Introduction
Laryngeal cancer is the commonest cancer in the
larynx. Squamous cell carcinoma is the predominant physiological
type of laryngeal cancer, as it originates from the glottic region
of the larynx. At present, to the best of the authors' knowledge,
effective treatment for laryngeal squamous cell carcinoma (LSCC)
primarily relies on radiotherapy and surgery. Tobacco and alcohol
consumption are the principal risk factors for the development of
laryngeal cancer (1). The annual
incidence of laryngeal cancer is ~5 per 100,000 individuals and the
5-year survival rate is 60.6% in the USA (2,3). It
was estimates that 23,400 new cases of laryngeal cancer occurred in
China in 2014 (4).
Patients with upper-airway malignancies exhibit an
increased risk of developing locoregional cancer recurrence and
second primary malignancies, and the larynx and nasopharynx are
among the most frequent sites of locoregional cancer recurrence
(5,6). Although the available therapeutic
strategies exhibit promising results, the clinical outcomes of
patients with LSCC remain poor (4). Understanding of the pathogenesis of
the disease may contribute to the development of novel and more
effective therapeutic strategies. Numerous studies have aimed to
investigate the molecular mechanisms underlying laryngeal cancer
development (7–9). Cell cycle proteins have been
identified to serve important roles in the carcinogenesis of
laryngeal cancer, and the upregulation of cellular tumor antigen
p53 (p53), p21 and cyclin dependent kinase 1 in the surgical margin
of early cancer is associated with local tumor recurrence (10). Recurrence rates in patients with
p27- and phosphatase and tensin homolog (PTEN)-negative carcinoma
were identified to be increased compared with patients with
increased expression levels of these factors (11).
Multiple types of RNA, including long noncoding RNAs
(lncRNAs), circular RNAs, microRNAs (miRNAs), pseudogenes and
protein-coding mRNAs may serve as key competing endogenous RNAs
(ceRNAs) to regulate the expression levels of various mRNAs in
mammalian cells (12,13). miR-196a may promote tumor
progression in numerous cancer types, and its expression level was
identified to be increased in laryngeal cancer (14). miR-221 may increase the cell
proliferation rate by inhibiting apoptotic protease activating
factor-1 in laryngeal cancer (15), whereas miRNA-299-3p targets the
transcript of human telomerase reverse transcriptase (16). NF-κB-interacting lncRNA was
identified to inhibit tumor cell viability and to promote apoptosis
(17). A previous study analyzed
an lncRNA expression dataset from the Gene Expression Omnibus (GEO)
database, and identified that two lncRNAs, RP11-169K16.4 and
RP11-107E5.3, were associated with the prediction of recurrence of
laryngeal cancer (18).
Although various previous studies have investigated
the molecular mechanisms underlying laryngeal cancer, the etiology
of this malignancy, and in particular of its recurrence, remains
unclear. Notably, the regulatory interactions among noncoding and
coding RNAs is only partially understood. The present study
investigated novel potential molecular biomarkers involved in the
recurrence of laryngeal cancer by constructing a ceRNA regulatory
network consisting of miRNAs, lncRNAs and mRNAs.
Materials and methods
Training and validation datasets and
data processing
Head and neck squamous cell carcinoma (HNSCC) mRNA
samples were downloaded from The Cancer Genome Atlas (TCGA)
database (https://gdc-portal.nci.nih.gov/) on August 8th, 2016.
The samples of laryngeal cancer were selected according to the
following criteria: i) Laryngeal origin; ii) recurrence status was
recorded; and iii) matched mRNAs and miRNAs were barcoded. In
total, 84 primary laryngeal cancer samples, including 19 recurrent
and 65 nonrecurrent samples, were selected from 501 HNSCC tissues.
The training dataset was constructed using the RNA sequencing
(RNA-seq) data downloaded from the TCGA database.
The gene expression datasets GSE27020 and GSE25727
were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and used as
validation datasets. GSE27020 dataset was obtained using the GPL96
platform (Affymetrix human genome U133A array; Affymetrix, Inc.;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) and included 34
recurrent and 75 non-recurrent samples. Patients included in this
dataset exhibited primary squamous cell laryngeal carcinoma and
underwent surgical tumor removal (19). The raw data of this dataset in
CIMFast Event Language format were downloaded and processed with
gene expression background correction and were normalized using the
oligo package version 1.46.0 (http://www.bioconductor.org/packages/release/bioc/html/oligo.html)
in R (version 3.1.0; R Foundation, Vienna, Austria; https://www.R project.org/) (20). GSE25727 dataset was generated using
the GPL8432 Illumina platform (Illumina, Inc., San Diego, CA, USA)
and included 17 recurrent and 39 non-recurrent samples. All the
patients were treated locally with surgery or radiotherapy, and
none of them received chemotherapy (21). The data from this dataset were
downloaded as text files, and the probe identification numbers were
converted into gene symbols. In the case of multiple probes
corresponding to one gene, the average value was considered as the
expression value of this gene using the limma package 3.22.1
(22).
Analysis of differentially expressed
lncRNAs, mRNAs and miRNAs
In the training dataset, the mRNAs and lncRNAs were
annotated based on the information recorded in the HUGO Gene
Nomenclature Committee (http://www.genenames.org/) database. Subsequently, the
differentially expressed RNAs, including differentially expressed
mRNAs (DEGs), differentially expressed lncRNAs (DE-lncRs) and
differentially expressed miRNAs (DE-miRs), between recurrent and
nonrecurrent laryngeal cancer samples were analyzed using the EdgeR
package version 3.8.5, which adopted an overdispersed Poisson model
and an empirical Bayesian approach to improve the reliability of
the prediction (23). The
thresholds for DEG and DE-lncR selection were based on a false
discovery rate (FDR; adjusted P-value) <0.05, whereas for
DE-miRs, P<0.05 was considered to indicate a statistically
significant difference. Hierarchical cluster analysis was used to
analyze sample similarity.
Analysis DE-lncRs associated with
recurrence
Samples in the training dataset were classified into
recurrent and nonrecurrent groups. The DE-lncRs that were
significantly differentially expressed between recurrent and
nonrecurrent samples were used to perform univariate Cox regression
analysis, which was used to select the lncRNAs associated with
recurrence. Kaplan-Meier analysis was performed to examine the
association between the upregulated or downregulated lncRNAs, and
recurrence and survival status.
Prediction of miRNA-regulated lncRNAs
and mRNAs
The regulatory interactions among miRNAs and lncRNAs
were investigated using the miRcode (http://www.mircode.org/) and starBase (http://starbase.sysu.edu.cn/) databases. All
miRNA-regulated mRNAs were collected from the miRTarBase
(http://mirtarbase.mbc.nctu.edu.tw)
database. Associations among coding genes were examined using
BioGRID (http://thebiogrid.org/), HPRD
(http://www.hprd.org/) and DIP (http://dip.doe-mbi.ucla.edu/) databases.
The identified DE-lncRs, DEGs and DE-miRs between
recurrent and nonrecurrent samples were included in these
regulatory interactions to obtain the lncRNA-miRNA and miRNA-mRNA
regulatory networks. These two networks were combined to construct
the ceRNA regulatory network, containing the associations among
lncRNAs, miRNAs and mRNAs. The ceRNA regulatory network was
constructed to investigate the regulatory mechanism underlying the
associations among various RNAs. The network was visualized using
Cytoscape software version 3.6.1 (24).
Analysis of feature coding genes in
the miRNA-mRNA regulatory network
Topological structure analysis was conducted to
identify the feature coding genes in the miRNA-mRNA network. The
betweenness centrality (BC) of genes, corresponding to the
importance of a certain node, or gene, in the network, was
calculated using the following formula (25):
CB(v)=∑t≠v≠u∈Vσst(v)σst
Where v, s and t represent the nodes in the network,
σst is the number of shortest paths from s to t, and
σst (v) is the number of shortest paths from s to t,
going through node v. BC values exhibited a range between 0 and 1,
and the BC value was correlated with the importance of that node in
the network. Therefore, the nodes exhibiting interactions with
numerous mRNAs, miRNAs or lncRNAs, were more important in the
construction of the network.
The DEGs in the miRNA-mRNA network presenting the
100 highest BC values were selected as candidate feature-coding
genes.
Construction of a support vector
machine (SVM) classifier for various samples
The candidate genes that were significantly
differentially expressed between recurrent and nonrecurrent samples
were selected, and the unsupervised clustering classification
method was used to validate the sample classification performance
of these feature-coding genes (26). The 100 DEGs exhibiting the highest
BC values were used to identify the optimal feature-coding gene set
using the recursive feature elimination (RFE) algorithm (27). Through the iterative random feature
combination, classification assessment and determination of the
performance of various samples (28), the optimal feature-coding gene set
was obtained. Subsequently, the set of optimal feature-coding genes
was used to construct an SVM classifier, which considered the
expression levels of the feature genes within the samples as the
feature value to classify and distinguish the samples. The
recurrence status of the samples was predicted using the SVM
classifier.
The robustness of the classifier were validated
using the GSE27020 and GSE25727 datasets. The classification
performance was evaluated using sensitivity (Se), specificity (Sp),
positive predictive value (PPV), negative predictive value (NPV)
and area under the receiver operating characteristic curve
(AUC).
Molecular function and pathway
enrichment analysis
The candidate feature coding genes selected from the
ceRNA network were analyzed using Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) (29) to investigate the enriched molecular
functions and signaling pathways in recurrent laryngeal cancer.
Fisher's exact test (Fisher's noncentral hypergeometric
distribution) was used to perform the enrichment analysis with the
following formula (30):
p=1-∑i=0x-1(Mi)(N-MK-i)(NK)
Where ‘N’ is the total number of genes, ‘M’ is the
number of genes in the enriched pathway, ‘K’ is the number of DEGs,
and ‘p’ is the probability that at least one DEG belongs to the
functional pathway. Multiple hypothesis testing correction was used
to identify the categories of GO molecular functions and KEGG
pathways.
Results
Screening of DE-lncRs, DEGs and
DE-miRs
Using the aforementioned methods for the analysis of
the RNA-seq data from the TCGA database, 853 lncRNAs, 18,924 mRNAs
and 1,047 human miRNAs were identified. A total of 21 DE-lncRs and
507 DEGs were identified between recurrent and nonrecurrent samples
(FDR<0.05). In addition, 55 DE-miRs were identified (P<0.05).
