Introduction
Cornea plana is a rare disorder characterized by
small flat corneas, marked hypermetropia, reduced visual activity,
iris abnormalities, a hazy corneal limbus and corneal clouding,
with or without associated accommodative esotropia. It is inherited
as either an autosomal dominant (CNA1; OMIM121400) or autosomal
recessive trait (CNA2; OMIM217300); The defective gene in CNA1 has
not been identified. CNA2 are associated with the more severe
phenotype and additional ocular manifestations, including the
presence of a degree of iris malformation and adhesions between the
cornea and iris (1–8). Corneal ectasia, corneal hydrops and
idiopathic corneal decompensation are very rare findings (2,9–11).
In humans, bi-allelic mutations in keratocan
(KERA) cause CNA2. The gene, which has been localized to
chromosome 12q22, contains two introns and three exons; a total of
352 amino acids are translated from the majority of exon 2 and a
small portion of exon 3 to form a precursor KERA protein, and exon
1 remains untranslated (12). The
gene encodes KERA, an evolutionarily-conserved small leucine-rich
proteoglycan (SLRP). The SLRPs are a family of highly conserved
extracellular matrix proteoglycans with core proteins composed of
leucine-rich repeats (LRRs). LRRs are composed of a series of
parallel β-strands, which stack into an arched β-sheet array. The
leucine-rich region LXXLXLXXNXL is highly conserved: N represents
cysteine or asparagine; L represents a hydrophobic amino acid or
leucine; and X represents any amino acid. The interior of the LRR
domain is composed of hydrophobic residues, with the exception of
the conserved asparagines (Asn, N) which create an Asn-ladder
stabilizing the structure, as reported in previous studies
(13–15). In addition, the KERA protein is
highly expressed in the cornea and is considered to be a key
regulator of collagen fibrin production, which regulates the
diameter and spacing of fibrils and determines corneal structure
and transparency (16). Mouse
studies indicate that this gene is exclusively expressed in the
cornea and serves an important role during eye development
(17). Narrow anterior chamber
angles, thin corneal stroma and large-diameter corneal fibers are
identified in KERA-knockout mice and no other systemic
abnormality (18). To date, 13
different disease-associated KERA mutations have been
documented in various populations, all of which manifest as various
