Introduction
Alzheimer's disease (AD) is a degenerative brain
disease and the most common cause of dementia (1,2). At
the microscopic level, the hallmarks of AD are amyloid β (Aβ)
deposition-induced senile plaques (SP), abnormal accumulation of
Tau protein-induced neurofibrillary tangles and extensive neuronal
loss (3–5). A key observation changing the
diagnostic and therapeutic approaches to AD is that the
pathological changes underlying brain degeneration and cognitive
loss in patients with AD begin 10–20 years prior to the onset of
dementia (6,7). Furthermore, the cognitive dysfunction
of AD patients may present years before the pathological changes
can be detected. Studies have demonstrated that the Aβ
oligomer-induced synaptic dysfunction is a major factor associated
with early cognitive dysfunction in AD (8,9).
Aβ is derived from a larger protein, amyloid
precursor protein (APP), and includes two main forms in
vivo: Amyloid β-peptide 1–42 (Aβ1-42) and amyloid β-peptide
1–40 (Aβ1-40) (9). Of these two
proteins, the proportion of Aβ1–42 is lower than that of Aβ1-40,
but Aβ1–42 polymerizes more readily and is more toxic (6). Furthermore, soluble Aβ1–42 oligomers
are also more neurotoxic than Aβ1–40 (10–12).
Studies on mouse hippocampal neurons overexpressing the APP gene
revealed that excitatory neuronal synaptic transmission was
impaired, reflected in the significant attenuation of miniature
excitatory postsynaptic currents (mEPSCs) (13). Synaptic dysfunction caused by Aβ
oligomers is an important cause of the cognitive decline observed
in AD, and synaptic dysfunction may affect synaptic plasticity
events associated with learning and memory, namely long-term
potentiation (LTP) and long-term depression (LTD) (14). Soluble Aβ1-42-perfused hippocampal
sections and in vivo experiments demonstrated that Aβ, and
its oligomers, can suppress the formation of LTP in the hippocampus
(15–17) or facilitate LTD (18), potentially leading to an early
decrease in cognitive function in AD. Furthermore, inhibitors of Aβ
oligomers can alleviate the damage of LTP caused by Aβ (19). Therefore, identifying factors that
can alleviate synaptic dysfunction is crucial for the prevention
and treatment of AD.
Endophilin A, which is composed of three homologous
cytosolic proteins (endophilin A1-3, also termed endophilin 1–3) is
highly expressed in the nervous system. Endophilin 1 is only
expressed in the brain, endophilin 2 is found in multiple tissues
and endophilin 3 is predominantly expressed in the brain and testis
(20,21). Endophilin A has been reported to
have a key role in endocytosis, including synaptic vesicle
endocytosis and member receptor endocytosis (22–25).
In the study of Ren et al (26), endophilin 1 was found to be highly
expressed in the brain of patients with AD and AD transgenic mice.
An increase in endophilin 1 levels in neurons is associated with an
increase in the activation of the stress kinase JNK, with
subsequent neuronal death (26).
The present study demonstrated that soluble Aβ can cause synaptic
dysfunction in cultured hippocampal neurons, and endophilin 1 is
highly expressed prior to the death of neurons. By coupling RNA
interference and electrophysiological recording techniques in
cultured hippocampal neurons, it was shown that knockdown of
endophilin 1 prevents synaptic dysfunction induced by Aβ.
Materials and methods
Plasmids and RNA interference
Total RNA was extracted from the brain of 1
1-month-old male Sprague-Dawley (SD) rat (purchased from the
Laboratory Animal Center of Sun Yat-Sen University and immediately
sacrificed upon arrival) using an RNeasy Mini Kit (Qiagen, Inc.).
The first-strand cDNA was generated using Superscript II reverse
transcriptase (Thermo Fisher Scientific, Inc.) and RT was performed
according to the manufacturer's protocol. Target DNA fragments were
amplified by PCR using the following primers designed and
synthetized according to the GenBank Database: Forward
5′-ATAGAATTCATGTCGGTGGCGGGG-3′ (containing an EcoRI target
sequence) and reverse, 5′-TAAGTCGACCTAATGGGGCAGAGC-3′ (containing a
SalI target sequence). PCR was conducted as follows: 95°C
for 3 min, followed by 34 cycles of 95°C for 50 sec, 58°C for 50
sec and 72°C for 2 min, and finally 72°C for 15 min. The
full-length endophilin 1 cDNA fragment was inserted into pEGFP-C1
plasmid (Clontech Laboratories, Inc.). The construct was verified
by sequencing. The methods used for constructing cDNA plasmids were
previously described in detail (27,28).
Validated endophilin 1 small interfering RNA (Endo1 siRNA;
5′-GGGCTAAACTCAGTATGAT-3′) and negative control (NC;
5′-AGCTAGGCATATGACTGTA-3′) were synthesized by Shanghai GenePharma
Co., Ltd.
Preparation of Aβ1–42 oligomers
Aβ1–42 (human) was purchased from Tocris Bioscience.
The lyophilized powder was solubilized in 50 mM Tris Buffer to 200
µM and stored at −20°C according to the manufacturer's
instructions. Prior to use, stock solutions were thawed and
incubated at 37°C for 24 h to induce peptide aggregation and then
diluted to the final concentration (1 or 50 µM) in culture medium
as previously described (29).
Cell culture and transfection
293 cells (a gift from Dr Mingtao Li, Zhongshan
School of Medicine, Sun Yat-Sen University, Guangzhou, China)
culture was performed as described previously (27,28).
Cells were maintained in DMEM (Gibco, Thermo Fisher Scientific,
Inc.) supplied with 10% FBS (Gibco, Thermo Fisher Scientific, Inc.)
and penicillin/streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.) in a 5% CO2 and 37°C incubator. To determine the
efficacy and specificity of Endo1 siRNA, 100 pmol Endo1 siRNA or NC
together with 2 µg endophilin 1-pEGFP-C1 plasmid were
co-transfected into 293 cells using calcium phosphate (Beyotime
Institute of Biotechnology). The calcium phosphate/DNA precipitate
were maintained in the culture for 4 h and washed with fresh
transfection media. Then fresh culture medium was added and further
analysis was performed 24–48 h after transfection.
