory MM, and, quickly, this boronic acid-based
reversible PI became recognized as the most effective anticancer
drug for MM treatment (36).
However, patients treated with bortezomib will ultimately develop
resistance and experience clinical progression, thus,
characterizing the mechanisms of PI resistance has become of great
interest.
To better understand the bortezomib resistance,
Chauhan et al (37)
provided the first evidence that heat shock protein 27 confers
bortezomib resistance in lymphoma cells. Besides this, cancer
testis antigens (CTA) appears to contribute to apoptosis
suppression, avoiding the effect of caspase inhibitors in MM cells
(38). MAGE C1/CT7 and MAGE A3
might play an important role protecting MM cells against
spontaneous apoptosis and their absence contributes to increased
bortezomib cytotoxic effects (39,40).
Moreover, Hu et al (41)
showed that CD9 expression increases MM cells sensitivity to
bortezomib treatment by inducing apoptosis. Additionally, Moschetta
et al (42) suggested that
cMet is a potential therapeutic target for MM because the presence
of this protein conferred a protective effect when MM cells were
treated with bortezomib.
Thus, our results are the first to suggest that MM
cells could become more susceptible to bortezomib cytotoxic effects
when DCC is absent. Along the same line, the miRNA hsa-miR-631,
which was able to re-sensitize bortezomib-resistant MM cell line
(43), was recently described as
targeting DCC transcript and inhibiting its transduction
(44). Further, our results showed
that the DCC biological function in MM should be related to cell
death regulation, exerting an anti-apoptotic role in these cells in
response to bortezomib treatment. In some way, these findings seem
to be contrary to our previous observations, which the presence of
DCC (no hypermethylation) was associated with better
prognosis (12). However, it is
worth mentioning that none of the patients included in the former
study were treated with bortezomib (they were all submitted to VAD,
melphalan/prednisone or thalidomide/prednisone therapy schemes).
Thus, the difference in the chemotherapy regimen adopted avoids a
direct comparison of the results observed in both studies.
In conclusion, our results indicate that the absence
of DCC increase the response to bortezomib in MM. Based on these
findings, we can speculate that drugs blocking DCC activity should
increase the efficacy of bortezomib-based therapy approaches,
although further studies are required to confirm this hypothesis
and to better discriminate the role of DCC in the MM cell response
to bortezomib.
Acknowledgements
Not applicable.
Funding
The present study was funded by São Paulo Research
Foundation (FAPESP; grant no. 2012/14837-7; 2010/20218-2). DMR-Jr
was recipient of scholarship from São Paulo Research Foundation
(FAPESP; grant no. 2012/01597-8).
Availability of data and materials
The dataset used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors contributions
DMR-J conducted the studies, analyzed the data and
drafted the manuscript. TPB, GEDA, VC, MVB and JM-J contributed to
the data analysis. ALV contributed to the design and coordination
of the study and helped to draft the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Morgan GJ: Advances in the biology and
treatment of myeloma. Br J Haematol. 105 (Suppl 1):S4–S6. 1999.
|
|
2
|
Smith ML and Newland AC: Treatment of
myeloma. QJM. 92:11–14. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Röllig C, Knop S and Bornhäuser M:
Multiple myeloma. Lancet. 385:2197–2208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mateos MV, Ocio EM and San Miguel JF:
Novel generation of agents with proven clinical activity in
multiple myeloma. Semin Oncol. 40:618–633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cvek B: Proteasome inhibitors. Prog Mol
Biol Transl Sci. 109:161–226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moreau P, Richardson PG, Cavo M, Orlowski
RZ, San Miguel JF, Palumbo A and Harousseau JL: Proteasome
inhibitors in multiple myeloma: 10 years later. Blood. 120:947–959.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McBride A and Ryan PY: Proteasome
inhibitors in the treatment of multiple myeloma. Expert Rev
Anticancer Ther. 13:339–358. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grosicki S, Barchnicka A, Jurczyszyn A and
Grosicka A: Bortezomib for the treatment of multiple myeloma.