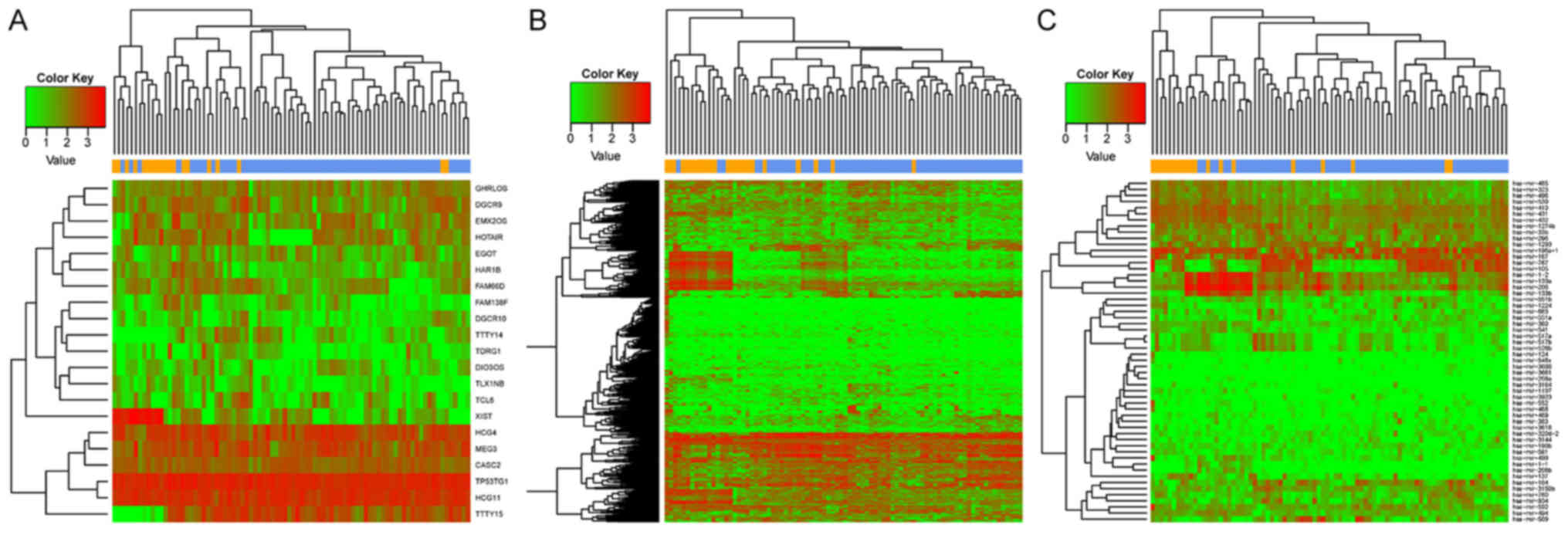
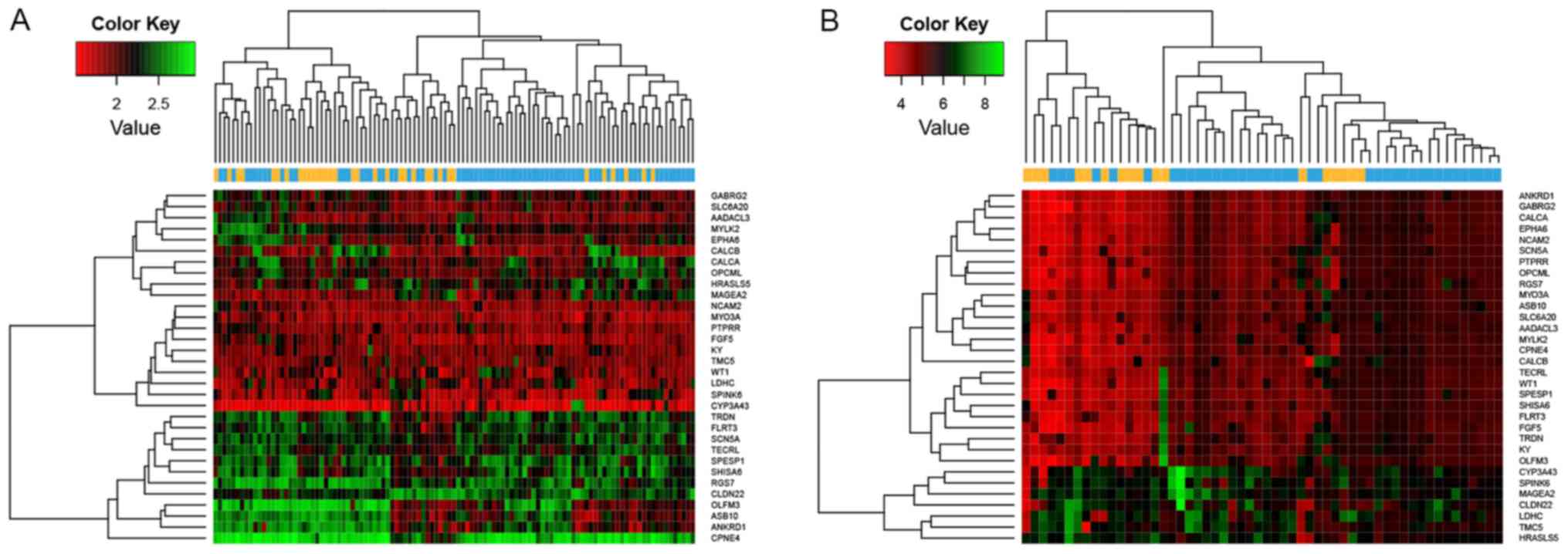
The results of the heat maps constructed for DEGs, DE-lncRs and
DE-miRs suggested that the differentially expressed RNAs clustered
according to the two sample types (Fig. 1). Numerous DEGs (507) were
identified between recurrent and nonrecurrent samples.
DE-lncRs associated with
recurrence
Following the selection of DE-lncRs between
recurrent and nonrecurrent samples, univariate Cox regression
analysis was performed to identify the lncRNAs associated with
recurrence. In total, five DE-lncRs, including testis-specific
transcript, Y-linked 15 (TTTY15), EMX2 opposite strand/antisense
RNA (EMX2OS), family with sequence similarity 138 member F, human
leukocyte antigen complex group 4 (HCG4) and HOX transcript
Antisense RNA (HOTAIR), were identified to be significantly
associated with recurrence-free survival time and survival ratio
(P<0.05). Survival analysis was conducted and the Kaplan-Meier
survival curves (Fig. 2) suggested
that the increased expression levels of the five DE-lncRs were
associated with poor prognosis.
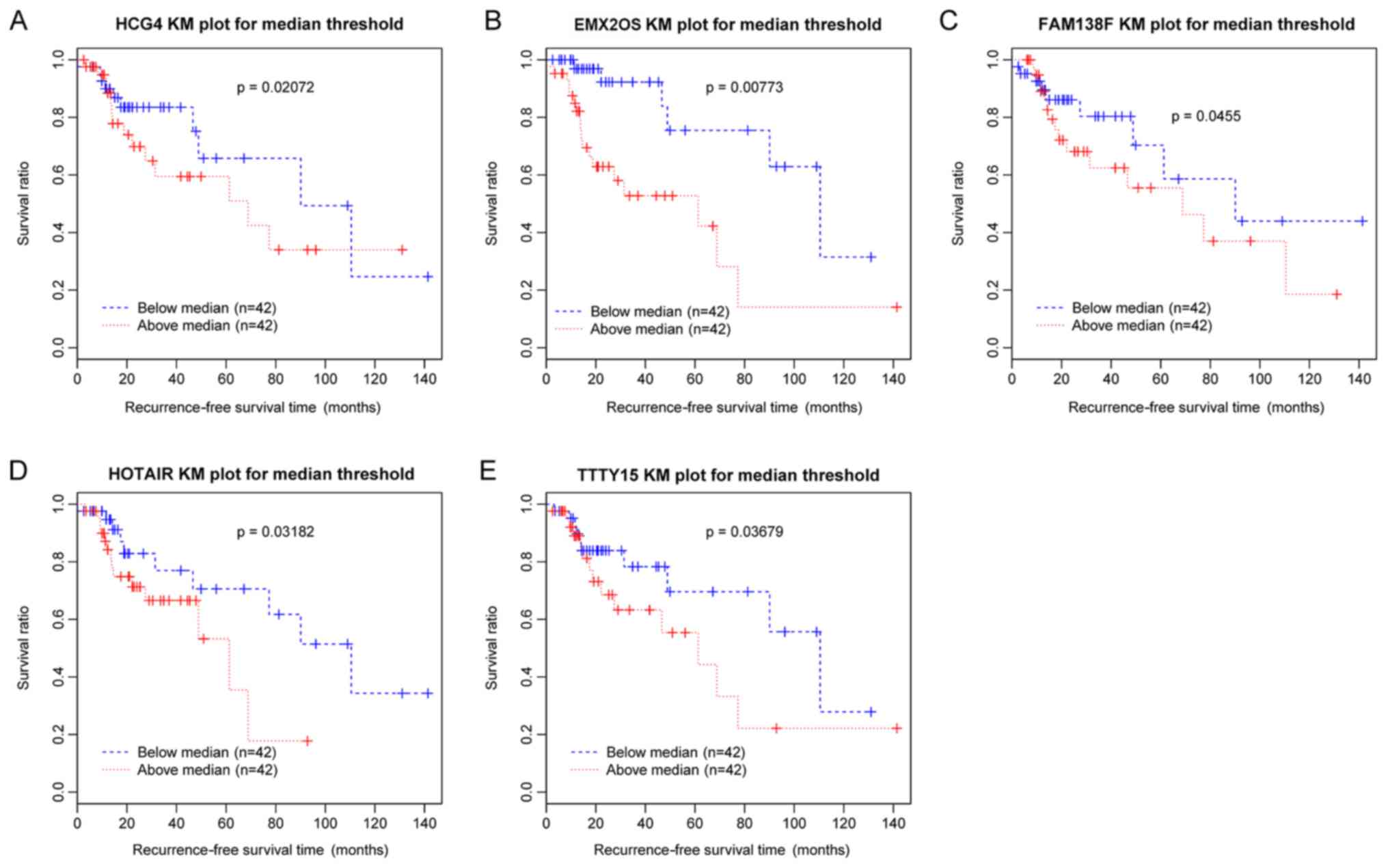 | Figure 2.KM survival curves for five
differentially expressed lncRNAs associated with laryngeal cancer
recurrence. Blue curves represent samples with downregulated
lncRNAs, and red curves represent samples with upregulated lncRNAs.