Western blotting
After co-transfection for 24–48 h, the 293 cells
were lysed in lysis buffer [150 mM NaCl, 20 mM Tris, 1 mM EDTA, 1%
Triton X-100, and protease inhibitors cocktail (Sigma-Aldrich;
Merck KGaA)] on ice for 30 min. The lysates were centrifuged at
20,000 × g for 15 min at 4°C. The supernatants were then recovered
and quantified using a bicinchoninic acid protein quantification
assay (Pierce; Thermo Fisher Scientific, Inc.). Proteins were
separated by SDS-PAGE, with ~30 µg protein loaded per lane of 10%
gels, which was then electrophoretically transferred onto PVDF
membranes (Pierce; Thermo Fisher Scientific, Inc.). The membranes
were blocked with 5% non-fat milk in TBS and 0.1% Tween-20 at room
temperature for 1 h, and then incubated with endophilin 1 antibody
(cat. no. sc-10874; Santa Cruz Biotechnology, Inc.) at a dilution
of 1:1,000 overnight at 4°C. After 3–4 washes with TBS and 0.1%
Tween-20, the membranes were incubated with horseradish
peroxidase-conjugated Donkey anti Goat IgG (H + L) secondary
antibody (cat. no. AS031; ABclonal Biotech Co., Ltd.) at a dilution
of 1:1,000 for 2 h at room temperature. The protein bands were
detected after developing the blots using SuperSignal West Femto
Maximum Sensitivity Substrate (Pierce; Thermo Fisher Scientific,
Inc.). Western blot bands were quantified by densitometric analysis
using Image-Pro Plus 6.0 (Media Cybernetics, Inc.)
Hippocampal neuronal culture and
transfection
Rat hippocampal neurons were cultured as described
previously (24). A total of 50
postnatal SD rat pups (days 0 to 1) were purchased from the
Laboratory Animal Center of Sun Yat-Sen University and immediately
sacrificed upon arrival. Hippocampi were dissected from SD rat
pups, and dissociated hippocampal neurons were obtained using
0.125% trypsin at 37°C for 10 min and plated at a density of
1×104 cells/cm2 onto poly-D-lysine coated
glass coverslips. Cultures were maintained in Neurobasal A medium
(Invitrogen; Thermo Fisher Scientific, Inc.) containing 2% B27
(Invitrogen; Thermo Fisher Scientific, Inc.) and 0.5 mM glutamine
supplement (Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C in
a 5% CO2 humidified incubator, and half of the culture
media was replaced every 3 days. The neurons were cultured in
vitro in 24-well culture plates for 8–10 days prior to
transfection. To transfect neurons in 24-well tissue culture
plates, 100 pmol of Endo1 siRNA or its control was combined with 37
µl of 2 M CaCl2 solution in sterile, deionized water to
a final volume of 300 µl, and then mixed well with 300 µl of 2X
HEPES-buffered saline. The mixtures were vortexed and incubated at
room temperature for ~4 min. In each well, 30 µl mixture was added
drop-wise to the cells. Additionally, 1 µg/well GFP plasmid was
co-transfected with the siRNA to mark the transfected cells. The
calcium phosphate/DNA precipitate were maintained in the culture
for 40 min and washed with fresh transfection media twice. At 24–48
h after transfection, 1 µM Aβ1–42 oligomer was added to the culture
medium.
Fluorescence immunostaining
Three days after Endo1 siRNA transfection, the
hippocampal neurons were fixed with 4% paraformaldehyde at room
temperature for 1 h (Sigma-Aldrich; Merck KGaA). Immunostaining was
then performed using a standard protocol, as described previously
(26). The primary anti-endophilin
1 antibody (cat no. sc-10874; Santa Cruz Biotechnology, Inc.) was
used at a dilution of 1:100, and Alexa Fluor® 594
AffiniPure Donkey anti-Goat IgG (H + L; cat no. 705-585-147;
Jackson ImmunoResearch Laboratories, Inc.) was used at a dilution
of 1:500. After staining, the cells were mounted on glass slides
using Fluoro Gel II with DAPI (Electron Microscopy Sciences) and
imaged with a Carl Zeiss LSM 710 confocal microscope (Carl Zeiss
AG). Images were acquired with the same optical slice thickness in
every channel using a 63× oil objective and a resolution of
1,024×1,024 pixels. The RNA interference efficiency in hippocampal
neurons was determined by calculating the percentage of endophilin
1-positive cells, as previously described (30).
Determination of neuronal survival
rate
To assess the survival rate of hippocampal neuronal,
hippocampal neurons were exposed to 0, 1 and 50 µM Aβ. At 24 h
after exposure, the hippocampal neurons were fixed with 4%
paraformaldehyde at room temperature for 1 h (Sigma-Aldrich; Merck
KGaA) and nuclear staining with DAPI was performed as described
above. After staining, the cells were mounted on glass slides using
Fluoro Gel II without DAPI (Electron Microscopy Sciences) and
imaged with Nikon Eclipse Ti-S fluorescence microscope (Nikon
Corporation). The number of live cells (as assessed by DAPI
staining; small bright blue nuclei indicated dead cells) per
coverslip at the completion of the experiment was counted. As a
small number of glial cells are mixed in the cultured neurons, only
the death of neurons was counted by also viewing cells under bright
field microscopy. The neuronal survival rate was calculated as the
number of living cells divided the total number of neurons. The
experiment was performed five times with three fields analyzed per
view.
Electrophysiology
Whole-cell patch-clamp recordings of mEPSCs were
obtained from transfected cultured hippocampal neurons treated with
Aβ oligomers on 8–12 days in vitro (DIV). During the
recordings, the cells were bathed in an external solution (pH 7.3)
containing 128 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM
MgCl2, 15 mM glucose, 20 mM HEPES, 1 mM tetrodotoxin,
and 100 µM picrotoxin. Recording pipettes were filled with the
intracellular solution containing 147 mM KCl, 5 mM
Na2-phosphocreatine, 2 mM EGTA, 10 mM HEPES, 2 mM MgATP
and 0.3 mM Na2GTP. Recordings were performed at room
temperature in voltage clamp mode, at a holding potential of −70
mV, using a Multiclamp 700 B amplifier (Molecular Devices, LLC) and
Clampex 10.5 software (Axon Instruments; Molecular Devices, LLC).
