Introduction
Cardiac fibrosis is a common feature of cardiac
remodeling that occurs in numerous types of cardiovascular
diseases, including hypertension, myocardial infarction and
cardiomyopathy (1). This condition
is characterized by the inappropriate interstitial deposition of
collagen and extracellular matrix (ECM) proteins, which impair
normal organ structure and function (2,3).
Cardiac fibroblasts (CFs) are the predominant cell type responsible
for physiological ECM homeostasis and tissue healing following
cardiac injury (4). During the
process of fibrosis, CFs transdifferentiate into myofibroblasts,
which are characterized by the expression of α-smooth muscle actin
(α-SMA), a contractile protein and marker of profibrogenic cardiac
fibroblast activation (5).
The renin-angiotensin system (RAS) is involved in
various aspects of pathological cardiac remodeling, including
cardiac hypertrophy and fibrosis (6,7).
Angiotensin II (Ang II), the key member of RAS, has been
demonstrated to promote myocardial remodeling and fibrosis via
interactions with the Ang II type 1 (AT1) receptor (8,9).
This process is distinguished by fibroblast cell proliferation and
fibrogenic gene expression (10,11).
Inhibition of the RAS with angiotensin-converting enzyme (ACE)
inhibitors or AT1 receptor blockers promotes anti-hypertrophic and
anti-fibrotic effects beyond reductions in blood pressure (BP)
(12). Importantly, knowledge of
the RAS and its inhibitors has substantially increased in recent
decades; however, numerous biological effects of these drugs cannot
be explained by the well-established mechanisms of action. Thus,
current understanding of the RAS is far from complete.
Alamandine (Ala) is a recently identified component
of the RAS. This heptapeptide differs from Ang (1–7) in
that it contains an N-terminal alanine, rather than an aspartate
residue (13). Functional studies
have revealed that Ala can induce endothelial-dependent
vasorelaxation, similar to the effect of Ang (1–7)
(10,11). On the contrary, Ala can counteract
the vasoconstriction induced by its precursor, angiotensin A (Ang
A), without affecting Ang II-induced vasoconstriction (14). Furthermore, it has been reported
that the vasodilatory effects of Ala are context dependent.
Specifically, in the thoracic aorta and iliac artery, Ala promotes
acetylcholine-mediated vasodilation; however, in the carotid
artery, Ala does not affect the levels of acetylcholine or vascular
tone (14). In the renal artery,
it suppresses acetylcholine-induced vasodilation (14). In a isoproterenol-treated rat model
of cardiac hypertrophy, the Mas-related G protein-coupled receptor
D (MrgD) has been reported to function as a major target for Ala
(13). Additionally, the role of
Ala in cardiac fibrosis induced by hypertension and RAS
overactivation remains unknown. In the present study, the potential
therapeutic effects of Ala on fibrosis in a spontaneous
hypertensive rat (SHR) model were investigated. We reported that
Ala significantly attenuated cardiac fibrosis in aged SHR rats and
that this effect may be independent of reductions in BP.
Materials and methods
Animals and grouping
Male rats (n=24; 50-weeks-old, mean weight, 380 g;
Beijing Vital River Laboratory Animal Technology Co., Ltd.,
Beijing, China), including six normotensive Wistar-Kyoto (WKY) and
18 SHRs, were housed in a temperature-controlled room (23°C; 40–60%
humidity; 21% oxygen) under a 12 h light/dark cycle, with free
access to standard chow and tap water. Three groups of SHRs (n=6
per group) were subjected to 6-weeks of intraperitoneal
administration of 50 µg/kg/day Ala (Phoenix Pharmaceuticals Inc.,
Burlingame, CA, USA), 10 mg/kg/day hydralazine (Hyd) or the
equivalent volume of saline. Following the completion of treatment,
all rats were examined via two-dimensional echocardiography,
weighed and sacrificed. Isolated hearts were dried and weighed, and
a portion of the left heart tissue was sectioned for staining. The
remaining tissue was used for western blotting and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
Measurement of tail artery BP
Systolic BP, diastolic BP and the mean arterial
pressure were measured every week for 6 weeks in the tails of
conscious rats, using a noninvasive computerized tail-cuff system
(Kent Scientific Corporation, Torrington, CT, USA). To minimize
stress-induced fluctuations, the rats were pre-trained by measuring
BP daily for at least 1 week prior to the start of the experiments,
and the total training duration was 2 weeks. The rats were
maintained under 28°C for 10–20 min prior to analysis to allow the
detection of tail artery pulsations and to achieve a steady pulse.