bilateral anterior segment lesions, including seven missense
mutations, three nonsense mutations, one frame shift mutation, one
splice-site mutation and one 3 base-pair deletion mutation.
Notably, mutations in KERA have not been reported in an
Asian population to date (1–4,6–10,19–24).
The present study reports two novel KERA
mutations in a Chinese family. The family history was consistent
with an autosomal recessive mode of inheritance. Clinical detection
and genetic analysis was conducted on five family members.
Materials and methods
Subject recruitment and clinical
examination
A family with CNA2 was recruited and identified at
the Hunan Jiahui Genetics Hospital (Changsha, China) in October
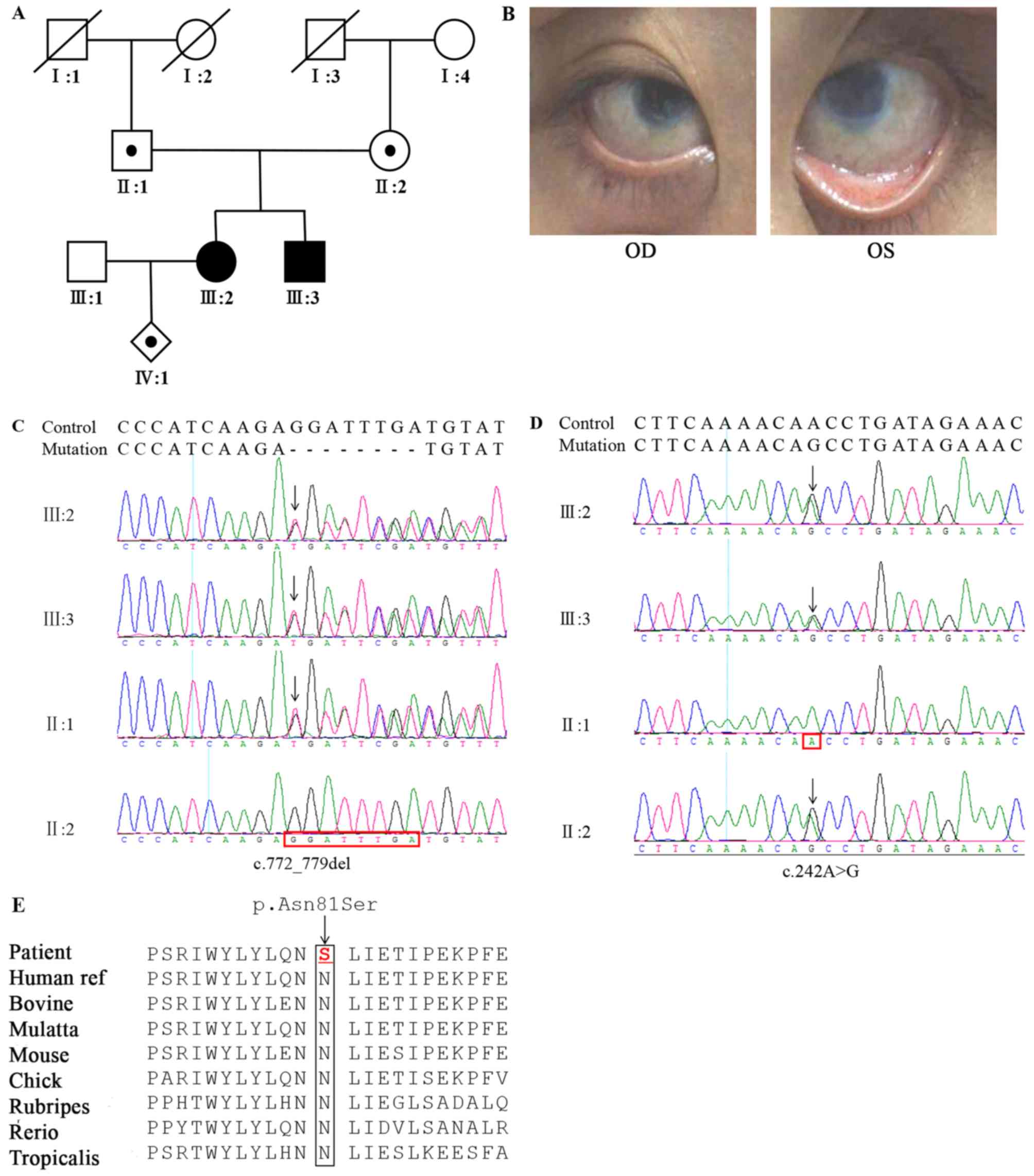
2017. A pedigree of the family is illustrated in Fig. 1A. The proband, III:2, developed
visual disorders shortly following birth, but hadn't been diagnosed
or treated. She visited the doctor at the age of 26 years due to a
considerable decrease in binocular visual acuity and accompanied by
photophobia. Clinical and eye examinations demonstrated temporal
iridocorneal synechiae, corneal clouding (Fig. 1B) and an unclear fundus. In
addition, she was pregnant at the same time. A similar phenotype
was observed in the younger brother of the proband, III:3.
Best-corrected visual acuity (BCVA) was established
using the Acta Ophthalmologica 2017 LogMAR charts. Visual acuity
was examined using the ETDRS chart. Intraocular pressure was
measured by Goldmann applanation tonometry. Anterior chamber depth
and axial length were measured with ocular A ultrasound. Motility,
pupillary reaction, ocular fundus, slit lamp examinations and
keratometry (Zeiss keratometer; Zeiss AG) were performed and
documented. The growth and development of III:2 and III:3 were in
the normal range. Physical examinations were performed to exclude
systemic diseases.
Ethical approval
The present study was performed according to the
guidelines approved by the Ethics Committee of Hunan Jiahui
Genetics Hospital and adhered to the tenets set out in the Helsinki
Declaration. Informed consent was obtained from all subjects.
Genomic DNA extraction and variant
screening
Venous blood samples were collected from the study
subjects (III:2 and III:3), their family members (II:1, II:2 and
III:1) and 200 randomly selected normal individuals without any
history of ophthalmic disease. In addition, amniotic fluid was
collected from III:2 to reflect the condition of the fetus (IV:1).