The series resistance was <30 MΩ, and data were acquired at 10
kHz and filtered at 1 kHz. mEPSCs were analyzed using MiniAnalysis
software (Synaptosoft, Inc.), and the experiments were repeated at
least three times.
Statistical analysis
Data are presented as the mean ± standard error. The
statistical significance of the differences between two groups was
analyzed using Student's t-test and comparisons between more than
two groups were performed using one-way analysis of variance with
Newman-Keuls post hoc tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
Oligomeric Aβ causes synaptic
dysfunction in cultured hippocampal neurons
Data from several studies has demonstrated that Aβ
causes synaptic dysfunction. For example, overexpression of APP in
hippocampal neurons depresses excitatory transmission (13). In addition, when brain slices were
incubated with 1 µM Aβ1–42 oligomers, the frequency and amplitude
of mEPSCs were markedly reduced (13,16).
To determine whether oligomeric Aβ can cause synaptic dysfunction
in cultured hippocampal neurons, cultured DIV8 hippocampal neurons
were incubated with 1 µM Aβ1–42 oligomers or control solvent for 24
h, then mEPSCs were recorded using the whole-cell patch-clamp
technique. The neurons selected for electrophysiological
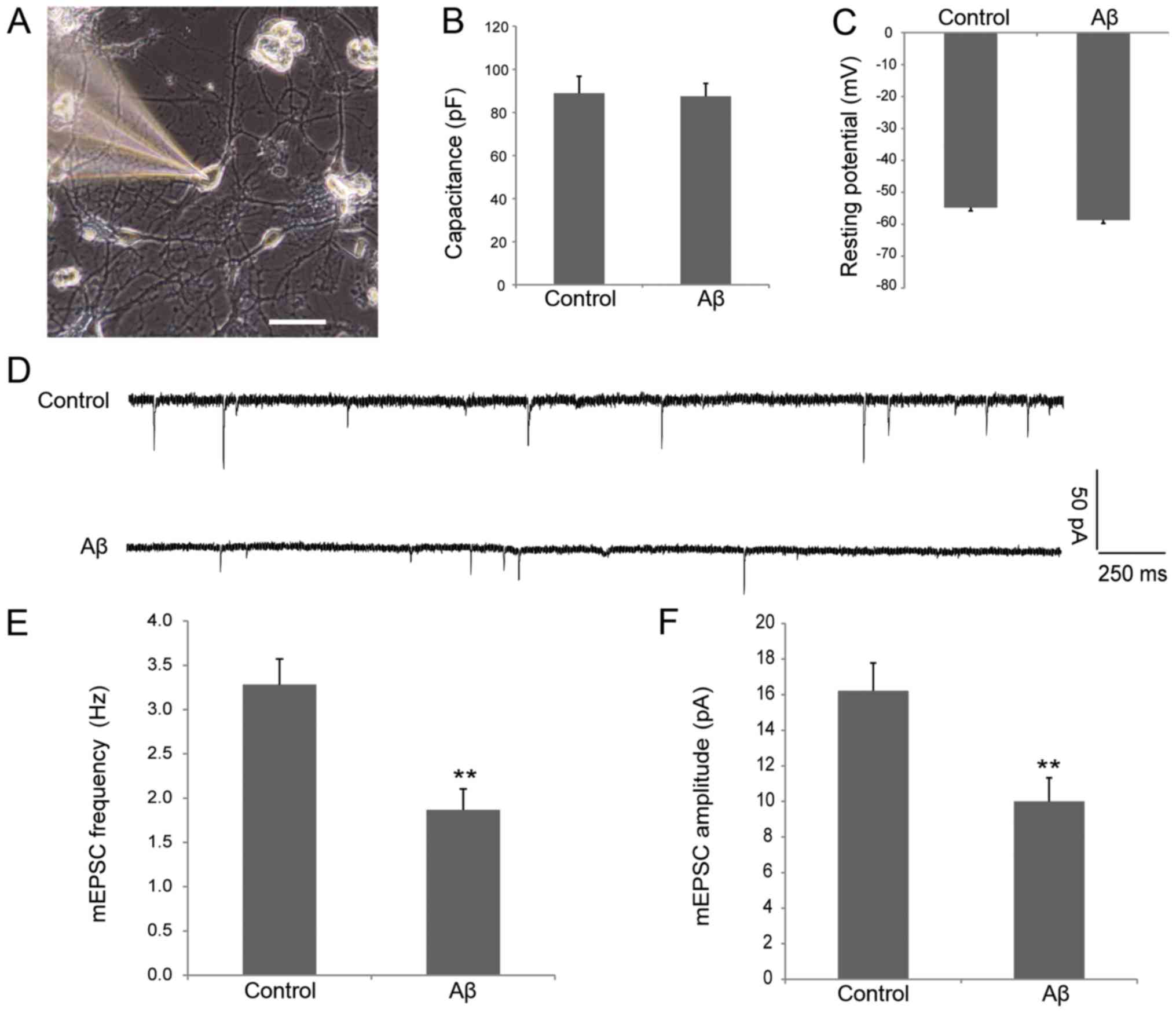
examination in the two groups are shown in Fig. 1A. The cells were healthy and
exhibited similar membrane capacitance (88.86±7.94 vs. 87.44±5.99
pF; Fig. 1B) and resting potential
(−54.8±2.06 vs. −58.7±1.83 mV; Fig.
1C) in control and Aβ groups, indicating that soluble Aβ does
not affect the intrinsic electrophysiological properties of
neurons. As shown in Fig. 1D-F,
the frequency (3.28±0.29 vs. 1.87±0.24 Hz; P<0.01; Fig. 1E) and amplitude (16.22±1.56 vs.
10.00±1.32 pA; P<0.01; Fig. 1F)
of the mEPSCs in neurons processed with oligomeric Aβ decreased
significantly when compared with the control group. These data
suggest that oligomeric Aβ can cause synaptic dysfunction in
cultured hippocampal neurons.