The reported tail artery BP data were obtained by averaging 10
measurements.
Echocardiography
After the 6-week course of drug or saline
administration, rats were anesthetized with continuous
administration of 2% isoflurane in 2 l/min of 100% oxygen via a
facemask. Then, transthoracic echocardiography was performed in all
rats using an ultrasound system (Vevo 2100, VisualSonics, Inc.,
Toronto, Canada), with a 21-MHz probe. LV diameter were assessed by
the measurement of the left ventricle (LV) mass and LV volumes in
diastole and systole, and the LV mass to body weight ratio. LV
ejection fraction (EF) and fractional shortening (FS) were
calculated to evaluate LV function. Measurements over three
consecutive cardiac cycles were averaged.
Masson and Sirius red staining
To assess the degree of fibrosis of cardiomyocytes,
5-µm cardiac sections were examined by Masson kit (cat. no. GP1016)
and Sirius red kit (cat. no. GP1017; Service Biological Technology
Co., Ltd, Wuhan, China, http://www.servicebio.cn/), according to the
manufacturer's protocols. Briefly, the tissues were fixed with 4%
paraformaldehyde at 4°C overnight, paraffin embedded and cut into
sections (5 µm thick). For trichrome Masson staining, sections were
de-paraffinized, rehydrated and rinsed with potassium dichromate
mordant solution overnight. Next, sections were sequentially
stained with iron hematoxylin for 3 min, acid ponceau fuchsin for
3–5 min, phosphomolybdic acid for 1–3 min, aniline blue for 3–6 min
and differentiated in 1% glacial acetic acid solution. The slides
were mounted with resin mounting medium. In addition, slides were
stained with Sirius red solution for 8–10 mins and subsequently
sealed with glycerol jelly. Three to five random fields (~30–50
cells per field) were selected from the three sections obtained
from each animal for observation under a light microscope (Zeiss
GmbH, Jena, Germany). Images were analyzed using Image-Pro Plus
software version 6.0 (Media Cybernetics, Inc., Rockville, MD,
USA).
Culture of cardiac fibroblasts
isolated from adult rats
Adult cardiac fibroblasts (CFs) were obtained from
male WKY rats. Ventricular tissue was dissected, washed, minced and
subjected to an initial digestion step in bacterial proteinase
solution (4 U/ml) at room temperature. for 15 min; seven rounds of
digestion were then performed at 37°C for 20 min in a solution
containing a mixture of 1 mg/ml collagenase A and 0.5 mg/ml
hyaluronidase. Following each cycle of digestion, the tissue was
mechanically dissociated using a wide-mouth pipet; the supernatant
containing dissociated cells was collected. Cells were resuspended
in Iscove's Modified Dulbecco's medium (IMDM; Sigma-Aldrich, Merck
KGaA, Darmstadt, Germany). Cells from all digestions were pooled
and resuspended in IMDM, supplemented with 20% fetal calf serum
(ScienCell Research Laboratories, Inc., San Diego, CA, USA),
penicillin (100 U/ml), streptomycin (100 µg/ml), nonessential amino
acids (1%), and 2-mercaptoethanol (0.1 mM). Cells (105)
were first plated and incubated for 2 h at 37°C to allow the
preferential attachment of fibroblasts. To confirm that the cells
were CFs, and exclude endothelial cells and cardiomyocytes from the
mixture, pure CF identification was performed by immunostaining
with anti-vimentin (1:1,000; cat. no. ab92547), anti-CD31 (1:500;
cat. no. ab28364; both Abcam, Cambridge, MA, USA) and anti-α-actin
(1:1,000; cat. no. A5228; Sigma-Aldrich; Merck KGaA) antibodies at
4°C overnight. Next, cells were incubated with an
Alexa-488-conjugated (1:2,000; cat. no. 111-545-003), or
Cy™3-conjugated (1:2,000; cat. no. 111-165-003; both Jackson
ImmunoResearch Laboratories, Inc., West Grove, PA, USA) secondary
antibodies, at room temperature for 1 h. CFs from passages 3–8 were
used for all experiments.