Genomic DNA from all subjects was extracted via the
phenol/chloroform method. Whole-exome sequencing data were screened
for pathological variants in the patients (III:2 and III:3), and
sanger sequencing of the candidate variants in KERA was performed
on all subjects.
The extracted DNA was fragmented with DNase and
purified via the magnetic bead method, followed by ligation of the
adaptor sequence and polymerase chain reaction (PCR) amplification.
PCR was conducted using a Phusion® High-Fidelity PCR
Master Mix (New England BioLabs, Inc.) and the follows primers:
5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′
and
5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT-3′.
PCR was performed under the following conditions: 98°C for 30 sec;
then 12 cycles of 98°C for 10 sec, 65°C for 30 sec and 72°C for 30
sec; and finally 72°C for 5 min. Library preparation was performed
as previously described (25). The
whole-exome was captured by Nimblegen SeqCap EZ Exome Probes v3.0
(Roche Diagnostics, Basel, Switzerland), extended, purified and
finally sequenced on an Illumina HiSeq sequencer (Illumina, Inc.).
The coverage of the target sequence was not less than 97% and the
average depth of the target area was 130.95.
The original data were obtained following sequencing
of all exons. The Burrows Wheeler Aligner algorithm was used to
compare the reference sequence (UCSChg19), GATK version 3.7.0
(https://software.broadinstitute.org/gatk/) and VarScan
version 2 software (http://dkoboldt.github.io/varscan/) were used to
identify mutations, e.g. single nucleotide polymorphisms (SNPs) and
InDels, including detection, annotation and statistical analysis.
Annovar (http://www.openbioinformatics.org/annovar/) was
utilized to catalog the detected variants. The list of mutations
was tabulated, with the variants being described according to the
HGVS guidelines. Combining this analysis with the public variation
database ExAc, mutations with frequencies <0.1% in the
population were identified. Mutations were scored for disease
effect using a range of predictive tools [e.g. SIFT (http://sift.bii.a-star.edu.sg/), PolyPhen-2
(http://genetics.bwh.harvard.edu/pph2), Provean
(http://provean.jcvi.org/) and MutationTaster
(http://www.mutationtaster.org/)]. Based
on the results of the data analysis, the mutations were identified
with reference to the 1000 Genomes Project (http://browser.1000genomes.org), the Exome Variant
Server (http://evs.gs.washington.edu/EVS/), dbSNP database
(https://www.ncbi.nlm.nih.gov/projects/SNP/index_old.html)
and the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). HomoloGene
(https://www.ncbi.nlm.nih.gov/homologene) and T-Coffee
multiple sequence alignment (http://www.tcoffee.org) were utilized to determine the
conservation of the mutated amino acid residues between different
species. A three-dimensional structure of keratocan was modeled
using SWISS-MODEL (https://swissmodel.expasy.org). The analysis was
performed in combination with assessment of the clinical
manifestations and the mode of inheritance. Finally, the nature of
the mutation sites were interpreted according to the guideline by
the American College of Medical Genetics and Genomics/the
Association for Molecular Pathology (ACMG) (26).
The two novel candidate variations were validated
using direct bidirectional sequencing methods. Based on the
relevant gene standard sequences listed at the NCBI, primers were
designed using Primer Premier 5 software (Premier Biosoft
International). Primers forward (5′-3′): CACCCAGTTTTCCTACTGCTTTATA,
primers reverse (5′-3′): GCACTGATTCGGGGAACCTT. PCR amplification
was performed on the target DNA fragment (700 bp product size, 20
µl reaction system, annealing temperature 62°C) and the product was
verified by 1.0% agarose gel electrophoresis. Direct sequencing was
performed on an ABI 3130 DNA Sequencer (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The sequenced results were aligned with
the reference sequence using SeqMan at the NCBI (no.
NM_007035).
Results
Clinical findings
III:2 exhibited a BCVA of 0.2 oculus dextrus (OD;
right eye) and 0.3 oculus sinister (OS; left eye) with hyperopic
refractive values. Additionally, she exhibited esotropia (right
eye), microcornea, corneal opacity, short axial length, shallow
anterior chamber, iridocorneal adhesion, pupillary irregularities
(1×2 mm irregular right pupil shifting to the nasal side;
needle-like changes on the left pupil) and slow light reflection.