KM curves corresponding to (A) HCG4, (B) EMX2OS, (C) FAM138F, (D)
HOTAIR and (E) TTTY15. KM, Kaplan-Meier; HCG4, HLA complex group 4;
EMX2OS, EMX2 opposite strand/antisense RNA; FAM138F, family with
sequence similarity 138 member F; HOTAIR, HOX transcript antisense
RNA; TTTY15, testis-specific transcript, Y-linked 15; lncRNA, long
noncoding RNA. |
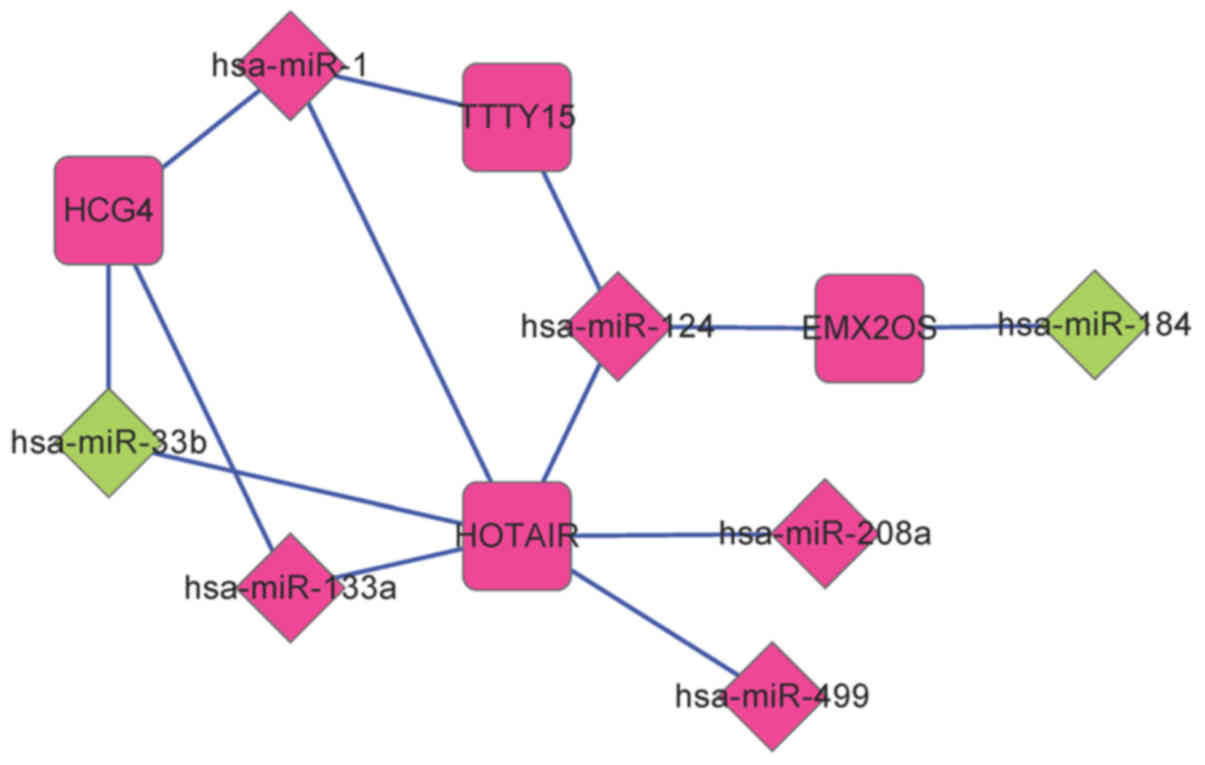
lncRNA-miRNA, miRNA-mRNA and mRNA-mRNA
regulatory networks
By investigating the miRcode and starBase databases,
a total of 268 lncRNA-miRNA regulatory interactions were screened.
Among these 268 interactions, 14 interactions were present among
DE-miRs and DE-lncRNAs. This lncRNA-miRNA regulatory network
comprised four DE-lncRs associated with recurrence and seven
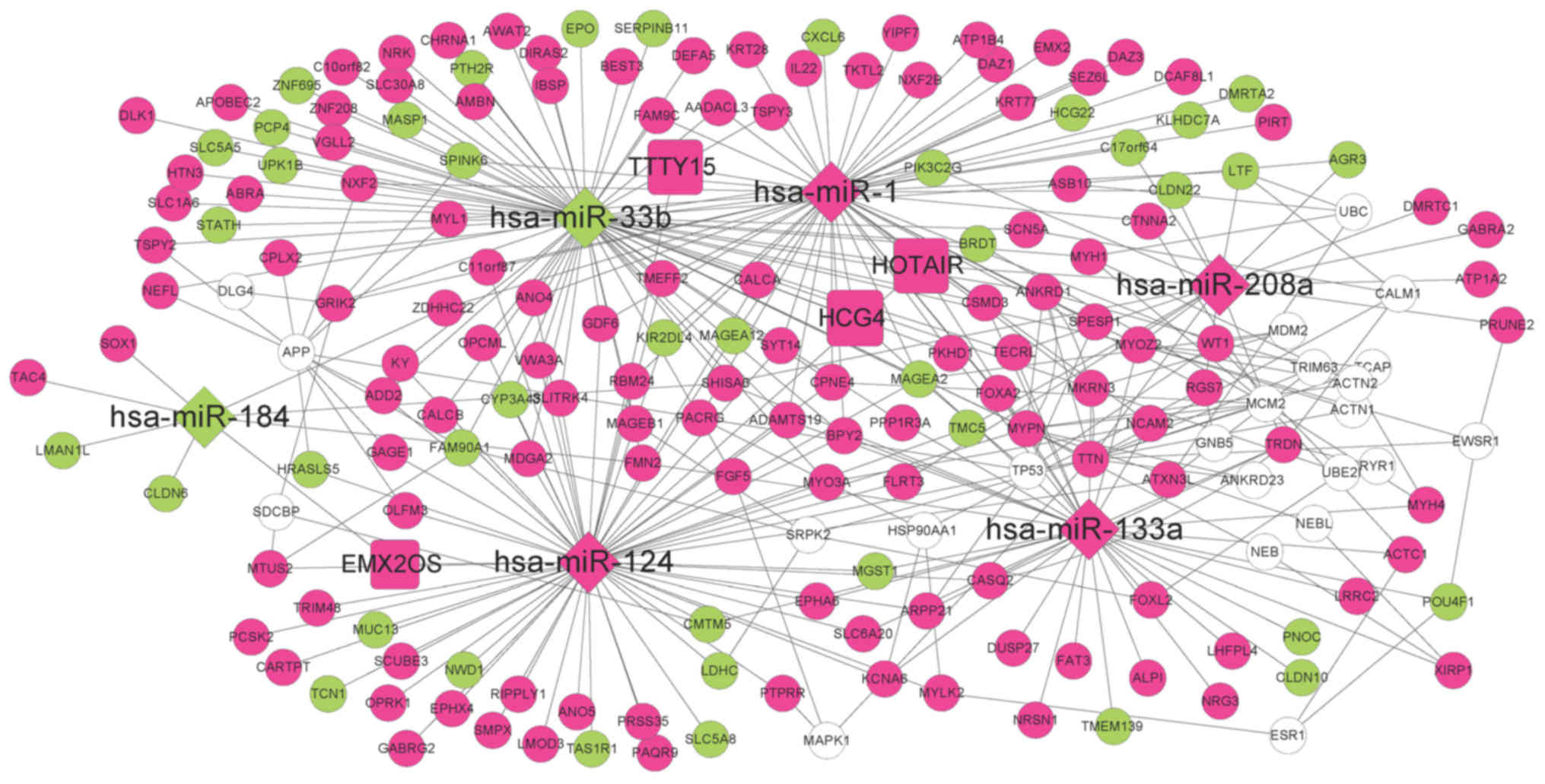
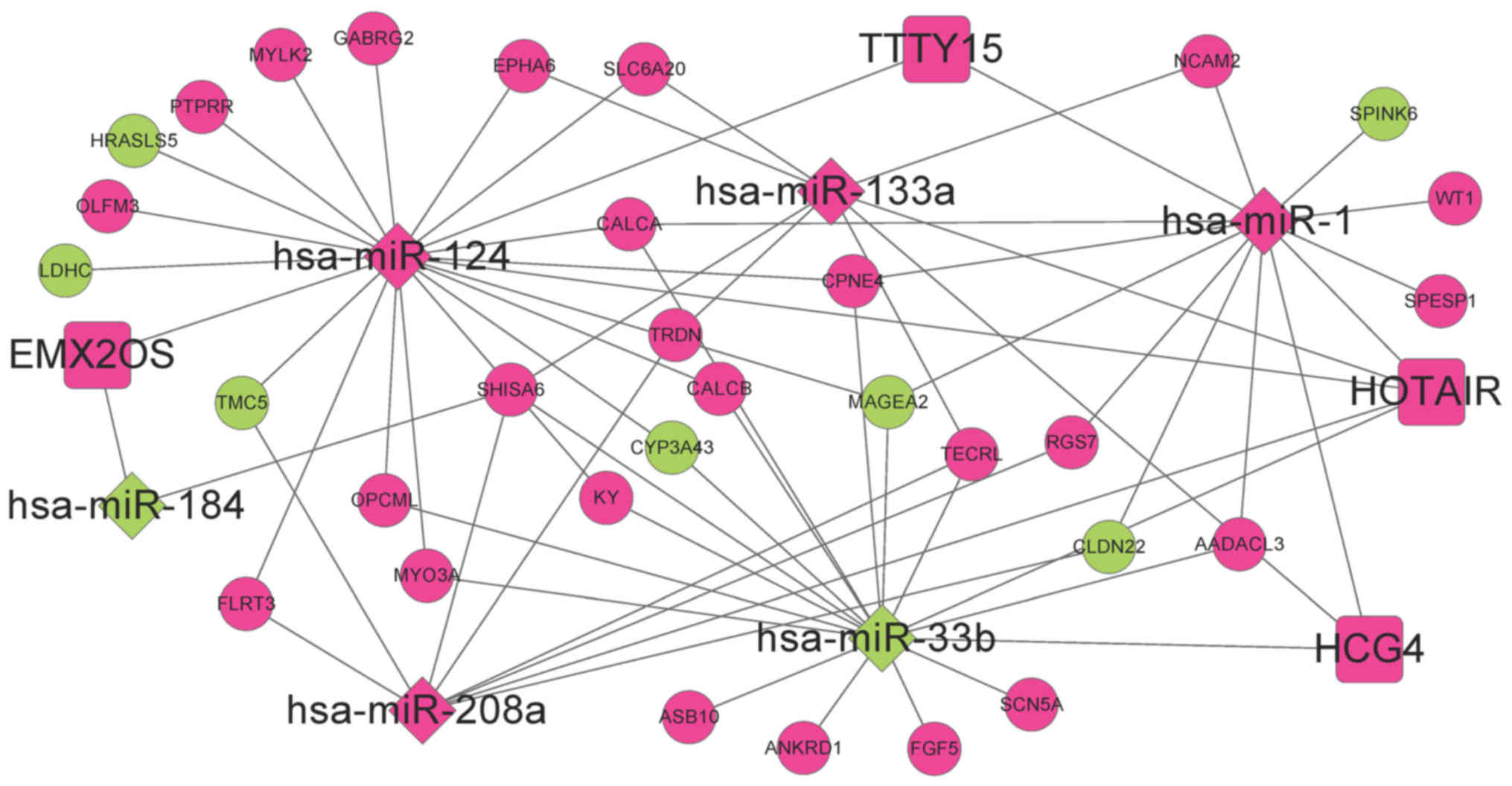
DE-miRs (Fig. 3).