CFs were divided into groups and treated with PBS,
PBS + Ang II, Ala, or Ala + Ang II. Pre-treatment with Ala (1 µM)
or PBS control was performed for 30 min at 37°C, after which the
PBS + Ang II and Ala + Ang II groups were treated at 37°C with
10−6 M Ang II (Sigma-Aldrich; Merck KGaA) for 24 h. For
inhibition experiments, CFs were divided into four groups and
treated with Ala (control), Ala + Ang II, Ala + PD123319 (10 µM;
cat. no. s7098; Selleck Chemicals, Houston, TX, USA), or Ala + Ang
II + PD123319. CF cell lysates were used for western blotting and
RT-qPCR.
Western blotting
Heart tissues or cultured cells were homogenized in
radioimmunoprecipitation assay lysis buffer (Thermo Fisher
Scientific, Inc.) and sonicated. Cell debris was removed, and the
supernatant was obtained by centrifugation at 12,000 × g for 10 min
at 4°C. Protein concentration was determined by a bicinchoninic
acid protein assay, and a total of 30–50 µg protein was separated
on 10–15% SDS-PAGE and transferred onto a polyvinylidene difluoride
membrane (EMD Millipore, Billerica, MA, USA). Following blocking
with 5% fat-free milk solution, the proteins of interest were
detected by using primary antibodies (4°C overnight) and a
corresponding horseradish peroxidase-linked secondary antibody at
room temperature for 1 h. The targets of the primary antibodies
used were as follows: Connective tissue growth factor (CTGF, 1:500;
cat. no. ab6992; Abcam); collagen I (COL-1; 1:500; Wanleibio Co.,
Ltd., Shanghai, China), matrix metalloproteinase 9 (MMP9; 1:1,000;
cat. no. 13667), protein kinase B (Akt; 1:1,000; cat. no. 469) and
phosphorylated (p)-Akt [(Ser 473) 1:1,000; all cat. no. 40601]; an
antibody against GAPDH (1:1,000; cat. no. 5174; all Cell Signaling
Technology, Inc., Danvers, MA, USA) was used as an internal
control. The blots were developed with enhanced chemiluminescence
reagent (Thermo Fisher Scientific, Inc.) and exposed on a ChemiDoc
MP imager (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Experiments were performed in triplicate for each indicated
protein. Images were analyzed using Image-Pro Plus software version
6.0.
RT-qPCR
Total RNA was isolated from CFs using TRIzol
(Invitrogen; thermos Fisher Scientific, Inc., Waltham, MA, USA) and
1 µg of the RNA was then converted to cDNA using Transcriptor First
Strand cDNA Synthesis kit (Roche Diagnostics, Basel, Switzerland)
according to the manufacturer's protocols. qPCR reactions were
performed on an ABI Prism 7900 system (Applied Biosystems; Thermo
Fisher Scientific, Inc.) using TaqMan™ probe qPCR kit (Roche
Diagnostics). The thermocycling conditions were as follows: 95°C
for 10 min, followed by 45 cycles of 95°C for 10 sec and 60°C for 1
min. Taqman probes to detect COL1A1, collagen type III α 1
chain (COL3A1), α-SMA and CTGF were purchased
from Roche Diagnostics. Each sample was analyzed in triplicate and
target genes were normalized to the reference housekeeping gene
GAPDH. Fold differences were then calculated for each treatment
group using normalized Cq values for the control, using the
2−∆∆Cq method (15).
Probes (5′-3′) used in the present study were: a-SMA
forward, CCCAGCACCATGAAGATCA and reverse, CGCCGATCCAGACAGAAT;
CTGF forward, GCTGACCTAGAGGAAAACATTAAGA and reverse,
CCGGTAGGTCTTCACACTGG; Col1a1 forward, TCCTGGCAAGAACGGAGAT
and reverse, CAGGAGGTCCACGCTCAC; Col3a1 forward,
TCCCCTGGAATCTGTGAATC and reverse, TGAGTCGAATTGGGGAGAAT.