Extraocular motility, confrontation fields and intraocular pressure
were normal, and the lens was transparent, although the fundus was
difficult to see clearly (Table
I). The younger brother of the proband (III:3) presented with
similar clinical symptoms, however the parents had not identified
any eye problems.
 | Table I.Keratocan gene mutations and clinical
characteristics of patient with cornea plana. |
Table I.
Keratocan gene mutations and clinical
characteristics of patient with cornea plana.
|
|
|
|
| Central corneal
haze | BCVA | Refraction (DS,
DC) | CCT (µm) | ACD (mm) | IOP (mmHg) | Axial length
(mm) | K1/K2 (D) |
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|---|
| Patient/origin | Gender/age (y) | Family history | Nucleotide
change | RE | LE | RE | LE | RE | LE | RE | LE | RE | LE | RE | LE | RE | LE | RE | LE |
|---|
| III:2/China | F/26 | Y | c.242A>G | Y | Y | 0.2 | 0.3 | +12.00/-2.0DC | +14.00 | 473 | 502 | 1.36 | 1.04 | 8.2 | 16.8 | 20.8 | 20.3 | 33.59/35.86 | 27.68/30.36 |
|
|
|
| c.772-779 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| delGGATTTGA |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Mutation screening and bioinformatics
analysis
The family of interest, originating from China,
displayed an autosomal recessive pattern of inheritance. A compound
heterozygous mutation c.242A>G (p.N81S) and
c.772_779delGGATTTGA(P.G258Cfs*30) in exon 2 of the KERA gene was
identified in III:2 and III:3, respectively derived from the
heterozygous father (c.772_779delGGATTTGA) and mother (c.242A>G;
Fig. 1C and D), although neither
of the two mutations were present in the normal controls. These
variants have not been previously reported in the 1000 Genomes
Project, the Human Gene Mutation Database, dbSNP or the Exome
Variant Server. The first variation, an N81S amino acid
substitution in the KERA protein, was predicted to be likely
pathogenic by the in-silico prediction programs, Mutation Taster,
Provean, n PolyPhen (27) and SIFT
(28) (Table II). The second mutation was a 7
base-pair deletion (G258Cfs*30), leading to the loss of LRR8-11 and
producing a strongly truncated protein, which may trigger
nonsense-mediated mRNA decay (−66 amino acid/>10% missing); the
prediction algorithms supported its pathogenic nature. Multiple
sequence alignment indicated that the residues at position 81 of
KERA are highly conserved across species (Fig. 1E) and the lost LRR8-11 regions due
to 258 residual changes are also highly conserved. According to
ACMG, p.N81S was judged likely to be damaging (PM1-3 and PP1-4) and
P.G258Cfs*30 was also judged as damaging (PVS1, PM2, PP1, PP3 and
PP4) (28).
| Table II.Predicted effect of 2 novel mutations
on function of KERA protein. |
Table II.
Predicted effect of 2 novel mutations
on function of KERA protein.
|
|
|
|
|
| Prediction |
|
|
|---|
|
|
|
|
|
|
|
|
|
|---|
| Mutation at
nucleotide level | Change at protein
level | Exon | Predicted protein
domain | Conservation |
PolyPhen2a | Mutation
taster |
Proveana | SIFTa | Frameshift and
PTC | ACMG/AMP variants
classificationb |
|---|
| c.242A>G | p.N81S | 2 | LRRs | Highly
conserved | Probably
damaging | Disease
causing | Deleterious | Damaging | No | Likely
Pathogenic |
|
c.772_779delGGATTTGA |
p.G258Cfs*30 | 2 | LRRs | Highly
conserved | NA | Disease
causing | NA | NA | Yes | damaging |
Primary sequence analysis indicated that the N81S
substitution occurs at an LRR motif, the first of 11 such motifs
present in the sequence (Fig. 2C)
(29). G258Cfs*30 indicates a 7
base-pair deletion leading to a frameshift and premature
termination codon (PTC), followed by the loss of LRR8-11. The
predicted three-dimensional structure of the KERA protein using the
SWISS-MODEL online software suggested that the current mutant
protein (P.G258Cfs*30) is significantly different from the wild
type and these changes may lead to abnormal protein function
(Fig. 2D) (30). The architecture of other known LRR
proteins reveals that the highly conserved LXXLXLXXNXL repetitive
motifs form a series of parallel β-strands and these β-strands form
a horseshoe-like concave shape. The asparagines (Asn, N) develop a
stable Asn-ladder structure. The present study identified that the
p.(Asn81Ser) mutation affects the conserved Asn81 residue of LRR1.