Endophilin 1 is highly expressed
during synaptic dysfunction induced by Aβ
It was previously demonstrated that endophilin 1 is
highly expressed in patients with AD patients and an AD mouse model
(26). To investigate whether the
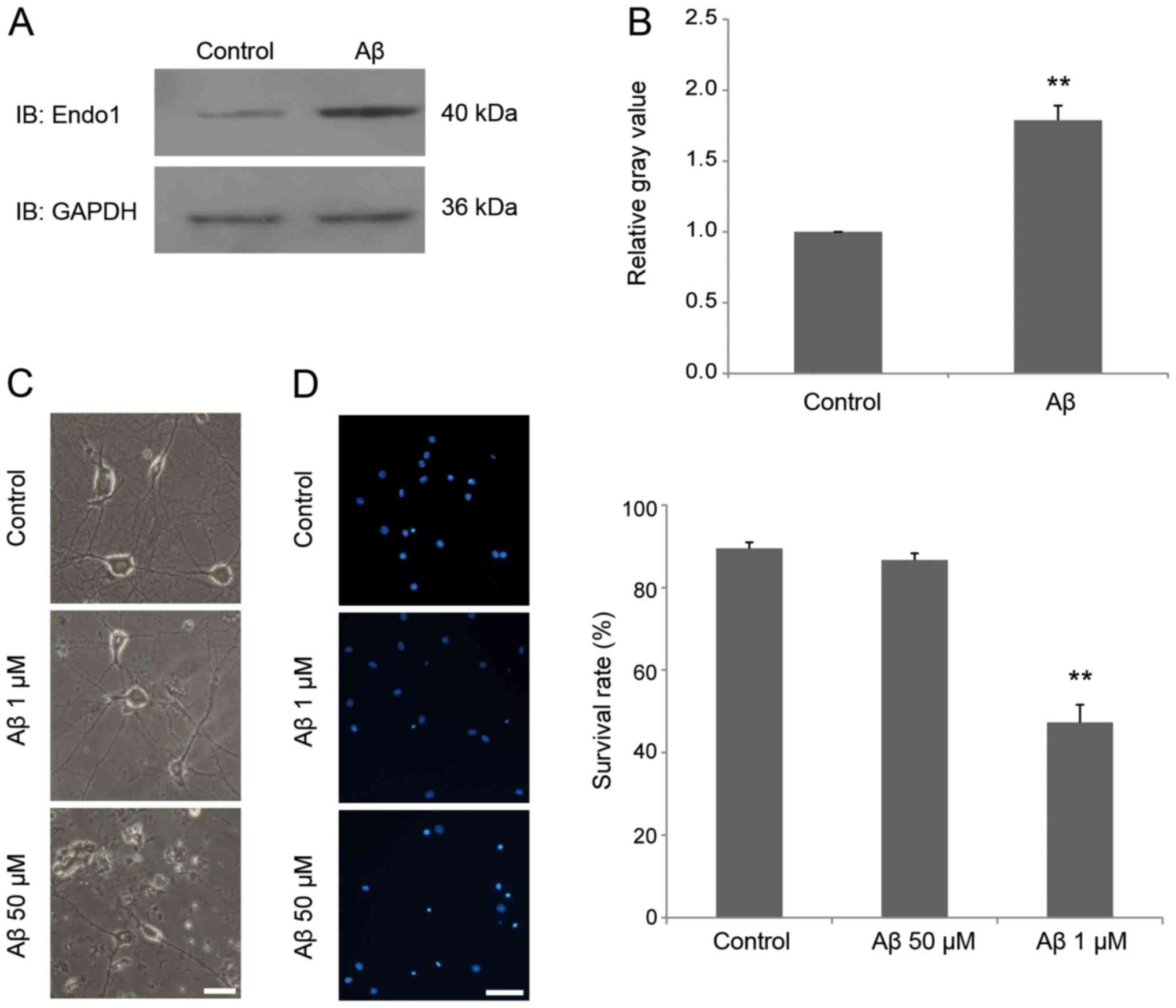
expression of endophilin 1 is also altered in Aβ-treated neurons,
cultured mature neurons were incubated in 1 µM Aβ1–42 for 24 h, and
then the expression of endophilin 1 was detected by western blot
analysis. The results demonstrated that the expression of
endophilin 1 in the Aβ-treated group was significantly higher than
in the control group (P<0.01; Fig.
2A and B). Furthermore, to assess whether 1 µM Aβ1–42 induced
the death of neurons, hippocampal neurons were treated with 0, 1
and 50 µM oligomeric Aβ1–42 for 24 h. As shown in Fig. 2C, neurons treated with 1 µM Aβ1–42
were healthy, as in the control group, whereas neurons treated with
50 µM Aβ1–42 exhibited broken cell bodies and collapsed
protrusions. DAPI staining further confirmed that there was no
neuronal death in the group treated with 1 µM Aβ1–42 for 24 h
(Fig. 2D). These results indicate
that oligomeric Aβ promotes endophilin 1 expression before it
causes neuronal death.
Interfering with the expression of
endophilin 1 attenuates the synaptic dysfunction caused by Aβ
To elucidate whether endophilin 1 is involved in
Aβ-induced synaptic dysfunction, RNA interference and
electrophysiological recording techniques were performed using
cultured hippocampal neurons.