GAPDH forward, GGCACAGTCAAGGCTGAGAATG and reverse,
ATGGTGGTGAAGACGCCAGTA.
Statistical analyses
All data are presented as mean ± standard error of
the mean of at least three independent experiments. Statistical
significance among multiple groups was evaluated by one-way
analysis of variance followed by a Bonferroni's post-hoc test,
using GraphPad Prism 5.0 (GraphPad Software Inc., CA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Aged SHRs were treated daily with Ala, Hyd, or
saline as a control, for six weeks and BP was measured weekly in
all animals. The systolic, diastolic and mean arterial BPs of Ala-
and Hyd-treated animals were significantly lower than those of the
saline-treated group (Fig. 1A-C).
Ala administration also resulted in a significantly decreased ratio
of LV mass to body weight compared with the control and Hyd-treated
SHRs (Fig. 1D). Assessment of LV
diameter and function by transthoracic echocardiography further
revealed a significantly decreased LV volume in the systolic and
diastolic phase compared with the control (Fig. 2A and B). On the contrary, Ala and
Hyd treatment did not affect LV diameter along the long axis view
(Fig. 2C and D). An improved EF
and FS in Ala-treated SHRs were observed compared with the control
and Hyd-treated SHRs (Fig. 2E and
F).
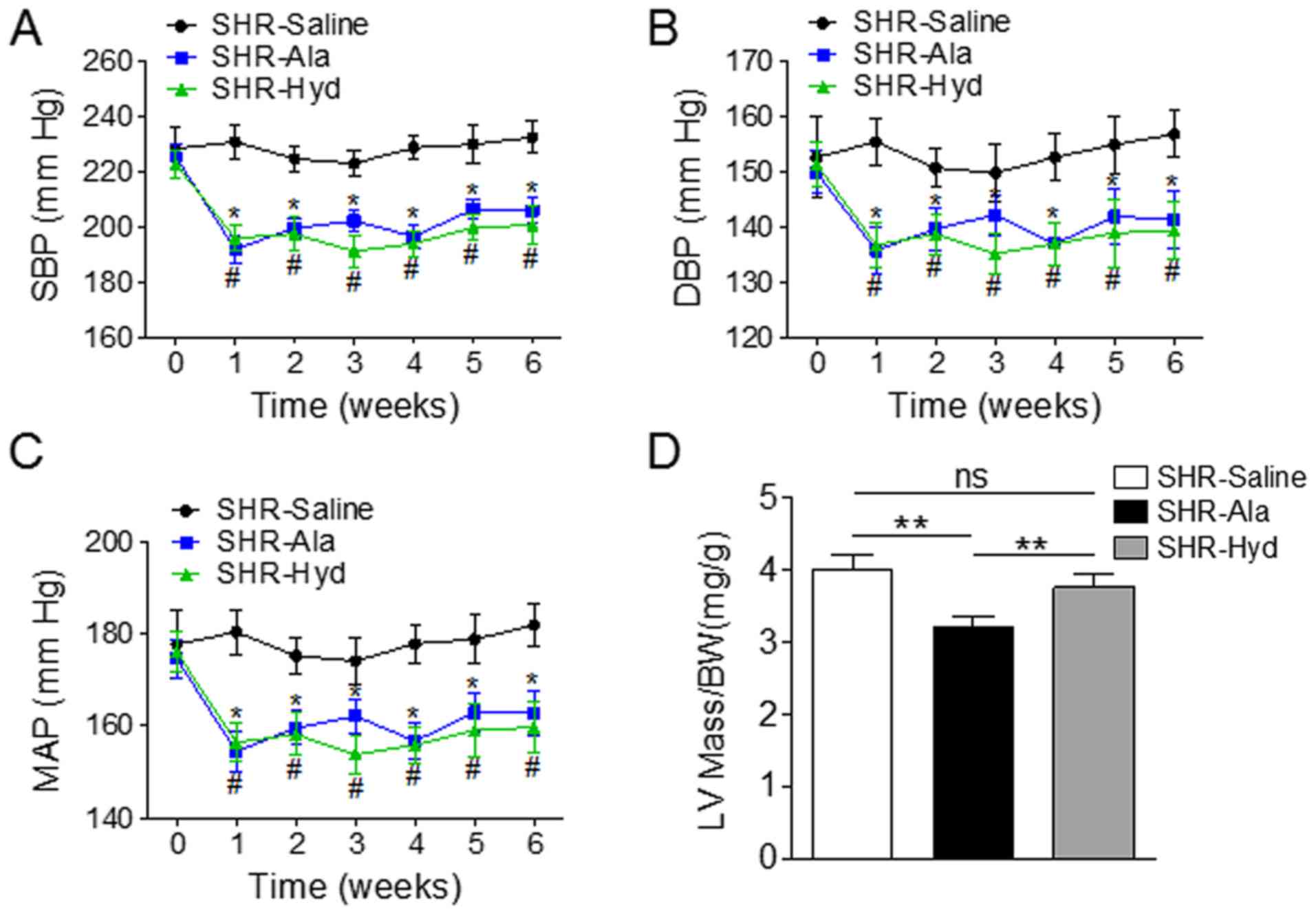 | Figure 1.Ala treatment decreases BP and LV mass
in a SHR model. SHRs were treated daily with Ala, Hyd or saline for
6 weeks, and tail arterial BP was measured weekly. (A-C)
Measurements of SBP, DBP and MAP; n=6 per group. *P<0.05 vs.
SHR-Ala, #P<0.05 vs. SHR-Hyd. (D) LV mass was
measured by echocardiography; the ratio of LV mass to BW was
compared among the three groups. **P<0.01. Data are presented as
the mean ± standard error of the mean. Ala, alamandine; BW, body
weight; DBP, diastolic blood pressure; Hyd, hydralazine; LV, left