This substitution to an aspartic acid residue will alter a hydrogen
bond donor to make it either an acceptor or neutral, possibly
resulting in the instability of the Asn-ladder structure or
abnormal interactions between the mutant KERA and other proteins,
including collagen. The second mutation, G258Cfs*30, that is
reported in the present study results in the loss of LRR8-11. This
deletion may form an abnormal truncated protein, thereby causing
disease.
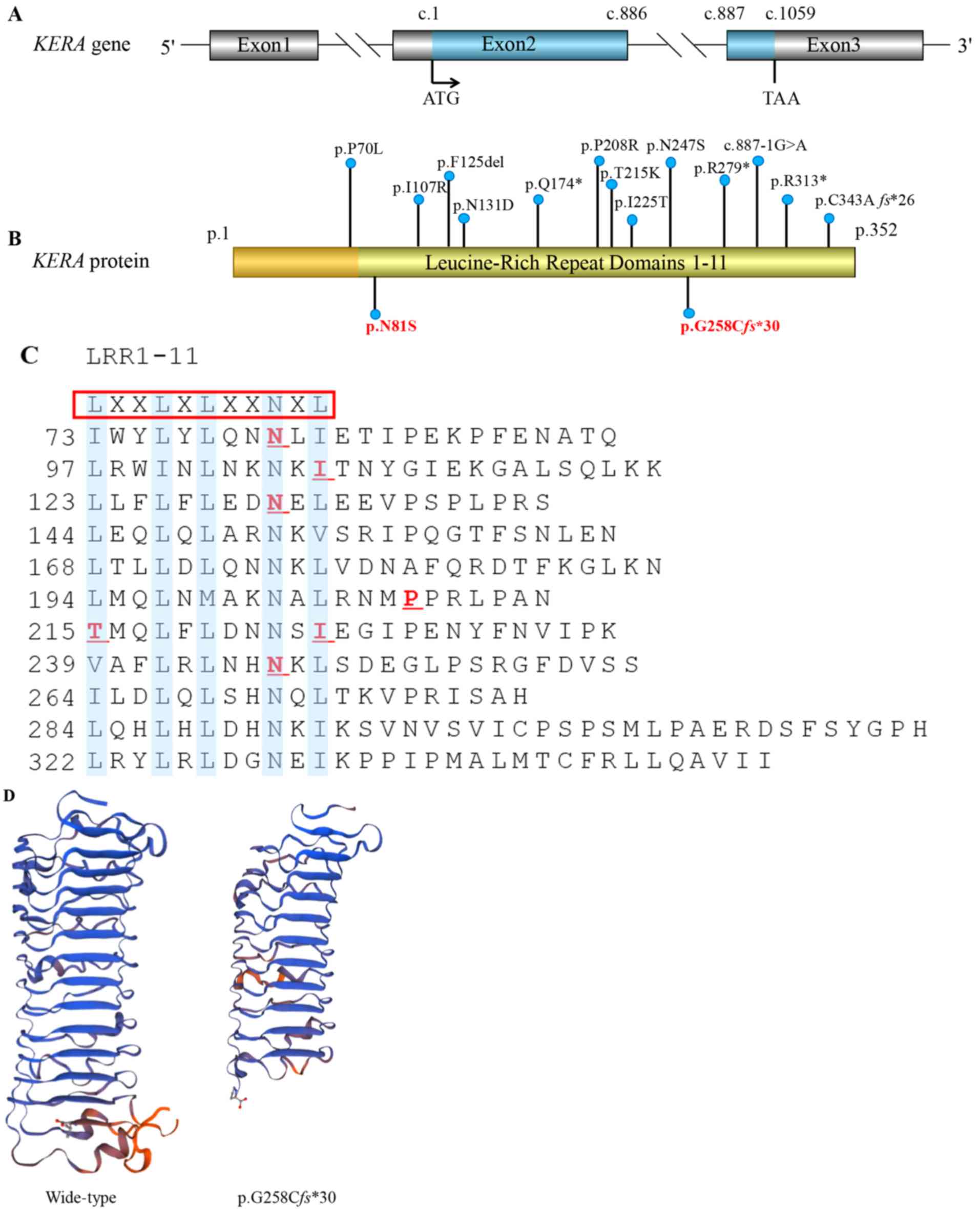 | Figure 2.KERA mutations associated with cornea
plana. (A) The structure of the KERA gene. It contains three exons
and two introns; the blue marker indicates the translation area,
which forms a precursor protein of 352 amino acids. ATG: Initiation
codon; TAA: Stop codon. (B) Kera protein schematic, including
locations of the variations that have previously been reported. In
the present study, amino acid position 81 was changed from
asparagine (Asn, N) to serine (Ser, S). The p.G258Cfs*30 mutation
leading to premature termination codon is indicated in bold red.