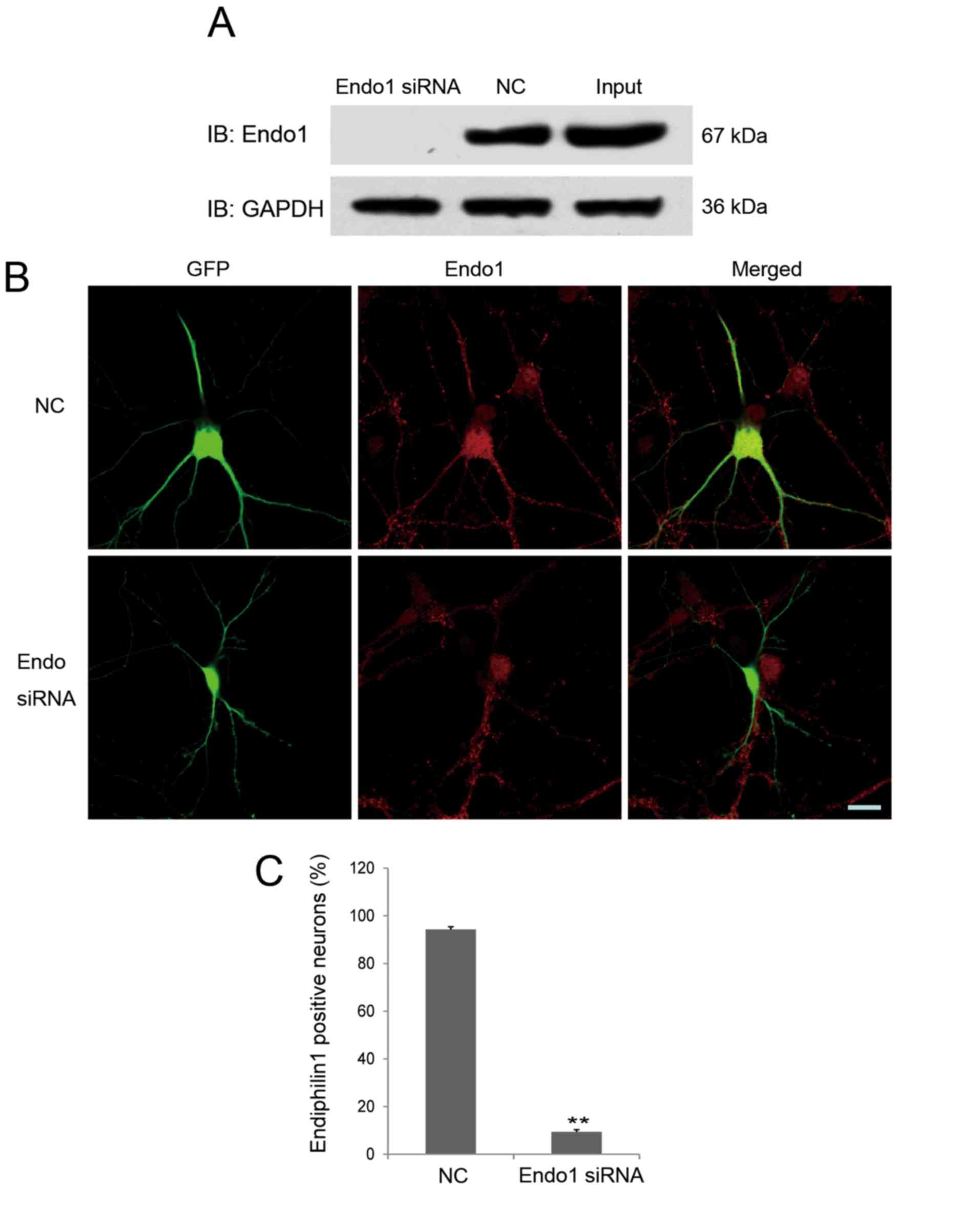
An siRNA targeting endophilin 1 was designed, and
its interference efficacy was determined by immunoblotting through
transfecting Endo1 siRNA or NC together with endophilin1-pEGFP-C1
plasmids into 293 cells. As shown in Fig. 3A, endophilin 1 expression was
barely detectable in 293 cells co-transfected with the Endo1 siRNA,
indicating that the interference fragment was effective.
The effectiveness of Endo1 siRNA in cultured
hippocampal neurons was also confirmed. As shown in Fig. 3B, compared with NC, the expression
of endogenous endophilin 1 was markedly knocked down in Endo1
siRNA-transfected neurons. As shown in Fig. 3B, endophilin 1 was expressed in
both hippocampal neurons transfected with NC (green and red
co-localized fluorescence) as well as untransfected neurons (red
fluorescence only), and red fluorescence was evenly distributed in
the cell body and processes of each neuron. However, invisible or
very weak red fluorescence was distributed in the cell body of
neurons transfected with Endo1 siRNA (identified by GFP
fluorescence). These results indicate that the expression of
endogenous endophilin 1 was reduced by the specific siRNA. To
further elucidate the effect of Endo1 siRNA on endogenous
endophilin 1, the experiment was repeated four times and the number
of endophilin 1-positive neurons in the NC and Endo1 siRNA groups
were counted; the result demonstrated that 9.47±0.91% neurons
transfected with Endo1 siRNA were positive for endophilin 1
staining, while 94.24±1.19% neurons transfected with NC were
positive for endophilin 1 staining. These results indicate that
Endo1 siRNA effectively reduced the expression of endogenous
endophilin 1 in cultured hippocampal neurons. This result was
consistent with previous validation results for subtype-specific
siRNAs targeting endophilin (30).