ventricle; MAP, mean arterial blood pressure; ns, no significance;
SHR, spontaneous hypertensive rat; SBP, systolic blood
pressure. |
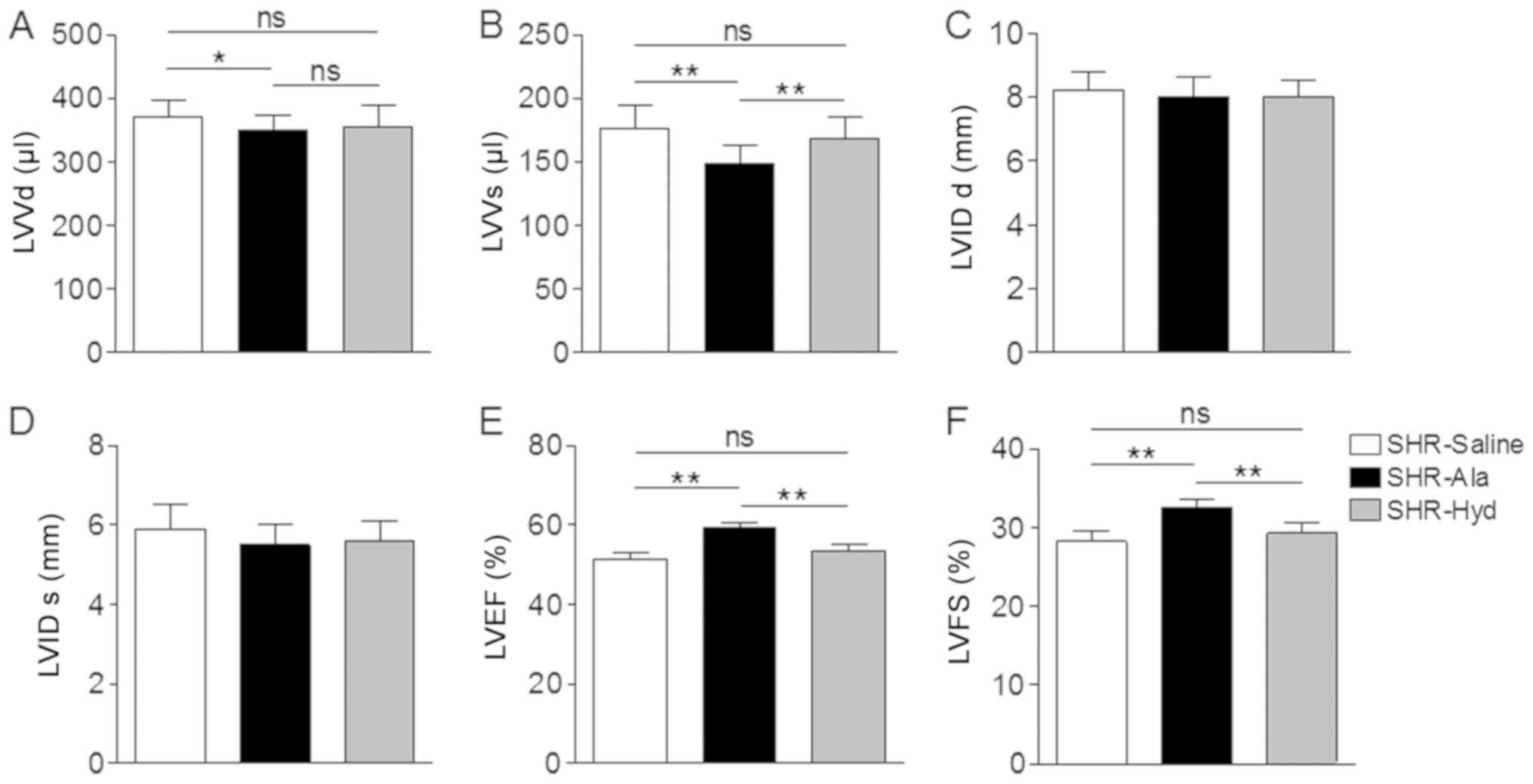 | Figure 2.Ala treatment attenuates cardiac
dysfunction in SHRs. (A and B) LV volume at diastolic phase and
systolic phase were measured by echocardiography. (C and D) LV
septal diameters at diastolic phase and systolic phase were
measured by echocardiography. (E and F) LVEF and LVFS were measured
by echocardiography; n=6 per group. Data are presented as the mean
± standard error of the mean. *P<0.05, **P<0.01. Ala,
alamandine; Hyd, hydralazine; LV, left ventricle; ns, no
significance; LVVd, LV volume in diastole; LVVs, LV volume in
systole; LVIDd, LV internal diameter in diastole; LVIDs, LV
internal diameter in systole; LVEF, LV ejection fraction; LVFS, LV
fractional shortening; SHR, spontaneous hypertensive rat. |
Subsequently, whether Ala affects the remodeling of