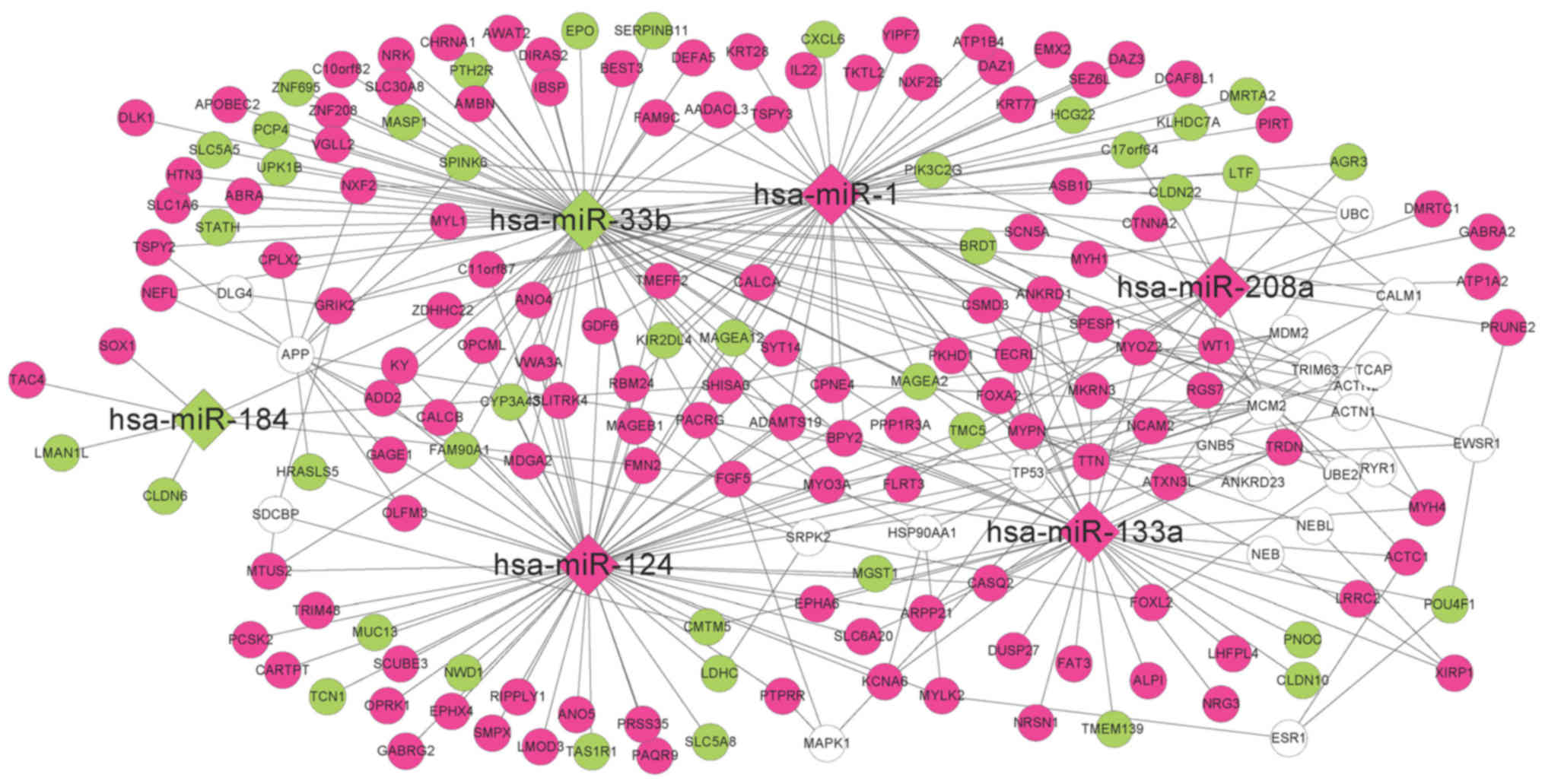
According to the information in the miRTarBase
database, the DEGs regulated by the seven DE-miRs were
investigated. A total of 55, 59, 34, 7, 17, 69 and 0 interacting
DEGs were reported for hsa-miR-1, hsa-miR-124, hsa-miR-133a,
hsa-miR-184, hsa-miR-208a, hsa-miR-33b and hsa-miR-499,
respectively. Therefore, an miRNA-mRNA regulatory network was
established comprising six miRNAs and 193 mRNAs (Fig. 4). Additionally to the DEGs, 22
coding genes exhibiting interactions with at least five DEGs were
included in the network, as determined using the BioGRID, HPRD and
DIP databases.
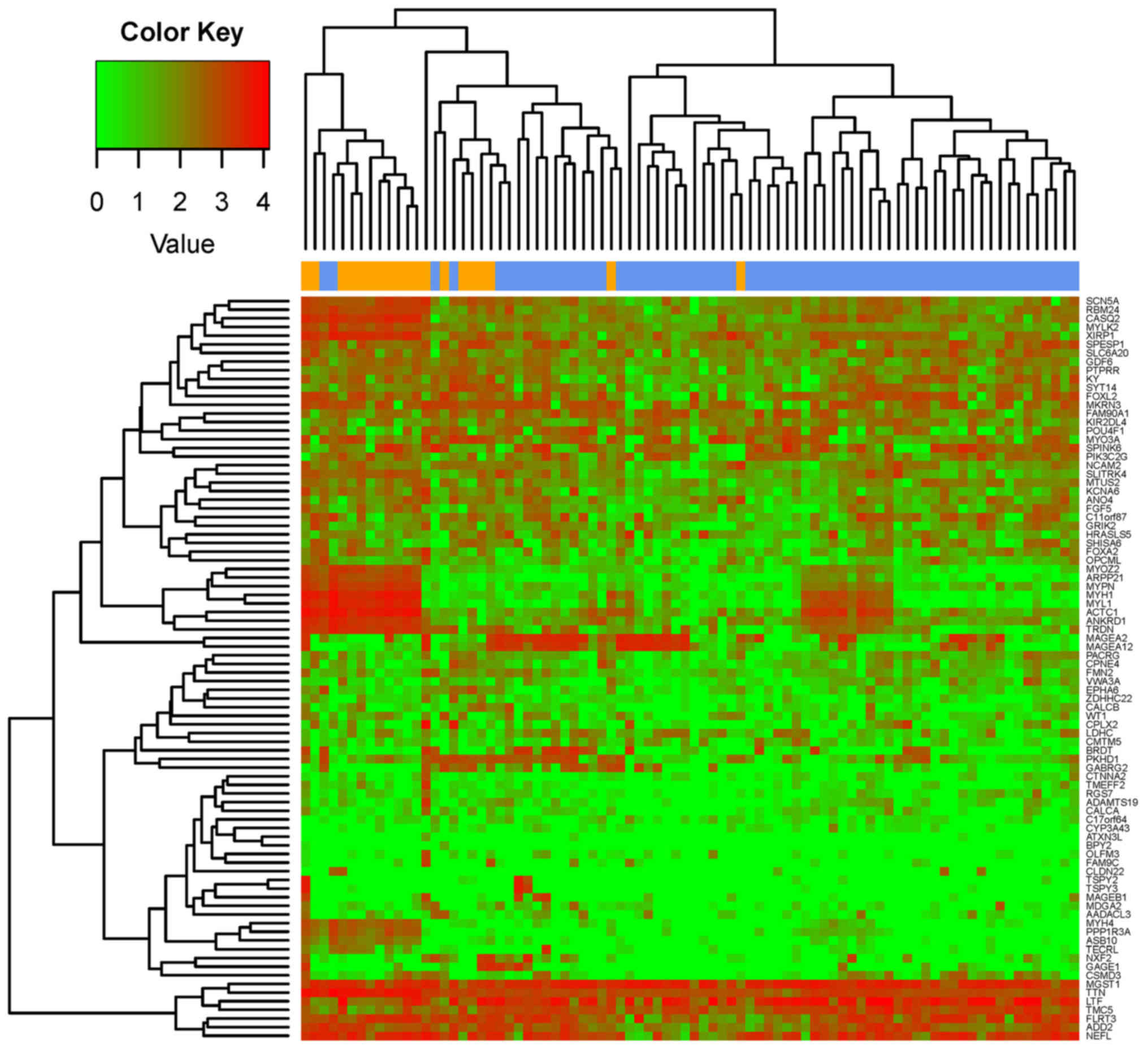
Feature coding gene
identification
The 100 coding genes, including 86 DEGs and 14
DEG-associated genes, exhibiting the highest BC values in the
miRNA-mRNA network were examined. The 86 DEGs were used to perform
clustering analysis, and the results suggested that the DEGs
clustered according to the two types of samples (Fig. 5). Similar results were identified
when all the DEGs were considered for the clustering analysis
(Fig. 2). The present results
suggested that the 86 DEGs may be used as feature-coding genes for
sample grouping.
SVM classifier
The RFE algorithm was used to refine the list of
feature-coding genes, and 32 optimal feature-coding genes
exhibiting the highest predictive accuracy (94.05%) for sample
grouping were selected. The 32 coding genes are presented in
Table I. The SVM classifier
constructed using the 32 feature-coding genes presented high
accuracy (94.05%, 79/84) in separating the recurrent samples from
the nonrecurrent samples.
 | Table I.Basic information of 32 optimal
feature-coding genes, among the genes significantly differentially
expressed between recurrent and nonrecurrent cancer samples. |
Table I.