Apart from p.P70L, all mutations associated with the disease are
located in the LRR domains. (C) The blue highlighting indicates the
consensus motifs LXXLXLXXNXL, which form 11 LRR domains. The red
underlining indicates the six missense mutations previously
identified. (D) Mutant Kera (p.G258Cfs*30) was predicted by
Swiss-Model online software to result in the loss of the partial
LRR domain and the C-terminal domain, compared with the wild type
protein. LRR, leucin-rich repeat; Kera, keratocan. |
Discussion
With all the previously reported KERA
mutations CNA2 displays a relatively homogeneous phenotype and
genetic heterogeneity. Although certain previous studies have
documented an atypical phenotype, including superior pellucid
corneal marginal degeneration, hypotrichosis and corneal edema
(2,9,11,21),
they retain a classically identifiable phenotype of CNA2. Any
alteration of a highly conserved encoded protein is likely to
result in CNA2 with specific characteristics.
The present study reports on the phenotype and
identification of suspected pathogenic variations in a Chinese
family with CNA2. The proband and her brother, with two novel
KERA mutations in a compound heterozygous form, exhibited
the typical corneal phenotype, including marked hypermetropia, flat
corneas with opacification and iridocorneal synechiae. In addition,
the proband exhibited a short axis length. This finding supports
the hypothesis proposed in previous studies that the KERA
mutation may lead to microphthalmia, although this has not been
proven (6,22). At present, there is no known effect
on endothelial cell function and no specific studies have been
conducted internationally. The unaffected parents in the present
case were mutation carriers and did not exhibit corneal
abnormalities, suggesting that in the presence of mutant gene
products there remains some normal KERA proteins, which allows the
cornea to develop normally.
Nearly all the conservative LRRs in the KERA
gene are encoded in the larger second exon. Original descriptions
of the gene mentioned 11 LRRs. A similar LRR motif exists in the
structure of a porcine RNase inhibitor, the crystal structure of
which has been elucidated (31).
The structure of other proteins containing similar LRR motifs also
mimic a β-sheet array and the KERA structural model presented in
the present study supports this. These LRRs are flanked by cysteine
clusters, a sulfur-containing amino acid that allows for disulfide
bonds and the maintenance of the three-dimensional conformation
that serves a key role in keratocan functioning.
The most reported mutations in KERA affect
the LRR structure/conformation. The present study uncovered a novel
missense mutation in KERA exon 2; an Asn81Ser replacement
occurs within first LRR motif and disrupts it, potentially altering
the stacking or spacing of the β-strand. Another novel deletion
mutation, p.G258Cfs*30, in exon 2 leads to a frame shift and PTC,
followed by the loss of LRR8-11. The protein is prematurely
truncated due to the destruction of 7th LRR and the deficiency of
the 8 to 11th LRR motifs, which contain 287 amino acids
instead of 353. Conventional LRR motif alignment is believed to be
important in the spacing of collagen fibrils on which corneal
transparency is dependent. Aberrant KERA in the corneal stroma may
not bind to collagen fibrils, therefore damaging the regulatory
effect of KERA during the development of the normal corneal
structure.