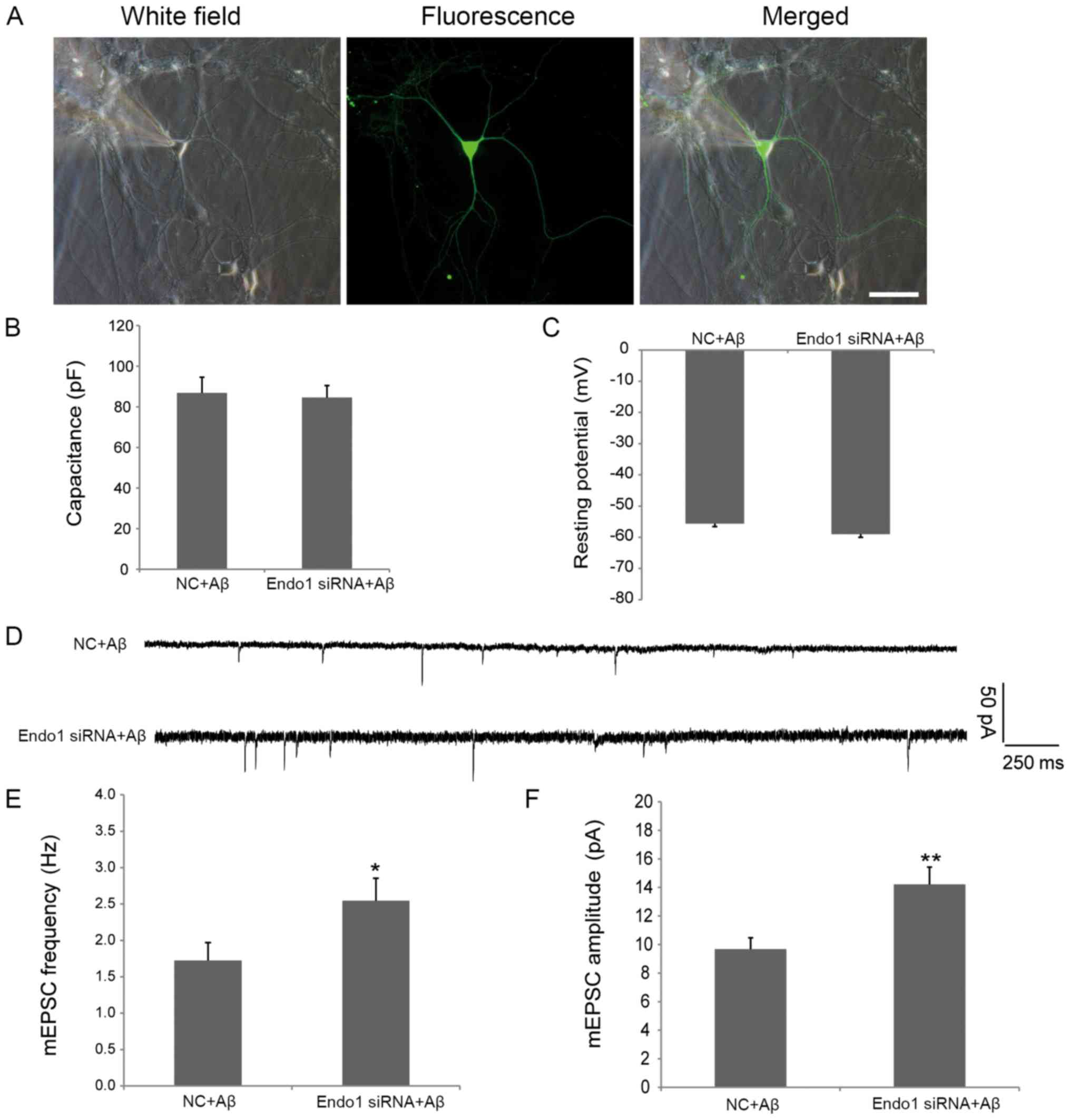
Finally, to explore the role of endophilin 1 in
Aβ-induced synaptic dysfunction, cultured DIV8 hippocampal neurons
were transfected with NC or Endo1 siRNAs for 48 h and incubated
with 1 µM Aβ1–42 oligomers for a further 24 h. Subsequently, the
mEPSCs of neurons transfected with NC and Endo1 siRNA were measured
using electrophysiological recording techniques. As shown in
Fig. 4A, healthy cells with the
same membrane capacitance and resting potential were selected to
determine the intrinsic electrophysiological properties of
transfected neurons (Fig. 4B and
C). As shown in Fig. 1, the
administration of oligomeric Aβ reduced both the frequency and
amplitude of mEPSCs. When endophilin 1 was silenced in cells
treated with Aβ, the frequency (2.55±0.31 vs. 1.72±0.25 Hz;
P<0.05; Fig. 4E) and amplitude
(14.22±1.21 vs. 9.68±0.79 pA; P<0.01; Fig. 4F) of mEPSCs were increased
significantly compared with the NC group. Of note, in the absence
of Aβ, there were no significant differences between the frequency
or amplitude of mEPSCs in NC- and Endo1 siRNA-transfected cells
(data not shown), indicating that the effects of knockdown of
endophilin1 on synaptic function were dependent on the presence of
Aβ. Therefore, the results suggested that knockdown of endophilin 1
expression can reduce oligomeric Aβ-induced synaptic
dysfunction.
Discussion
Accumulating evidence indicates that oligomeric Aβ
has an important role in the cognitive impairment of patients with
AD (3,4). During the early stages of AD, before
a large number of neurons are lost, oligomeric Aβ can induce
synaptic dysfunction, which is an important cause of cognitive
decline in patients with AD (6).
Therefore, using various methods to prevent synaptic damage and
rescuing damaged synapses may help prevent AD-related cognitive
impairment, and improve learning and memory ability.
Synaptic disorders include changes in synaptic
numbers, abnormal synaptic transmission and synaptic plasticity.
The present study demonstrated that 1 µM Aβ1–42 can cause synaptic
dysfunction in cultured hippocampal neurons, which is manifested by
a decrease in the frequency and amplitude of mEPSCs, which is
consistent with the results of a previous study on APP
overexpression in mouse hippocampal neurons (13). Alteration of mEPSCs is closely
associated with the inhibition of presynaptic vesicle transport and
postsynaptic glutamate receptor transport induced by Aβ (13). The effect of Aβ on postsynaptic
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type
glutamate receptors has a more important role (13). It has been demonstrated that Aβ can
inhibit the induction and maintenance of LTP by directly or
indirectly interfering with the insertion and localization of AMPA
receptors in the postsynaptic membrane (17,31).
Furthermore, Aβ can enhance LTD by promoting endocytosis and
enzymatic hydrolysis of AMPA receptors (18,32).
The frequency and amplitude of mEPSCs mediated by AMPA receptors in
hippocampal sections was reported to be markedly attenuated
following perfusion with 1 µM Aβ1–42 (12).
Endophilin is an important contributor to
clathrin-dependent endocytosis, and includes three subtypes,
endophilins 1–3 (20,21). Among the three endophilin subtypes,
which exhibit high homology, endophilin 1 is specifically expressed
in the central nervous system and distributed in neuronal cell
bodies and at synaptic sites (21,25).
Previous studies have demonstrated that endophilin 1 has an
important role in the regulation of synaptic vesicle endocytosis
(30,33). Knockdown or mutation of endophilin
1 inhibits neuronal synaptic vesicle endocytosis, resulting in
synaptic transmission disorders (30,33).
In recent years, Ren et al (26) reported that endophilin 1 is highly
expressed in patients with AD and AD transgenic mice. The increase
in endophilin 1 levels in neurons is associated with an increase in
the activation of the stress kinase JNK, with subsequent neuronal
death (26). The present study
demonstrated that endophilin 1 is highly expressed when synaptic
dysfunction occurs. We hypothesize that synaptic dysfunction occurs
earlier than neuronal death, because endophilin 1 is mainly
distributed at the synaptic site (25). The initial increase in endophilin 1
caused by Aβ may induce synaptic transmission disorder via
regulation of postsynaptic receptors. With the increased
neurotoxicity, the increasing endophilin 1 level will alter JNK
activation, with the subsequent death of neurons. However, the more
detailed mechanisms via which endophilin 1 causes synaptic
inhibition and neuronal death require further research.