Basic information of 32 optimal
feature-coding genes, among the genes significantly differentially
expressed between recurrent and nonrecurrent cancer samples.
| Gene symbol | Betweenness
centrality | Number of
interactions | P-value | Log2
fold-change |
|---|
| SHISA6 | 0.240 | 4 | 0.028 |
0.544 |
| MAGEA2 | 0.215 | 5 | 0.047 | −0.394 |
| TRDN | 0.156 | 6 | 0.002 |
0.601 |
| RGS7 | 0.149 | 4 | 0.001 |
1.331 |
| CPNE4 | 0.142 | 4 | 0.020 |
0.657 |
| CALCA | 0.111 | 3 | 0.004 |
1.214 |
| GABRG2 | 0.101 | 2 | 0.035 |
0.599 |
| TECRL | 0.083 | 3 | 0.043 |
0.846 |
| MYLK2 | 0.073 | 3 | 0.005 |
0.543 |
| MYO3A | 0.058 | 3 | 0.048 |
0.365 |
| ASB10 | 0.057 | 3 | 0.001 |
1.261 |
| WT1 | 0.039 | 4 | 0.015 |
0.712 |
| CALCB | 0.038 | 2 | 0.006 |
0.986 |
| OPCML | 0.038 | 2 | 0.011 |
0.693 |
| KY | 0.038 | 2 | 0.011 |
0.502 |
| CYP3A43 | 0.038 | 2 | 0.027 | −2.312 |
| EPHA6 | 0.038 | 2 | 0.001 | 1.216 |
| SLC6A20 | 0.038 | 2 | 0.001 |
0.583 |
| NCAM2 | 0.036 | 2 | 0.026 |
0.451 |
| FLRT3 | 0.033 | 2 | 0.022 |
0.312 |
| TMC5 | 0.033 | 2 | 0.034 | −0.334 |
| AADACL3 | 0.033 | 2 | 0.011 |
0.983 |
| ANKRD1 | 0.026 | 6 | 0.005 |
0.520 |
| CLDN22 | 0.020 | 2 | 0.005 | −1.662 |
| FGF5 | 0.018 | 2 | 0.015 |
0.593 |
| SCN5A | 0.016 | 2 | 0.009 |
0.475 |
| PTPRR | 0.016 | 2 | 0.005 |
0.561 |
| LDHC | 0.015 | 2 | 0.000 | −1.366 |
| OLFM3 | 0.012 | 2 | 0.036 |
1.283 |
| HRASLS5 | 0.012 | 2 | 0.042 | −0.560 |
| SPINK6 | 0.010 | 2 | 0.011 | −0.533 |
| SPESP1 | 0.008 | 2 | 0.004 |
0.499 |
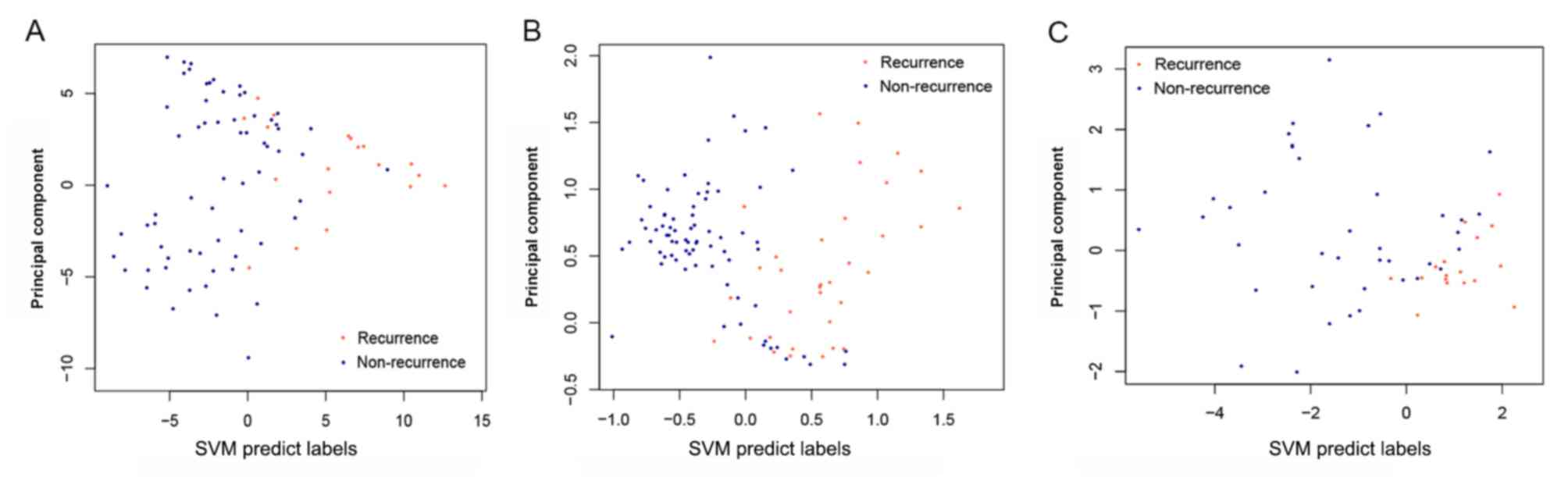
By examining the two validation datasets, the SVM
classifier was able to distinguish 101 samples (30 recurrent and 71
non-recurrent) and 51 samples (16 recurrent and 35 non-recurrent)
from the GSE27020 and GSE25727 datasets (Fig. 6) with an accuracy of 92.66 and
91.07%, respectively. The scatter plot of sample classifications is
presented in Fig. 7. The five
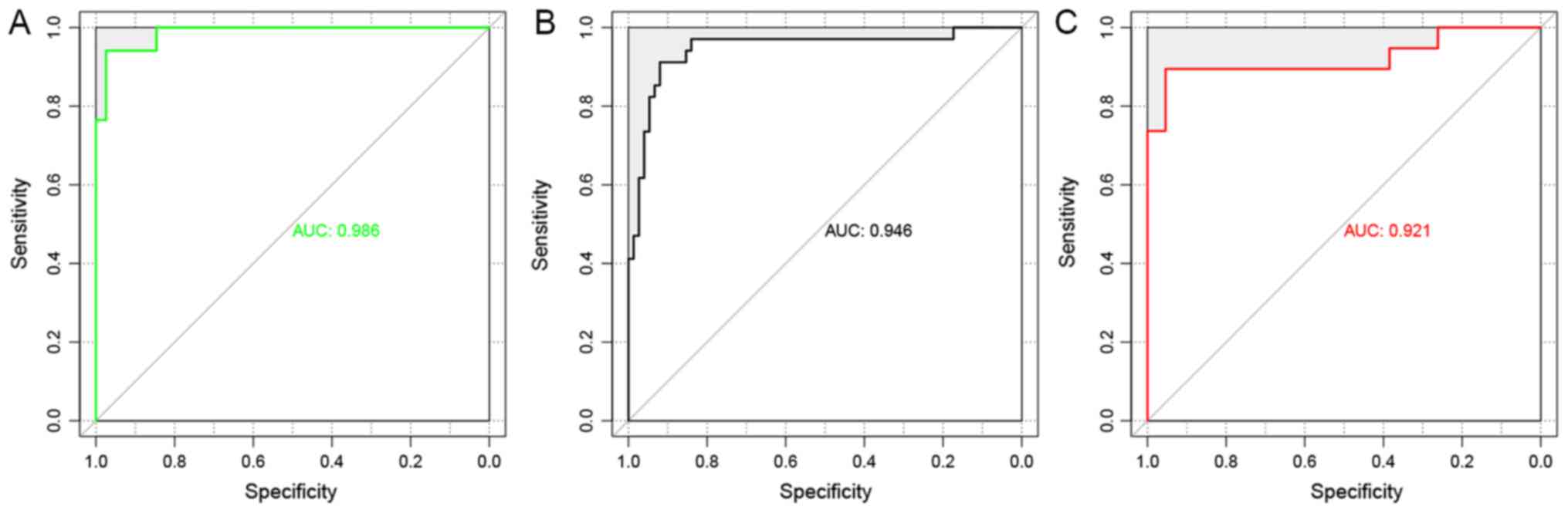
indicators (Se, Sp, PPV, NPV and AUC) presented high scores
(Table II). AUC exhibited the
highest score among the five indicators (Fig. 8). The present results suggested
that the SVM classifier exhibited high accuracy and reliability in
grouping samples from various datasets.
| Table II.Classifying parameters of the support
vector machine classifier for the three datasets analyzed. |
Table II.
Classifying parameters of the support
vector machine classifier for the three datasets analyzed.
| Dataset | Number of
samples | Accuracy | Sensitivity | Specificity | Positive predictive
value | Negative predictive
value | Area under receiver
operating characteristic curve |
|---|
| TCGA | 84 | 0.9410 | 0.947 | 0.938 | 0.818 | 0.984 | 0.986 |
| GSE27020 | 109 | 0.9266 | 0.882 | 0.947 | 0.882 | 0.947 | 0.946 |
| GSE25727 | 56 | 0.9107 | 0.941 | 0.897 | 0.8 | 0.972 | 0.921 |
Molecular function and pathway
enrichment analysis
The ceRNA regulatory network comprised 203 nodes and
346 lines and was constructed by combining the lncRNA-miRNA
regulatory network and the miRNA-mRNA regulatory network (Fig. 9). In the network, a node indicated
a transcript (mRNA, miRNA or lncRNA), whereas a line indicated the
association between two nodes. The coding genes in the ceRNA
network were significantly enriched in 11 pathway categories,
including ‘neuroactive ligand-receptor interaction’, ‘salivary
secretion’ and ‘tight junction’ (Table III), and 16 molecular function
categories, including ‘muscle contraction’, ‘muscle system process’
and ‘blood circulation’ (Table
IV). The multiple hypotheses testing correction results for the
GO molecular function and KEGG pathway categories were not
statistically significant (data not shown).
| Table III.Significantly enriched pathways in
the competing endogenous RNA regulatory network. |
Table III.
Significantly enriched pathways in
the competing endogenous RNA regulatory network.
| KEGG pathway
ID | KEGG pathway
name | Gene count | P-value | Differentially
expressed genes |
|---|
| hsa04530 | Tight junction | 5 | 0.001 | MYH1, CLDN6, MYH4,
CTNNA2, CLDN10 |
| hsa04970 | Salivary
secretion | 4 | 0.001 | HTN3, ATP1B4,
ATP1A2, STATH |
| hsa04080 | Neuroactive
ligand-receptor interaction | 6 | 0.004 | GABRA2, GABRG2,
PTH2R, OPRK1, CHRNA1, GRIK2 |
| hsa04918 | Thyroid hormone
synthesis | 3 | 0.007 | 6528, ATP1B4,
ATP1A2 |
| hsa04964 | Proximal tubule
bicarbonate reclamation | 2 | 0.008 | ATP1B4, ATP1A2 |
| hsa04960 |
Aldosterone-regulated sodium
reabsorption | 2 | 0.020 | ATP1B4, ATP1A2 |
| hsa05033 | Nicotine
addiction | 2 | 0.020 | GABRA2, GABRG2 |
| hsa04973 | Carbohydrate
digestion and absorption | 2 | 0.025 | ATP1B4, ATP1A2 |
| hsa04670 | Leukocyte
transendothelial migration | 3 | 0.026 | CLDN6, CTNNA2,
CLDN10 |
| hsa04978 | Mineral
absorption | 2 | 0.032 | ATP1B4, ATP1A2 |
| hsa04514 | Cell adhesion
molecules (CAMs) | 3 | 0.041 | CLDN6, NCAM2,
CLDN10 |
| Table IV.Significantly enriched molecular
functions in the competing endogenous RNA regulatory network. |
Table IV.