Reviewing the literature, it was identified that
CNA2 is a rare disorder with a worldwide distribution and a high
prevalence in particular populations, e.g. those of Finnish
(19), Saudi (3) and Czech descent (23). Almost no mutations have been
identified in the Asian population, except for a Chinese American
individual who was mentioned in a previous study, although this
case was not documented in detail (8). In historically isolated populations,
including Saudi and Finnish populations, the occurrence of CNA2 is
not only due to a single common founder effect; certain cultural
preferences, including intermarriage or endogamy, may also increase
the incidence of recessively inherited diseases (8).
In conclusion, the present study identified two
novel KERA variants in a Chinese family with cornea plana.
These findings, together with the previously reported mutations,
demonstrate the occurrence of CNA2 in different populations and
expand the variant spectrum of the KERA gene; therefore, the
results of the present study may be valuable for the diagnosis of
and genetic counseling for cornea plana.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Key R&D Program of China (grant no. 2017YFC1001802)
and the National Natural Science Foundation of China (grant nos.
81501268, 31601035 and 81571450).
Availability of data and materials
All data generated and analyzed during the present
study are included in this published article.
Authors' contributions
CZH, XGL, HT collected clinical data from the CNA2
family. CZH, XGL, LQW and PSYL performed molecular genetics
experiments on the family and the controls. CZH, XGL, HU TAN and CP
conducted the bioinformatics analysis. CZH, CP and LQW designed and
supervised the study. CZH drafted the manuscript. WGL contributed
in data analysis. CP and PSYL, WGL and LQW revised the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Informed consent for this investigation was obtained
from all participating CNA2 patients and parents, and the
principles outlined in the Declaration of Helsinki were followed.
The study was conducted in agreement with the Ethical Committee of
the Center for Medical Genetics, School of Life Sciences, Central
South University (Changsha, China).
Patient consent for publication
The publication of images from the CNA2 patient has
the support and informed consent of the patient.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CNA2
|
autosomal recessive cornea plana
|
|
L
|
hydrophobic amino acid or leucine
|
|
X
|
any amino acid
|
|
N
|
cysteine or asparagine
|
|
SLRP
|
small leucine-rich proteoglycan
|
|
LRRs
|
leucine-rich repeats
|
|
BCVA
|
best-corrected visual acuity
|
|
ACD
|
anterior chamber depth
|
|
PTC
|
premature termination codon
|
References
|
1
|
Pellegata NS, Dieguez-Lucena JL, Joensuu
T, Lau S, Montgomery KT, Krahe R, Kivelä T, Kucherlapati R, Forsius
H and de la Chapelle A: Mutations in KERA, encoding keratocan,
cause cornea plana. Nat Genet. 25:91–95. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khan AO, Aldahmesh M and Meyer B: Corneal
ectasia and hydrops in a patient with autosomal recessive cornea
plana. Ophthalmic Genet. 27:99–101. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Khan AO, Aldahmesh M and Meyer B:
Recessive cornea plana in the Kingdom of Saudi Arabia.
Ophthalmology. 113:1773–1778. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khan AO, Aldahmesh MA, Al-Gehedan S, Meyer
BF and Alkuraya FS: Corneal decompensation in recessive cornea
plana. Ophthalmic Genet. 30:142–145. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tahvanainen E, Forsius H, Kolehmainen J,
Damsten M, Fellman J and de la Chapelle A: The genetics of cornea
plana congenita. J Med Genet. 33:116–119. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lehmann OJ, El-ashry MF, Ebenezer ND,
Ocaka L, Francis PJ, Wilkie SE, Patel RJ, Ficker L, Jordan T, Khaw
PT and Bhattacharya SS: A novel keratocan mutation causing
autosomal recessive cornea plana. Invest Ophthalmol Vis Sci.
42:3118–3122. 2001.PubMed/NCBI
|
|
7
|
Khan AO, Al-Saif A and Kambouris M: A
novel KERA mutation associated with autosomal recessive cornea
plana. Ophthalmic Genet. 25:147–152. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ebenezer ND, Patel CB, Hariprasad SM, Chen
LL, Patel RJ, Hardcastle AJ and Allen RC: Clinical and molecular
characterization of a family with autosomal recessive cornea plana.