Expert Rev Hematol. 7:173–185. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brenner H, Gondos A and Pulte D: Recent
major improvement in long-term survival of younger patients with
multiple myeloma. Blood. 111:2521–2526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumar SK, Rajkumar SV, Dispenzieri A, Lacy
MQ, Hayman SR, Buadi FK, Zeldenrust SR, Dingli D, Russell SJ, Lust
JA, et al: Improved survival in multiple myeloma and the impact of
novel therapies. Blood. 111:2516–2520. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pulte D, Redaniel M, Brenner H, Jansen L
and Jeffreys M: Recent improvement in survival of patients with
multiple myeloma: Variation by ethnicity. Leuk Lymphoma.
55:1083–1089. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
de Carvalho F, Colleoni GW, Almeida MS,
Carvalho AL and Vettore AL: TGFbetaR2 aberrant methylation is a
potential prognostic marker and therapeutic target in multiple
myeloma. Int J Cancer. 125:1985–1991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hedrick L, Cho KR, Fearon ER, Wu TC,
Kinzler KW and Vogelstein B: The DCC gene product in cellular
differentiation and colorectal tumorigenesis. Genes Dev.
8:1174–1183. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Keino-Masu K, Masu M, Hinck L, Leonardo
ED, Chan SS, Culotti JG and Tessier-Lavigne M: Deleted in
colorectal cancer (DCC) encodes a netrin receptor. Cell.
87:175–185. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vogelstein B, Fearon ER, Hamilton SR, Kern
SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM and Bos
JL: Genetic alterations during colorectal-tumor development. N Engl
J Med. 319:525–532. 1998. View Article : Google Scholar
|
|
16
|
Fearon ER, Cho KR, Nigro JM, Kern SE,
Simons JW, Ruppert JM, Hamilton SR, Preisinger AC, Thomas G,
Kinzler KW, et al: Identification of a chromosome 18q gene that is
altered in colorectal cancers. Science. 247:49–56. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cho KR and Fearon ER: DCC: Linking tumour
suppressor genes and altered cell surface interactions in cancer?
Eur J Cancer 31A. 1055–1060. 1995. View Article : Google Scholar
|
|
18
|
Fearon ER: DCC: Is there a connection
between tumorigenesis and cell guidance molecules? Biochim Biophys
Acta. 1288:M17–M23. 1996.PubMed/NCBI
|
|
19
|
Fazeli A, Dickinson SL, Hermiston ML,
Tighe RV, Steen RG, Small CG, Stoeckli ET, Keino-Masu K, Masu M,
Rayburn H, et al: Phenotype of mice lacking functional Deleted in
colorectal cancer (Dcc) gene. Nature. 386:796–804. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Forcet C, Ye X, Granger L, Corset V, Shin
H, Bredesen DE and Mehlen P: The dependence receptor DCC (deleted
in colorectal cancer) defines an alternative mechanism for caspase
activation. Proc Natl Acad Sci USA. 98:3416–3421. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bamias AT, Bai MC, Agnantis NJ, Michael
MC, Alamanos YP, Stefanaki SV, Razi ED, Skarlos DV, Kappas AM and
Pavlidis NA: Prognostic significance of the deleted in colorectal
cancer gene protein expression in high-risk resected gastric
carcinoma. Cancer Invest. 21:333–340. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Andrade VC, Vettore AL, Panepucci RA,
Almeida MS, Yamamoto M, De Carvalho F, Caballero OL, Zago MA and
Colleoni GW: Number of expressed cancer/testis antigens identifies
focal adhesion pathway genes as possible targets for multiple
myeloma therapy. Leuk Lymphoma. 51:1543–1549. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vidal DO, Paixão VA, Brait M, Souto EX,
Caballero OL, Lopes LF and Vettore AL: Aberrant methylation in
pediatric myelodysplastic syndrome. Leuk Res. 31:175–181. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rettori MM, de Carvalho AC, Bomfim Longo
AL, de Oliveira CZ, Kowalski LP, Carvalho AL and Vettore AL:
Prognostic significance of TIMP3 hypermethylation in post-treatment
salivary rinse from head and neck squamous cell carcinoma patients.