Endophilin has been reported to have an important
role in endocytosis of postsynaptic AMPA receptor (25). AMPA receptor endocytosis is mainly
achieved via a clathrin-dependent pathway (34). Two subtypes of endophilin A
(endophilin 2 and endophilin 3) have been demonstrated to be
involved in AMPA receptor endocytosis mediated by the immediate
early gene activity regulated cytoskeleton associated protein
(Arc/Arg3.1). Endocytosis proteins, such as endophilin 2/3 and
dynamin, form complexes with Arc/Arg3.1 to mediate AMPA receptor
endocytosis (25). However, none
of these proteins were shown to have a direct interaction with AMPA
receptor (25). It was
subsequently demonstrated that endophilin 2 interacts with
glutamate ionotropic receptor AMPA type subunit 1, which is one of
the subunits of the AMPA receptor, to mediate AMPA receptor
endocytosis (35). In addition,
endophilin 1 and endophilin 2 are predominantly found as stable
dimers interacting via a coiled-coil domain in their conserved
NH2-terminal moiety (36), and they have similar effects on
calcium-dependent interactions with other proteins and synaptic
vesicle endocytosis (30).
Therefore, it was hypothesized that endophilin 1, which has a
similar function to endophilin 2, may be involved in Aβ-mediated
AMPA receptor endocytosis. Endophilin B also has been reported be
associated with AD; however, unlike endophilin 1, loss of the
endophilin B1 subunit exacerbates AD pathology. In mouse primary
cortical neuron cultures, overexpression of the neuron-specific
endophilin B1 isoform protected against Aβ-induced apoptosis and
mitochondrial dysfunction (37).
These studies suggest that isoform specificity results in different
roles within the mechanism of AD. The current study demonstrated
the effect of endophilin 1 knockdown on the frequency and amplitude
of mEPSCs in cultured neurons incubated with Aβ, and concluded that
knockdown of endophilin 1 prevents synaptic dysfunction induced by
Aβ. However, further research is required to determine whether this
process is due to inhibition of AMPA receptor endocytosis.
In summary, the present study revealed that
silencing of endophilin 1 expression alleviates oligomeric
Aβ-mediated synaptic dysfunction. These findings may provide
experimental evidence for identifying targets for AD prevention and
treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 31300885,
81571191 and 81771144), the Natural Science Foundation of Guangdong
Province, China (grant no. 2017B030311002), the Science and
Technology Planning Project of Guangdong Province (grant no.
2013B021800036), the Medical Research Foundation of Guangdong
Province, China (grant no. A2017154) and the Guangzhou Institute of
Pediatrics/Guangzhou Women and Children's Medical Center (grant no.
IP-2018-010).
Availability of data and materials
The datasets generated and analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
YY, CC, JZ and GG conceived and designed the study.
YY and CC performed the experiments on hippocampal neuronal
culture, whole-cell patch-clamp recordings and analyzed the data.
FW, JL, SL and XZ performed the western blotting, fluorescence
immunostaining, 293 cell culture and transfection. JZ drafted the
manuscript. GG reviewed and edited the manuscript. All the authors
have read and approved the final version of the manuscript.
Ethics approval and consent to
participate
All animal procedures were performed in strict
accordance with the recommendations in the Guide for the Care and
Use of Laboratory Animals (38).
The protocol was approved by the Institutional Animal Care and Use
Committee at Jinan University. All efforts were made to minimize
suffering and the number of animals used.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
delEtoile J and Adeli H: Graph theory and
brain connectivity in Alzheimer's disease. Neuroscientist.
23:616–626. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lane CA, Hardy J and Schott JM:
Alzheimer's disease. Eur J Neurol. 25:59–70. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Luo J, Wärmländer SK, Graslund A and
Abrahams JP: Cross-interactions between the alzheimer disease
amyloid-β peptide and other amyloid proteins: A further aspect of
the amyloid cascade hypothesis. J Biol Chem. 291:16485–16493. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jamasbi E, Wade JD, Separovic F and
Hossain MA: Amyloid Beta (Aβ) Peptide and factors that play
important roles in Alzheimer's disease. Curr Med Chem. 23:884–892.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pini L, Pievani M, Bocchetta M, Altomare
D, Bosco P, Cavedo E, Galluzzi S, Marizzoni M and Frisoni GB: Brain
atrophy in Alzheimer's disease and aging. Ageing Res Rev. 30:25–48.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Holtzman DM, Morris JC and Goate AM:
Alzheimer's disease: The challenge of the second century. Sci
Transl Med. 3:77sr12011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hong S, Beja-Glasser VF, Nfonoyim BM,
Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A,
Barres BA, et al: Complement and microglia mediate early synapse
loss in Alzheimer mouse models. Science. 352:712–716. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mucke L and Selkoe DJ: Neurotoxicity of
amyloid β-protein: Synaptic and network dysfunction. Cold Spring
Harb Perspect Med. 2:a0063382012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tu S, Okamoto S, Lipton SA and Xu H:
Oligomeric Aβ-induced synaptic dysfunction in Alzheimer's disease.
Mol Neurodegener. 9:482014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Selkoe DJ: Soluble oligomers of the
amyloid beta-protein impair synaptic plasticity and behavior. Behav
Brain Res. 192:106–113. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Laurén J, Gimbel DA, Nygaard HB, Gilbert
JW and Strittmatter SM: Cellular prion protein mediates impairment
of synaptic plasticity by amyloid-beta oligomers. Nature.
457:1128–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Parameshwaran K, Sims C, Kanju P,
Vaithianathan T, Shonesy BC, Dhanasekaran M, Bahr BA and
Suppiramaniam V: Amyloid beta-peptide Abeta(1–42) but not
Abeta(1–40) attenuates synaptic AMPA receptor function. Synapse.