Arch Ophthalmol. 123:1248–1253. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khan AO, Aldahmesh M, Al-Saif A and Meyer
B: Pellucid marginal degeneration coexistent with cornea plana in
one member of a family exhibiting a novel KERA mutation. Br J
Ophthalmol. 89:1538–1540. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liskova P, Hysi PG, Williams D, Ainsworth
JR, Shah S, de la Chapelle A, Tuft SJ and Bhattacharya SS: Study of
p.N247S KERA mutation in a British family with cornea plana. Mol
Vis. 13:1339–1347. 2007.PubMed/NCBI
|
|
11
|
AlBakri A and Khan AO: Regarding corneal
decompensation in recessive cornea plana. Ophthalmic Genet.
37:350–351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tasheva ES, Funderburgh JL, Funderburgh
ML, Corpuz LM and Conrad GW: Structure and sequence of the gene
encoding human keratocan. DNA. 10:67–74. 1999.
|
|
13
|
Kobe B and Deisenhofer J: The leucine-rich
repeat: A versatile binding motif. Trends Biochem Sci. 19:415–421.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kobe B and Deisenhofer J: Proteins with
leucine-rich repeats. Curr Opin Struct Biol. 5:409–416. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bella J, Hindle KL, McEwan PA and Lovell
SC: The leucine-rich repeat structure. Cell Mol Life Sci.
65:2307–2333. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kao WW and Liu CY: Roles of lumican and
keratocan on corneal transparency. Glycoconj J. 19:275–285. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu CY, Shiraishi A, Kao CW, Converse RL,
Funderburgh JL, Corpuz LM, Conrad GW and Kao WW: The cloning of
mouse keratocan cDNA and genomic DNA and the characterization of
its expression during eye development. J Biol Chem.
273:22584–22588. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu CY, Birk DE, Hassell JR, Kane B and
Kao WW: Keratocan-deficient mice display alterations in corneal
structure. J Biol Chem. 278:21672–21677. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Forsius H, Damsten M, Eriksson AW, Fellman
J, Lindh S and Tahvanainen E: Autosomal recessive cornea plana. A
clinical and genetic study of 78 cases in Finland. Acta Ophthalmol
Scand. 76:196–203. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dudakova L, Palos M, Hardcastle AJ and
Liskova P: Corneal endothelial findings in a Czech patient with
compound heterozygous mutations in KERA. Ophthalmic Genet.
35:252–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roos L, Bertelsen B, Harris P, Bygum A,
Jensen H, Grønskov K and Tümer Z: Case report: A novel KERA
mutation associated with cornea plana and its predicted effect on
protein function. BMC Med Genet. 16:402015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kumari D, Tiwari A, Choudhury M, Kumar A,
Rao A and Dixit M: A novel KERA mutation in a case of autosomal
recessive cornea plana with primary angle-closure glaucoma. J
Glaucoma. 25:e106–e109. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dudakova L, Vercruyssen JHJ, Balikova I,
Postolache L, Leroy BP, Skalicka P and Liskova P: Analysis of KERA
in four families with cornea plana identifies two novel mutations.
Acta Ophthalmol. 96:e87–e91. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Khan AO: Corneal ectasia in a boy with
homozygous KERA mutation. Ophthalmic Genet. 39:141–143. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen R, Im H and Snyder M: Whole-exome
enrichment with the Roche nimbleGen SeqCap EZ exome library SR
platform. Cold Spring Harb Protoc. 2015:634–641. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ng PC and Henikoff S: Predicting
deleterious amino acid substitutions. Genome Res. 11:863–874. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Offord V, Coffey TJ and Werling D:
LRRfinder: A web application for the identification of leucine-rich
repeats and an integrative Toll-like receptor database. Dev Comp
Immunol. 34:1035–1041. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Arnold K, Bordoli L, Kopp J and Schwede T:
The SWISS-MODEL workspace: A web-based environment for protein
structure homology modelling. Bioinformatics. 22:195–201. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kobe B and Deisenhofer J: Crystal
structure of porcine ribonuclease inhibitor, a protein with
leucine-rich repeats. Nature. 366:751–756. 1993. View Article : Google Scholar : PubMed/NCBI
|