Carcinogenesis. 34:20–27. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Palumbo A and Anderson K: Multiple
myeloma. N Engl J Med. 364:1046–1060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Garinis GA, Patrinos GP, Spanakis NE and
Menounos PG: DNA hypermethylation: When tumour suppressor genes go
silent. Hum Genet. 111:115–127. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mehlen P and Fearon ER: Role of the
dependence receptor DCC in colorectal cancer pathogenesis. J Clin
Oncol. 22:3420–3428. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Meimei L, Peiling L, Baoxin L, Changmin L,
Rujin Z and Chunjie H: Lost expression of DCC gene in ovarian
cancer and its inhibition in ovarian cancer cells. Med Oncol.
28:282–289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cai Y, Hu CJ, Wang J and Wang ZH:
Influence of deleted in colorectal carcinoma gene on proliferation
of ovarian cancer cell line SKOV-3 in vivo and in vitro. Chin Med
Sci J. 26:175–181. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Roush W: Putative cancer gene shows up in
development instead. Science. 276:534–535. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Crawford LJ, Walker B, Ovaa H, Chauhan D,
Anderson KC, Morris TC and Irvine AE: Comparative selectivity and
specificity of the proteasome inhibitors BzLLLCOCHO, PS-341, and
MG-132. Cancer Res. 66:6379–6386. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kisselev AF, van der Linden WA and
Overkleeft HS: Proteasome inhibitors: An expanding army attacking a
unique target. Chem Biol. 19:99–115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nawrocki ST, Carew JS, Dunner K Jr, Boise
LH, Chiao PJ, Huang P, Abbruzzese JL and McConkey DJ: Bortezomib
inhibits PKR-like endoplasmic reticulum (ER) kinase and induces
apoptosis via ER stress in human pancreatic cancer cells. Cancer
Res. 65:11510–11519. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rajkumar SV and Kyle RA: Multiple myeloma:
Diagnosis and treatment. Mayo Clin Proc. 80:1371–1382. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
San Miguel J, Bladé J, Boccadoro M,
Cavenagh J, Glasmacher A, Jagannath S, Lonial S, Orlowski RZ,
Sonneveld P and Ludwig H: A practical update on the use of
bortezomib in the management of multiple myeloma. Oncologist.
11:51–61. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chauhan D, Li G, Podar K, Hideshima T,
Shringarpure R, Catley L, Mitsiades C, Munshi N, Tai YT, Suh N, et
al: The bortezomib/proteasome inhibitor PS-341 and triterpenoid
CDDO-Im induce synergistic anti-multiple myeloma (MM) activity and
overcome bortezomib resistance. Blood. 103:3158–3166. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang B, OHerrin SM, Wu J, Reagan-Shaw S,
Ma Y, Bhat KM, Gravekamp C, Setaluri V, Peters N, Hoffmann FM, et
al: MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1
and suppress p53-dependent apoptosis in MAGE-positive cell lines.
Cancer Res. 67:9954–9962. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Atanackovic D, Hildebrandt Y, Jadczak A,
Cao Y, Luetkens T, Meyer S, Kobold S, Bartels K, Pabst C, Lajmi N,
et al: Cancer-testis antigens MAGE-C1/CT7 and MAGE-A3 promote the
survival of multiple myeloma cells. Haematologica. 95:785–793.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
de Carvalho F, Costa ET, Camargo AA,
Gregorio JC, Masotti C, Andrade VC, Strauss BE, Caballero OL,
Atanackovic D and Colleoni GW: Targeting MAGE-C1/CT7 expression
increases cell sensitivity to the proteasome inhibitor bortezomib
in multiple myeloma cell lines. PLoS One. 6:e277072011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hu X, Xuan H, Du H, Jiang H and Huang J:
Down-regulation of CD9 by methylation decreased bortezomib
sensitivity in multiple myeloma. PLoS One. 9:e957652014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Moschetta M, Basile A, Ferrucci A,
Frassanito MA, Rao L, Ria R, Solimando AG, Giuliani N, Boccarelli
A, Fumarola F, et al: Novel targeting of phospho-cMET overcomes
drug resistance and induces antitumor activity in multiple myeloma.
Clin Cancer Res. 19:4371–4382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xi H, Li L, Du J, An R, Fan R, Lu J, Wu
YX, Wu SX, Hou J and Zhao LM: hsa-miR-631 resensitizes
bortezomib-resistant multiple myeloma cell lines by inhibiting
UbcH10. Oncol Rep. 37:961–968. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:12–Aug;2015.doi: 10.7554/eLife.05005. View Article : Google Scholar
|