61:367–374. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ting JT, Kelley BG, Lambert TJ, Cook DG
and Sullivan JM: Amyloid precursor protein overexpression depresses
excitatory transmission through both presynaptic and postsynaptic
mechanisms. Proc Natl Acad Sci USA. 104:353–358. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Benilova I, Karran E and De Strooper B:
The toxic Aβ oligomer and Alzheimer's disease: An emperor in need
of clothes. Nat Neurosci. 15:349–357. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zeng Y, Zhao D and Xie CW: Neurotrophins
enhance CaMKII activity and rescue amyloid-β-induced deficits in
hippocampal synaptic plasticity. J Alzheimers Dis. 21:823–831.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schmid AW, Freir DB and Herron CE:
Inhibition of LTP in vivo by beta-amyloid peptide in different
conformational states. Brain Res. 1197:135–142. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Walsh DM, Klyubin I, Fadeeva JV, Cullen
WK, Anwyl R, Wolfe MS, Rowan MJ and Selkoe DJ: Naturally secreted
oligomers of amyloid beta protein potently inhibit hippocampal
long-term potentiation in vivo. Nature. 416:535–539. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li S, Hong S, Shepardson NE, Walsh DM,
Shankar GM and Selkoe D: Soluble oligomers of amyloid beta protein
facilitate hippocampal long-term depression by disrupting neuronal
glutamate uptake. Neuron. 62:788–801. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Walsh DM, Townsend M, Podlisny MB, Shankar
GM, Fadeeva JV, El Agnaf O, Hartley DM and Selkoe DJ: Certain
inhibitors of synthetic amyloid beta-peptide (Abeta)
fibrillogenesis block oligomerization of natural A beta and thereby
rescue long-term potentiation. J Neurosci. 25:2455–2462. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ringstad N, Nemoto Y and De Camilli P: The
SH3p4/Sh3p8/SH3p13 protein family: Binding partners for
synaptojanin and dynamin via a Grb2-like Src homology 3 domain.
Proc Natl Acad Sci USA. 94:8569–8574. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Giachino C, Lantelme E, Lanzetti L,
Saccone S, Bella Valle G and Migone N: A novel SH3-containing human
gene family preferentially expressed in the central nervous system.
Genomics. 41:427–434. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gad H, Ringstad N, Löw P, Kjaerulff O,
Gustafsson J, Wenk M, Di Paolo G, Nemoto Y, Crun J, Ellisman MH, et
al: Fission and uncoating of synaptic clathrin-coated vesicles are
perturbed by disruption of interactions with the SH3 domain of
endophilin. Neuron. 27:301–312. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huttner WB and Schmidt A: Lipids, lipid
modification and lipid-protein interaction in membrane budding and
fission-insights from the roles of endophilin A1 and synaptophysin
in synaptic vesicle endocytosis. Curr Opin Neurobiol. 10:543–551.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Milosevic I, Giovedi S, Lou X, Raimondi A,
Collesi C, Shen H, Paradise S, O'Toole E, Ferguson S, Cremona O and
De Camilli P: Recruitment of endophilin to clathrin-coated pit
necks is required for efficient vesicle uncoating after fission.
Neuron. 72:587–601. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chowdhury S, Shepherd JD, Okuno H, Lyford
G, Petralia RS, Plath N, Kuhl D, Huganir RL and Worley PF:
Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA
receptor trafficking. Neuron. 52:445–459. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ren Y, Xu HW, Davey F, Taylor M, Aiton J,
Coote P, Fang F, Yao J, Chen D, Chen JX, et al: Endophilin I
expression is increased in the brains of Alzheimer disease
patients. J Biol Chem. 283:5685–5691. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang J, Fan J, Tian Q, Song Z, Zhang J
and Chen Y: Characterization of two distinct modes of endophilin in
clathrin-mediated endocytosis. Cell Signal. 24:2043–2050. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tian Q, Zhang J, Fan J, Song Z and Chen Y:
Endophilin isoforms have distinct characteristics in interactions
with N-type Ca2+ channels and dynamin I. Neurosci Bull. 28:483–492.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Doherty GH, Beccano-Kelly D, Yan SD,
Gunn-Moore FJ and Harvey J: Leptin prevents hippocampal synaptic
disruption and neuronal cell death induced by amyloid β. Neurobiol
Aging. 34:226–237. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang J, Tan M, Yin Y, Ren B, Jiang N, Guo
G and Chen Y: Distinct functions of endophilin isoforms in synaptic
vesicle endocytosis. Neural Plast. 2015:3714962015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cleary JP, Walsh DM, Hofmeister JJ,
Shankar GM, Kuskowski MA, Selkoe DJ and Ashe KH: Natural oligomers
of the amyloid-beta protein specifically disrupt cognitive
function. Nat Neurosci. 8:79–84. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hsieh H, Boehm J, Sato C, Iwatsubo T,
Tomita T, Sisodia S and Malinow R: AMPAR removal underlies
Abeta-induced synaptic depression and dendritic spine loss. Neuron.
52:831–843. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Verstreken P, Kjaerulff O, Lloyd TE,
Atkinson R, Zhou Y, Meinertzhagen IA and Bellen HJ: Endophilin
mutations block clathrin-mediated endocytosis but not
neurotransmitter release. Cell. 109:101–112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Anggono V and Huganir RL: Regulation of
AMPA receptor trafficking and synaptic plasticity. Curr Opin
Neurobiol. 22:461–469. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang J, Yin Y, Ji Z, Cai Z, Zhao B, Li J,
Tan M and Guo G: Endophilin2 Interacts with GluA1 to Mediate AMPA
receptor endocytosis induced by oligomeric amyloid-β. Neural Plast.
2017:81970852017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ringstad N, Nemoto Y and De Camilli P:
Differential expression of endophilin 1 and 2 dimers at central
nervous system synapses. J Biol Chem. 276:40424–40430. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang DB, Kinoshita Y, Kinoshita C, Uo T,
Sopher BL, Cudaback E, Keene CD, Bilousova T, Gylys K, Case A, et
al: Loss of endophilin-B1 exacerbates Alzheimer's disease
pathology. Brain. 138:2005–2019. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Care NRCU, Animals AUOL. Guide for the
care use of laboratory animals. Washington (DC): National Academies
Press (US); 2011
|