Significantly enriched molecular
functions in the competing endogenous RNA regulatory network.
| GO term ID | GO term name | Gene count | P-value | Genes |
|---|
| GO:0006936 | Muscle
contraction | 11 |
4.17×10−7 | TRDN, ACTC1, MYH1,
MYL1, MYLK2, MYH4, SMPX, TTN, CHRNA1, SCN5A, CASQ2 |
| GO:0003012 | Muscle system
process | 11 |
9.88×10−7 | TRDN, ACTC1, MYH1,
MYL1, MYLK2, MYH4, SMPX, TTN, CHRNA1, SCN5A, CASQ2 |
| GO:0008015 | Blood
circulation | 10 |
1.89×10−5 | CALCA, CALCB,
ACTC1, MYL1, MYLK2, CARTPT, ATP1A2, TAC4, SCN5A, EPO |
| GO:0003013 | Circulatory system
process | 10 |
1.89×10−5 | CALCA, CALCB,
ACTC1, MYL1, MYLK2, CARTPT, ATP1A2, TAC4, SCN5A, EPO |
| GO:0007267 | Cell-cell
signaling | 16 |
9.03×10−5 | FGF5, GABRG2,
GABRA2, GRIK2, OPRK1, MYLK2, ATP1A2, CXCL6, IL22, CTNNA2, CALCA,
PNOC, SLC1A6, CARTPT, SLC30A8, CHRNA1 |
| GO:0006811 | Ion transport | 18 |
1.32×10−4 | GABRG2, SLC5A5,
GABRA2, GRIK2, ATP1B4, KCNA6, ATP1A2, TCN1, BEST3, SLC1A6, SLC5A8,
LTF, ANO5, ANO4, CHRNA1, SLC30A8, SCN5A, ADD2 |
| GO:0007268 | Synaptic
transmission | 11 |
1.44×10−4 | GABRG2, GABRA2,
PNOC, GRIK2, OPRK1, SLC1A6, MYLK2, CARTPT, ATP1A2, CHRNA1,
CTNNA2 |
| GO:0030182 | Neuron
differentiation | 13 |
2.09×10−4 | NCAM2, SLITRK4,
SOX1, OPCML, FOXA2, PKHD1, EMX2, MDGA2, PTPRR, POU4F1, OLFM3, NEFL,
CTNNA2 |
| GO:0019226 | Transmission of
nerve impulse | 11 |
5.20×10−4 | GABRG2, GABRA2,
PNOC, GRIK2, OPRK1, SLC1A6, MYLK2, CARTPT, ATP1A2, CHRNA1,
CTNNA2 |
| GO:0055082 | Cellular chemical
homeostasis | 10 |
3.60×10−3 | CALCA, CALCB,
XIRP1, GRIK2, SLC1A6, LTF, CARTPT, ATP1A2, SLC30A8, CHRNA1 |
| GO:0019725 | Cellular
homeostasis | 10 |
1.29×10−2 | CALCA, CALCB,
XIRP1, GRIK2, SLC1A6, LTF, CARTPT, ATP1A2, SLC30A8, CHRNA1 |
| GO:0048878 | Chemical
homeostasis | 10 |
2.24×10−2 | CALCA, CALCB,
XIRP1, GRIK2, SLC1A6, LTF, CARTPT, ATP1A2, SLC30A8, CHRNA1 |
| GO:0007155 | Cell adhesion | 12 |
2.54×10−2 | IBSP, FLRT3, CALCA,
NCAM2, AMBN, OPCML, FAT3, PKHD1, CLDN6, CLDN22, CLDN10, CTNNA2 |
| GO:0022610 | Biological
adhesion | 12 |
2.56×10−2 | IBSP, FLRT3, CALCA,
NCAM2, AMBN, OPCML, FAT3, PKHD1, CLDN6, CLDN22, CLDN10, CTNNA2 |
| GO:0050877 | Neurological system
process | 17 |
3.28×10−2 | GABRG2, GABRA2,
MYO3A, GRIK2, OPRK1, MYLK2, ATP1A2, TAS1R1, CTNNA2, CALCA, NCAM2,
PNOC, SLC1A6, CARTPT, POU4F1, CHRNA1, NEFL |
| GO:0042592 | Homeostatic
process | 12 |
3.94×10−2 | CALCA, CALCB,
XIRP1, GRIK2, PKHD1, SLC1A6, LTF, CARTPT, ATP1A2, SLC30A8, CHRNA1,
EPO |
ceRNA regulatory network of 32 optimal
feature-coding genes
The ceRNA regulatory network corresponding to the 32
optimal feature coding genes was established and consisted of six
DE-miRs, four DE-lncRs and 32 feature-coding genes (Fig. 10). In the ceRNA network, the
interactions between HCG4-miR-33b, HOTAIR-miR-1-MAGE family member
A2 (MAGEA2), EMX2OS-miR-124-calcitonin related polypeptide α
(CALCA) and EMX2OS-miR-124-γ-aminobutyric acid type A receptor γ2
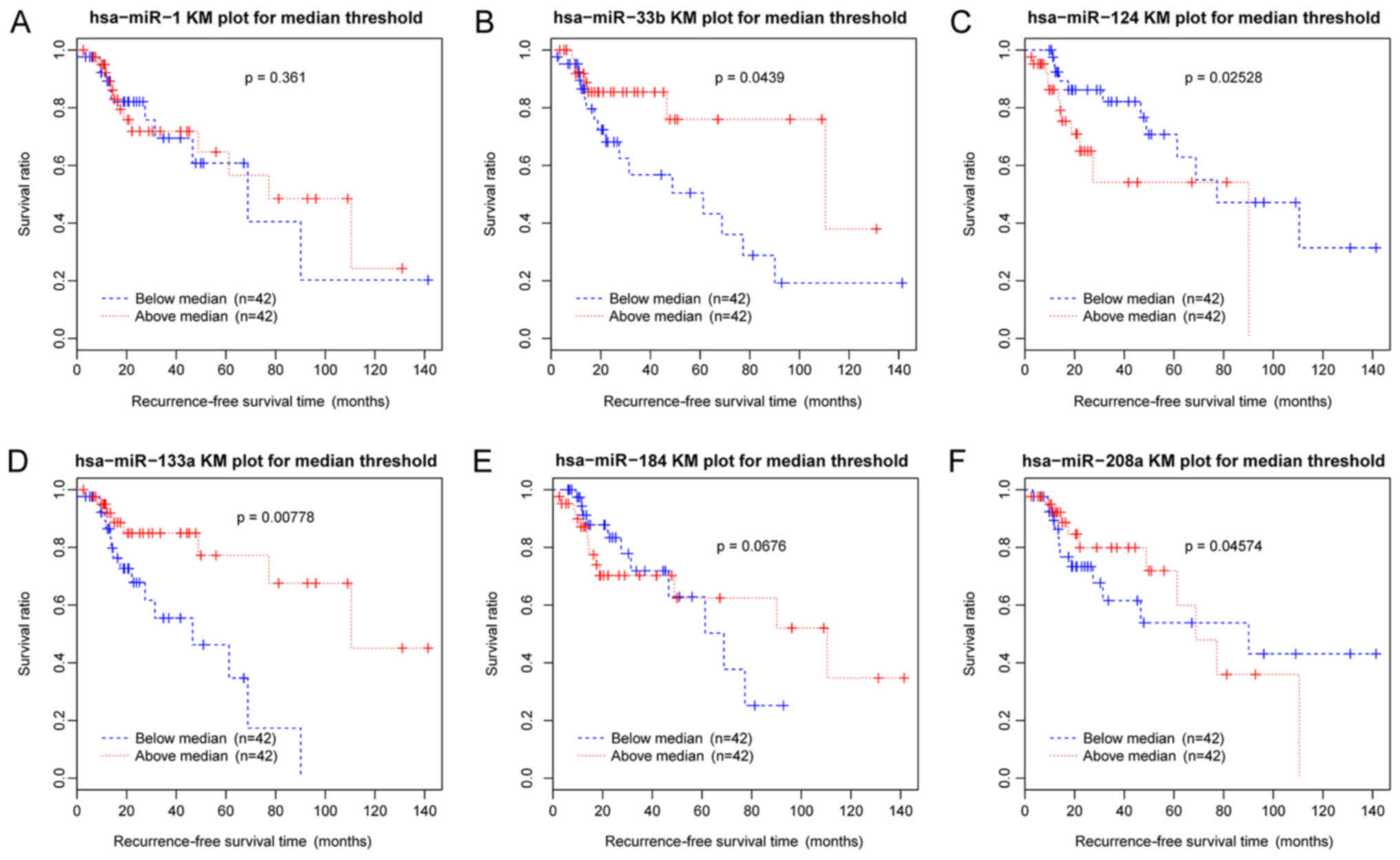
subunit (GABRG2) exhibited the highest scores. The Kaplan-Meier
survival curves of these six DE-miRs suggested a significant
association between recurrence-free survival times and the
expression levels of the miRNAs examined, including hsa-miR-33b,
hsa-miR-124, hsa-miR-133a and hsa-miR-208a (P<0.05; Fig. 11).
Discussion
By analyzing the RNA-seq data downloaded from TCGA,
the DE-miRs, DEGs and DE-lncRs between recurrent and nonrecurrent
laryngeal cancer samples were identified. Based on the associations
identified in various databases, numerous networks (miRNA-mRNA,
lncRNA-mRNA and ceRNA networks) were constructed to examine the
potential interactions among feature-coding genes, miRNAs and
lncRNAs. The aim of the present study was to identify the
regulatory mechanisms underlying the recurrence of laryngeal
cancer. In the ceRNA network, the interactions between
HCG4-miR-33b, HOTAIR-miR-1- MAGEA2, EMX2OS-miR-124- CALCA and
EMX2OS-miR-124-GABRG2 exhibited the highest scores. According to
the BC values in the miRNA-mRNA network and the RFE algorithm, 32
optimal feature-coding genes, including MAGEA2, CALCA and GABRG2
were identified to be associated with recurrent laryngeal cancer
samples. Since the 32 feature-coding genes were identified by
analyzing the miRNA-mRNA network, all 32 coding genes were
associated with DE-miRs. The SVM classifier constructed using these
feature-coding genes exhibited high accuracy in grouping samples
from the TCGA dataset (94.05%) and from the two validation datasets
(GSE27020: 92.66%; GSE25727: 91.07%).
miR-206 serves an important role in the inhibition
of tumor proliferation and metastasis in numerous cancer types
(31–33). In laryngeal cancer, miR-206 may
serve as a tumor suppressor by targeting vascular endothelial
growth factor (34). miR-1, a
homolog of miR-206 (35), was
identified as a DE-miR in the present ceRNA network and may serve a
role in the control of laryngeal cancer recurrence. The present
bioinformatics analyses identified that miR-1 may be associated
with the lncRNA HOTAIR in laryngeal cancer. In LSCC, HOTAIR is
upregulated and associated with the risk of lymphatic metastasis,
and with poor prognosis (36,37).