cardiac tissues in SHRs was investigated in the present study.
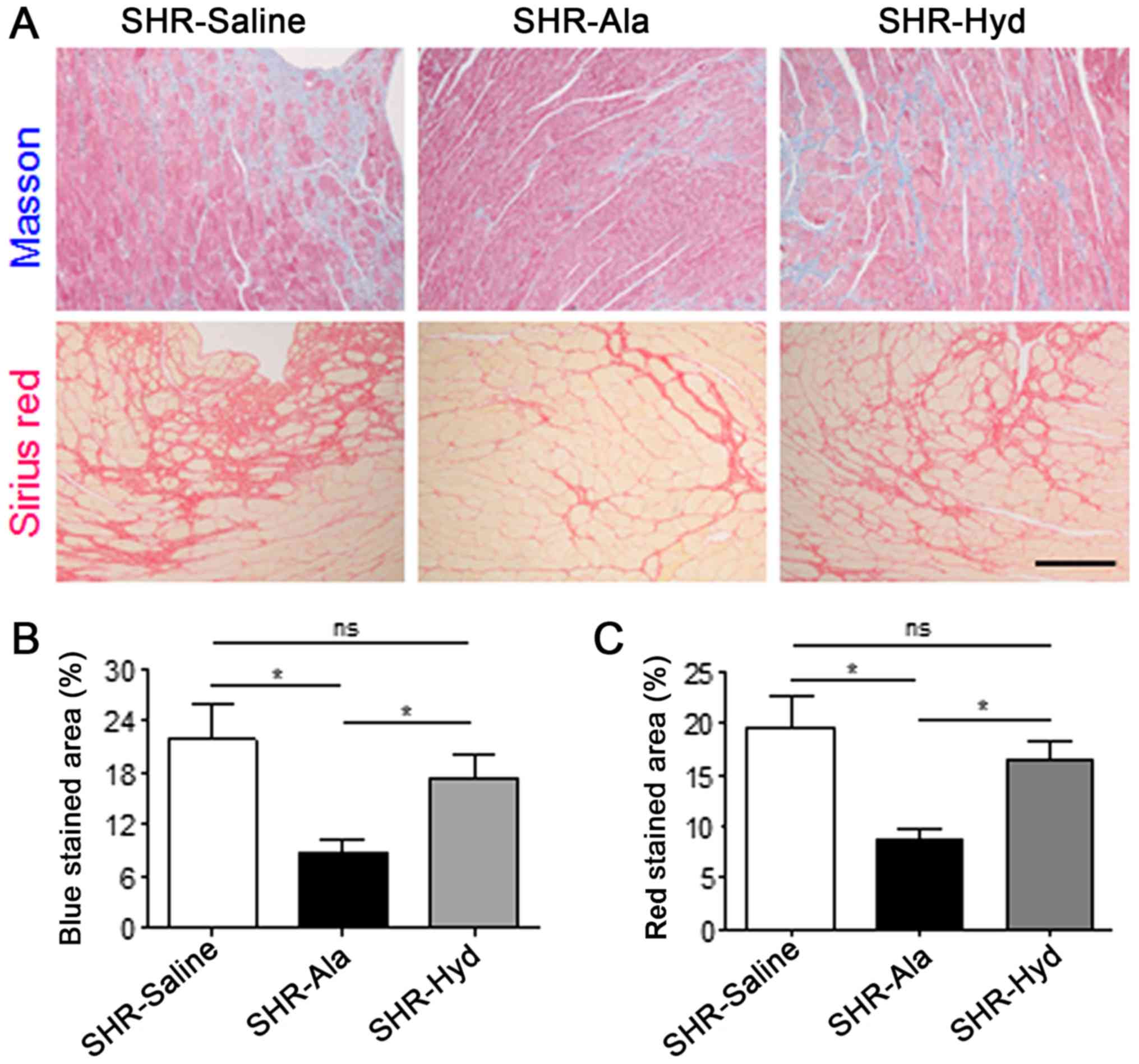
Masson and Sirius red staining of LV sections indicated that the
degree of fibrosis in Ala-treated animals was significantly lower
than in the control group; a marked reduction in fibrosis was
observed in response to Hyd treatment (Fig. 3A-C). Furthermore, analysis of the
expression of fibrosis-associated and remodeling marker proteins,
including CTGF, COL-1 and MMP-9 in LV tissue, by western blotting
revealed that, compared with the control group, Ala treatment
significantly decreased expression levels of all three proteins.
The expression levels of these markers were markedly lower in the
Ala-treated group than the Hyd-treated group; however, the
expression of MMP-9 exhibited a significant difference. Conversely,
we reported that the protein expression levels of these markers
were notably different between the control and Hyd-treated groups
(Fig. 4A-D).