In addition, the oncogenic role of HOTAIR may be associated with
PTEN methylation (38). To the
best of the authors' knowledge, no previous study has investigated
the regulatory association between miR-1 and HOTAIR in laryngeal
cancer. However, miR-1 was demonstrated to be a direct downstream
target of HOTAIR in thyroid cancer cells, and miR-1 inhibition may
promote HOTAIR-mediated tumor progression (39). In bladder cancer cells, the
increased expression of miR-1 is negatively associated with HOTAIR
(40). In the present ceRNA
network, MAGEA2 was identified to be a potential target of miR-1.
In HNSCC, MAGEA2 is upregulated and promotes the growth of normal
oral keratinocytes, partly via the p53 pathway (41). Collectively, the interaction
HOTAIR-miR-1-MAGEA2 may serve a regulatory role in the progression
of laryngeal cancer.
miR-33b belongs to the miR-33 family and regulates
cholesterol homeostasis, and lipid and glucose metabolism (42). A previous study observed that
miR-33b may inhibit the metastasis of breast cancer (43). In gastric cancer, curcumin exerts
its inhibitory activity by upregulating the expression level of
miR-33b (44). In laryngeal
cancer, a previous study identified that miR-33a may exert
antiproliferative effects by targeting pim-1 proto-oncogene,
serine/threonine kinase (45). In
the present ceRNA network, miR-33b was associated with the lncRNA
HCG4, which was identified to be a candidate gene in multiple
sclerosis (46); however, its role
in laryngeal cancer remains unclear. The present results suggested
that HCG4 was a DE-lncR associated with the recurrence of laryngeal
cancer, which interacted with miR-33b. Therefore, these two
molecular factors may be associated with and may serve important
functional roles during the recurrence of laryngeal cancer.
miR-124 has been identified to serve a role in
numerous cancer types (47,48).
Hypermethylation of miR-124 may result in the development of colon,
breast and lung cancer (49). The
downregulation of miR-124 expression may suppress nasopharyngeal
carcinoma (50). Furthermore, the
expression level of miR-124 is associated with recurrence in
patients with lung cancer (51).
In HNSCC, miR-124 serves as a tumor suppressor gene (52) and inhibits the
epithelial-restricted with serine box/epidermal growth factor
receptor signaling pathway (53).
In the present study, miR-124 was identified to be differentially
expressed between recurrent and nonrecurrent laryngeal cancer
samples. Therefore, miR-124 may serve as a tumor suppressor gene in
laryngeal cancer and may be associated with laryngeal cancer
recurrence. The present ceRNA network results suggested that EMX2OS
and TTTY15 were two lncRNAs that may interact with miR-124. EMX2OS
was previously identified to be associated with brain diseases
(54). In comparison with healthy
controls, patients with myalgic encephalomyelitis/chronic fatigue
syndrome exhibited an increase in the expression level of EMX2OS
(55). EMX2OS is expressed in the
central nervous system, similarly to empty spiracles homeobox 2
(54). The gene fusion zinc finger
DHHC-type containing 2-TTTY15 was identified as one of four fusion
genes in a patient with acute myeloid leukemia, by whole genome
sequencing (56). The fusion gene
TTTY15-ubiquitin specific peptidase 9 Y-linked (USP9Y) exhibited
high prevalence among Chinese patients with prostate cancer, and
the TTTY15-USP9Y fusion is an independent predictor of prostate
cancer (57). To the best of the
authors' knowledge, no previous studies have reported the
association between TTTY15 or EMX2OS and laryngeal cancer.
Therefore, these lncRNAs may represent novel biomarkers or may
exhibit gene fusion in laryngeal cancer. In addition, miR-184
expression is upregulated in the majority of HNSCC tumors (58). In the present study, miR-124 and
miR-184 were associated with the lncRNA EMX2OS, suggesting a
potential role EMX2OS in the regulation of laryngeal cancer. Among
the genes interacting with miR-124, the present study identified
that CALCA and GABRG2 were associated with recurrent laryngeal
carcinoma. A previous study demonstrated that methylation of CALCA
was identified to be detected in 70% of patients with recurrent
thyroid cancer (59). Therefore,
CALCA and GABRG2 may serve important roles in the recurrence of
laryngeal cancer, and the regulatory mechanism underlying laryngeal
cancer recurrence may involve the EMX2OS-miR-124-CALCA/GABRG2
axis.
The SVM classifier is an effective classification
method. The SVM classifier has been used to classify various cancer
samples, in cancer subtypes including melanoma (60), metastatic breast cancer (61) and lung cancer (62). The SVM classifier was constructed
to identify the feature-coding genes that may be able to
discriminate recurrent from nonrecurrent laryngeal cancer samples.
In the present study, the SVM classifier comprising 32 feature
coding genes exhibited high accuracy in sample classification.
Specifically, the SVM classifier exhibited high accuracy in the two
validation datasets, suggesting that this method was a reliable
classification method.
Although the present bioinformatics results were
tested using validation datasets, the present study exhibited two
limitations. Since the samples were derived from TCGA and GEO
databases, the sample size may be limited. Furthermore, the present
results require validation in vitro and in vivo.
Therefore, further experiments are required to be performed in the
future to confirm the present study. Nevertheless, the present
results may provide useful insights for improving the understanding
of the pathogenesis of laryngeal cancer recurrence.
In conclusion, an SVM classifier composed of 32
feature-coding genes classified recurrent and nonrecurrent
laryngeal cancer samples with high accuracy. miR-1, miR-33b,
miR-124, HOTAIR, HCG4 and EMX2OS may represent a signature of
noncoding RNAs in recurrent laryngeal cancer. The interactions
HCG4-miR-33b, HOTAIR-miR-1-MAGEA2 and EMX2OS-miR-124-CALCA/GABRG2
may be important in the regulation of the molecular mechanisms
underlying the development of recurrent laryngeal cancer.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZT and GW performed data analyses and wrote the
manuscript. LZ and ZX conceived and designed the study. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Piccirillo JF: Importance of comorbidity
in head and neck cancer. Laryngoscope. 125:22422015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hoffman HT, Porter K, Karnell LH, Cooper
JS, Weber RS, Langer CJ, Ang KK, Gay G, Stewart A and Robinson RA:
Laryngeal cancer in the United States: Changes in demographics,
patterns of care, and survival. Laryngoscope. 116 (Suppl 111):1–13.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tamaki A, Miles BA, Lango M, Kowalski L
and Zender CA: AHNS Series: Do you know your guidelines? Review of
current knowledge on laryngeal cancer. Head Neck. 40:170–181. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wei KR, Zheng RS, Liang ZH, Sun KX, Zhang
SW, Li ZM, Zeng HM, Zou XN, Chen WQ and He J: Incidence and
mortality of laryngeal cancer in China, 2014. Zhonghua Zhong Liu Za
Zhi. 40:736–743. 2018.(In Chinese). PubMed/NCBI
|
|
5
|
Haas I, Hauser U and Ganzer U: The dilemma
of follow-up in head and neck cancer patients. Eur Arch
Otorhinolaryngol. 258:177–183. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Johansen LV, Grau C and Overgaard J:
Glottic carcinoma--patterns of failure and salvage treatment after
curative radiotherapy in 861 consecutive patients. Radiother Oncol.
63:257–267. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dai MY, Wang Y, Chen C, Li F, Xiao BK,
Chen SM and Tao ZZ: Phenethyl isothiocyanate induces apoptosis and
inhibits cell proliferation and invasion in Hep-2 laryngeal cancer
cells. Oncol Rep. 35:2657–2664. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miao S, Mao X, Zhao S, Song K, Xiang C, Lv
Y, Jiang H, Wang L, Li B, Yang X, et al: miR-217 inhibits laryngeal
cancer metastasis by repressing AEG-1 and PD-L1 expression.
Oncotarget. 8:62143–62153. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saito K, Inagaki K, Kamimoto T, Ito Y,
Sugita T, Nakajo S, Hirasawa A, Iwamaru A, Ishikura T, Hanaoka H,
et al: MicroRNA-196a is a putative diagnostic biomarker and
therapeutic target for laryngeal cancer. PLoS One. 8:e714802013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang JQ, Liu HX, Liang Z, Sun YM and Wu M:
Over-expression of p53, p21 and Cdc2 in histologically negative
surgical margins is correlated with local recurrence of laryngeal
squamous cell carcinoma. Int J Clin Exp Pathol. 7:4295–4302.
2014.PubMed/NCBI
|
|
11
|
Yang JQ, Liang Z, Wu M, Sun YM and Liu HX:
Expression of p27 and PTEN and clinical characteristics in early
laryngeal squamous cell carcinoma and their correlation with
recurrence. Int J Clin Exp Pathol. 8:5715–5720. 2015.PubMed/NCBI
|
|
12
|
Memczak S, Jens M, Elefsinioti A, Torti F,
Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer
M, et al: Circular RNAs are a large class of animal RNAs with
regulatory potency. Nature. 495:333–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hansen TB, Jensen TI, Clausen BH, Bramsen
JB, Finsen B, Damgaard CK and Kjems J: Natural RNA circles function
as efficient microRNA sponges. Nature. 495:384–388. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jin C, Zhang Y and Li J: Upregulation of
MiR-196a promotes cell proliferation by downregulating p27kip1 in
laryngeal cancer. Biol Res. 49:402016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sun X, Liu B, Zhao XD, Wang LY and Ji WY:
MicroRNA-221 accelerates the proliferation of laryngeal cancer cell
line Hep-2 by suppressing Apaf-1. Oncol Rep. 33:1221–1226. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li M, Chen SM, Chen C, Zhang ZX, Dai MY,
Zhang LB, Wang SB, Dai Q and Tao ZZ: microRNA-299-3p inhibits
laryngeal cancer cell growth by targeting human telomerase reverse
transcriptase mRNA. Mol Med Rep. 11:4645–4649. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang T, Li S, Liu J, Yin D, Yang X and
Tang Q: lncRNA-NKILA/NF-κB feedback loop modulates laryngeal cancer
cell proliferation, invasion, and radioresistance. Cancer Med.
7:2048–2063. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bai Z, Shi E, Wang Q, Dong Z and Xu P: A
potential panel of two-long non-coding RNA signature to predict
recurrence of patients with laryngeal cancer. Oncotarget.
8:69641–69650. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fountzilas E, Kotoula V, Angouridakis N,
Karasmanis I, Wirtz RM, Eleftheraki AG, Veltrup E, Markou K,
Nikolaou A, Pectasides D, et al: Identification and validation of a
multigene predictor of recurrence in primary laryngeal cancer. PLoS
One. 8:e704292013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carvalho B, Bengtsson H, Speed TP and
Irizarry RA: Exploration, normalization, and genotype calls of
high-density oligonucleotide SNP array data. Biostatistics.
8:485–499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fountzilas E, Markou K, Vlachtsis K,
Nikolaou A, Arapantoni-Dadioti P, Ntoula E, Tassopoulos G, Bobos M,
Konstantinopoulos P, Fountzilas G, et al: Identification and
validation of gene expression models that predict clinical outcome
in patients with early-stage laryngeal cancer. Ann Oncol.
23:2146–2153. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kohl M, Wiese S and Warscheid B:
Cytoscape: Software for visualization and analysis of biological
networks. Methods Mol Biol. 696:291–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Uoh KI, Kahng B and Kim D: Universal
behavior of load distribution in scale-free networks. Phys Rev
Lett. 87:2787012001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eisen MB, Spellman PT, Brown PO and
Botstein D: Cluster analysis and display of genome-wide expression
patterns. Proc Natl Acad Sci USA. 95:14863–14868. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baur B and Bozdag S: A Feature Selection
Algorithm to Compute Gene Centric Methylation from Probe Level
Methylation Data. PLoS One. 11:e01489772016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang X, Lu X, Shi Q, Xu XQ, Leung HC,
Harris LN, Iglehart JD, Miron A, Liu JS and Wong WH: Recursive SVM
feature selection and sample classification for mass-spectrometry
and microarray data. BMC Bioinformatics. 7:1972006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rivals I, Personnaz L, Taing L and Potier
MC: Enrichment or depletion of a GO category within a class of
genes: Which test? Bioinformatics. 23:401–407. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ge X, Lyu P, Cao Z, Li J, Guo G, Xia W and
Gu Y: Overexpression of miR-206 suppresses glycolysis,
proliferation and migration in breast cancer cells via PFKFB3
targeting. Biochem Biophys Res Commun. 463:1115–1121. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wei C, Wang S, Ye ZQ and Chen ZQ: miR-206
inhibits renal cell cancer growth by targeting GAK. J Huazhong Univ
Sci Technolog Med Sci. 36:852–858. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ren XL, He GY, Li XM, Men H, Yi LZ, Lu GF,
Xin SN, Wu PX, Li YL, Liao WT, et al: MicroRNA-206 functions as a
tumor suppressor in colorectal cancer by targeting FMNL2. J Cancer
Res Clin Oncol. 142:581–592. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang T, Liu M, Wang C, Lin C, Sun Y and
Jin D: Down-regulation of MiR-206 promotes proliferation and
invasion of laryngeal cancer by regulating VEGF expression.
Anticancer Res. 31:3859–3863. 2011.PubMed/NCBI
|
|
35
|
Yang Q, Zhang C, Huang B, Li H, Zhang R,
Huang Y and Wang J: Downregulation of microRNA-206 is a potent
prognostic marker for patients with gastric cancer. Eur J
Gastroenterol Hepatol. 25:953–957. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zheng J, Xiao X, Wu C, Huang J, Zhang Y,
Xie M, Zhang M and Zhou L: The role of long non-coding RNA HOTAIR
in the progression and development of laryngeal squamous cell
carcinoma interacting with EZH2. Acta Otolaryngol. 137:90–98. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang J, Zhou Y, Lu J, Sun Y, Xiao H, Liu M
and Tian L: Combined detection of serum exosomal miR-21 and HOTAIR
as diagnostic and prognostic biomarkers for laryngeal squamous cell
carcinoma. Med Oncol. 31:1482014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li D, Feng J, Wu T, Wang Y, Sun Y, Ren J
and Liu M: Long intergenic noncoding RNA HOTAIR is overexpressed
and regulates PTEN methylation in laryngeal squamous cell
carcinoma. Am J Pathol. 182:64–70. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Di W, Li Q, Shen W, Guo H and Zhao S: The
long non-coding RNA HOTAIR promotes thyroid cancer cell growth,
invasion and migration through the miR-1-CCND2 axis. Am J Cancer
Res. 7:1298–1309. 2017.PubMed/NCBI
|
|
40
|
Yu D, Zhang C and Gui J: RNA-binding
protein HuR promotes bladder cancer progression by competitively
binding to the long noncoding HOTAIR with miR-1. OncoTargets Ther.
10:2609–2619. 2017. View Article : Google Scholar
|
|
41
|
Glazer CA, Smith IM, Bhan S, Sun W, Chang
SS, Pattani KM, Westra W, Khan Z and Califano JA: The role of
MAGEA2 in head and neck cancer. Arch Otolaryngol Head Neck Surg.
137:286–293. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rayner KJ, Suárez Y, Dávalos A, Parathath
S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ and
Fernández-Hernando C: MiR-33 contributes to the regulation of
cholesterol homeostasis. Science. 328:1570–1573. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lin Y, Liu AY, Fan C, Zheng H, Li Y, Zhang
C, Wu S, Yu D, Huang Z, Liu F, et al: MicroRNA-33b Inhibits Breast
Cancer Metastasis by Targeting HMGA2, SALL4 and Twist1. Sci Rep.
5:99952015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sun Q, Zhang W, Guo Y, Li Z, Chen X, Wang
Y, Du Y, Zang W and Zhao G: Curcumin inhibits cell growth and
induces cell apoptosis through upregulation of miR-33b in gastric
cancer. Tumour Biol. 37:13177–13184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Karatas OF: Antiproliferative potential of
miR-33a in laryngeal cancer Hep-2 cells via targeting PIM1. Head
Neck. 40:2455–2461. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Burfoot RK, Jensen CJ, Field J, Stankovich
J, Varney MD, Johnson LJ, Butzkueven H, Booth D, Bahlo M, Tait BD,
et al: SNP mapping and candidate gene sequencing in the class I
region of the HLA complex: Searching for multiple sclerosis
susceptibility genes in Tasmanians. Tissue Antigens. 71:42–50.
2008.PubMed/NCBI
|
|
47
|
Wang M, Meng B and Liu Y, Yu J, Chen Q and
Liu Y: MiR-124 Inhibits Growth and Enhances Radiation-Induced
Apoptosis in Non-Small Cell Lung Cancer by Inhibiting STAT3. Cell
Physiol Biochem. 44:2017–2028. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen Z, Liu S, Tian L, Wu M, Ai F, Tang W,
Zhao L, Ding J, Zhang L and Tang A: miR-124 and miR-506 inhibit
colorectal cancer progression by targeting DNMT3B and DNMT1.
Oncotarget. 6:38139–38150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Roman-Gomez J, Agirre X, Jiménez-Velasco
A, Arqueros V, Vilas-Zornoza A, Rodriguez-Otero P, Martin-Subero I,
Garate L, Cordeu L, San José-Eneriz E, et al: Epigenetic regulation
of microRNAs in acute lymphoblastic leukemia. J Clin Oncol.
27:1316–1322. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Peng XH, Huang HR, Lu J, Liu X, Zhao FP,
Zhang B, Lin SX, Wang L, Chen HH, Xu X, et al: MiR-124 suppresses
tumor growth and metastasis by targeting Foxq1 in nasopharyngeal
carcinoma. Mol Cancer. 13:1862014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Patnaik SK, Kannisto E, Knudsen S and
Yendamuri S: Evaluation of microRNA expression profiles that may
predict recurrence of localized stage I non-small cell lung cancer
after surgical resection. Cancer Res. 70:36–45. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao Y, Ling Z, Hao Y, Pang X, Han X,
Califano JA, Shan L and Gu X: MiR-124 acts as a tumor suppressor by
inhibiting the expression of sphingosine kinase 1 and its
downstream signaling in head and neck squamous cell carcinoma.
Oncotarget. 8:25005–25020. 2017.PubMed/NCBI
|
|
53
|
Zhang M, Piao L, Datta J, Lang JC, Xie X,
Teknos TN, Mapp AK and Pan Q: miR-124 Regulates the
Epithelial-Restricted with Serine Box/Epidermal Growth Factor
Receptor Signaling Axis in Head and Neck Squamous Cell Carcinoma.
Mol Cancer Ther. 14:2313–2320. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Spigoni G, Gedressi C and Mallamaci A:
Regulation of Emx2 expression by antisense transcripts in murine
cortico-cerebral precursors. PLoS One. 5:e86582010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yang CA, Bauer S, Ho YC, Sotzny F, Chang
JG and Scheibenbogen C: The expression signature of very long
non-coding RNA in myalgic encephalomyelitis/chronic fatigue
syndrome. J Transl Med. 16:2312018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang L, Sun Y, Sun Y, Meng L and Xu X:
First case of AML with rare chromosome translocations: A case
report of twins. BMC Cancer. 18:4582018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhu Y, Ren S, Jing T, Cai X, Liu Y, Wang
F, Zhang W, Shi X, Chen R, Shen J, et al: Clinical utility of a
novel urine-based gene fusion TTTY15-USP9Y in predicting prostate
biopsy outcome. Urol Oncol. 33:384.e9–384.e20. 2015. View Article : Google Scholar
|
|
58
|
Kao SY, Tsai MM, Wu CH, Chen JJ, Tseng SH,
Lin SC and Chang KW: Co-targeting of multiple microRNAs on
factor-Inhibiting hypoxia-Inducible factor gene for the
pathogenesis of head and neck carcinomas. Head Neck. 38:522–528.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hu S, Ewertz M, Tufano RP, Brait M,
Carvalho AL, Liu D, Tufaro AP, Basaria S, Cooper DS, Sidransky D,
et al: Detection of serum deoxyribonucleic acid methylation
markers: A novel diagnostic tool for thyroid cancer. J Clin
Endocrinol Metab. 91:98–104. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Afifi S, Gholamhosseini H and Sinha R: SVM
classifier on chip for melanoma detection. In: Proceedings of the
39th Annual International Conference of the IEEE Engineering in
Medicine and Biology Society (EMBC). IEEE; Seogwipo: pp. 270–274.
2017, PubMed/NCBI
|
|
61
|
Tuo Y, An N and Zhang M: Feature genes in
metastatic breast cancer identified by MetaDE and SVM classifier
methods. Mol Med Rep. 17:4281–4290. 2018.PubMed/NCBI
|
|
62
|
Zhao J, Cheng W, He X, Liu Y, Li J, Sun J,
Li J, Wang F and Gao Y: Construction of a specific SVM classifier
and identification of molecular markers for lung adenocarcinoma
based on lncRNA-miRNA-mRNA network. OncoTargets Ther. 11:3129–3140.
2018. View Article : Google Scholar
|