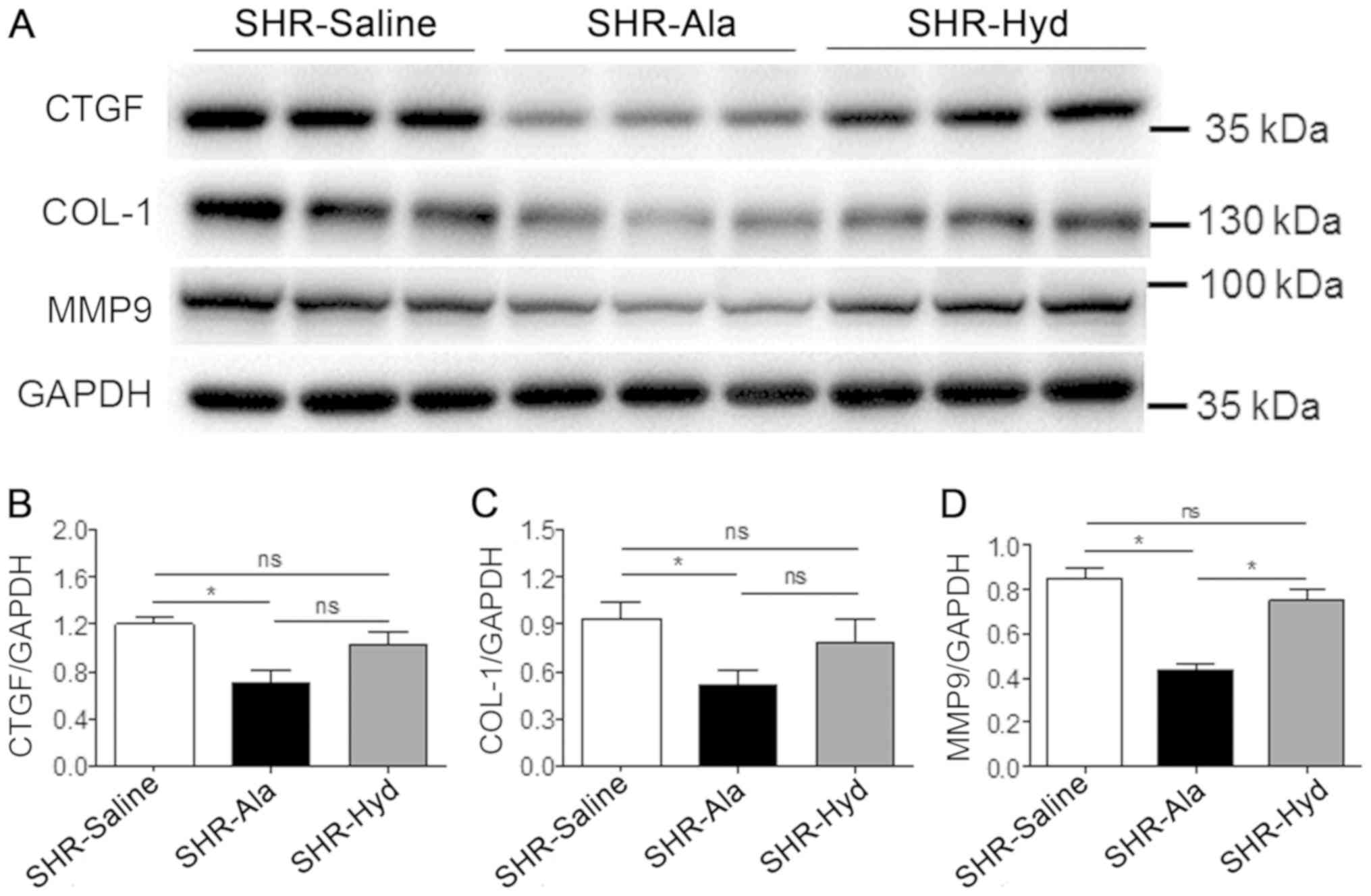 | Figure 4.Ala treatment decreases fibrotic
marker protein expression. (A) Protein expression levels of CTGF,
COL-1 and MMP9 were assessed in LV tissue from Ala-, Hyd- and
saline control-treated SHRs by western blotting with the indicated
antibodies. (B-D) Quantification of relative CTGF, COL-1 and MMP9
expression levels, which were normalized to that of GAPDH; n=6 per
group. Data are presented as mean ± standard error of the mean.
*P<0.05. Ala, alamandine; COL-1, collagen I; CTGF, connective
tissue growth factor; Hyd, hydralazine; MMP9, matrix
metalloproteinase 9; ns, no significance; SHR, spontaneous
hypertensive rat. |
To further investigate the mechanism underlying the
antifibrotic effects of Ala in vitro, primary CFs isolated
from WKY rats were employed and treated with Ang II to mimic the
cardiac fibrosis observed in SHRs. We observed that, compared with
Ang II treatment alone, Ala pre-treatment significantly decreased
the mRNA levels of several genes associated with fibrosis,
including COL1A1, COL3A1, α-SMA and CTGF (Fig. 5A), which were induced in response
to Ang II. In addition, Ala pre-treatment significantly decreased
Ang II-induced protein expression of COL-1 and Akt phosphorylation,
which are associated with fibrosis (Fig. 5B-D).
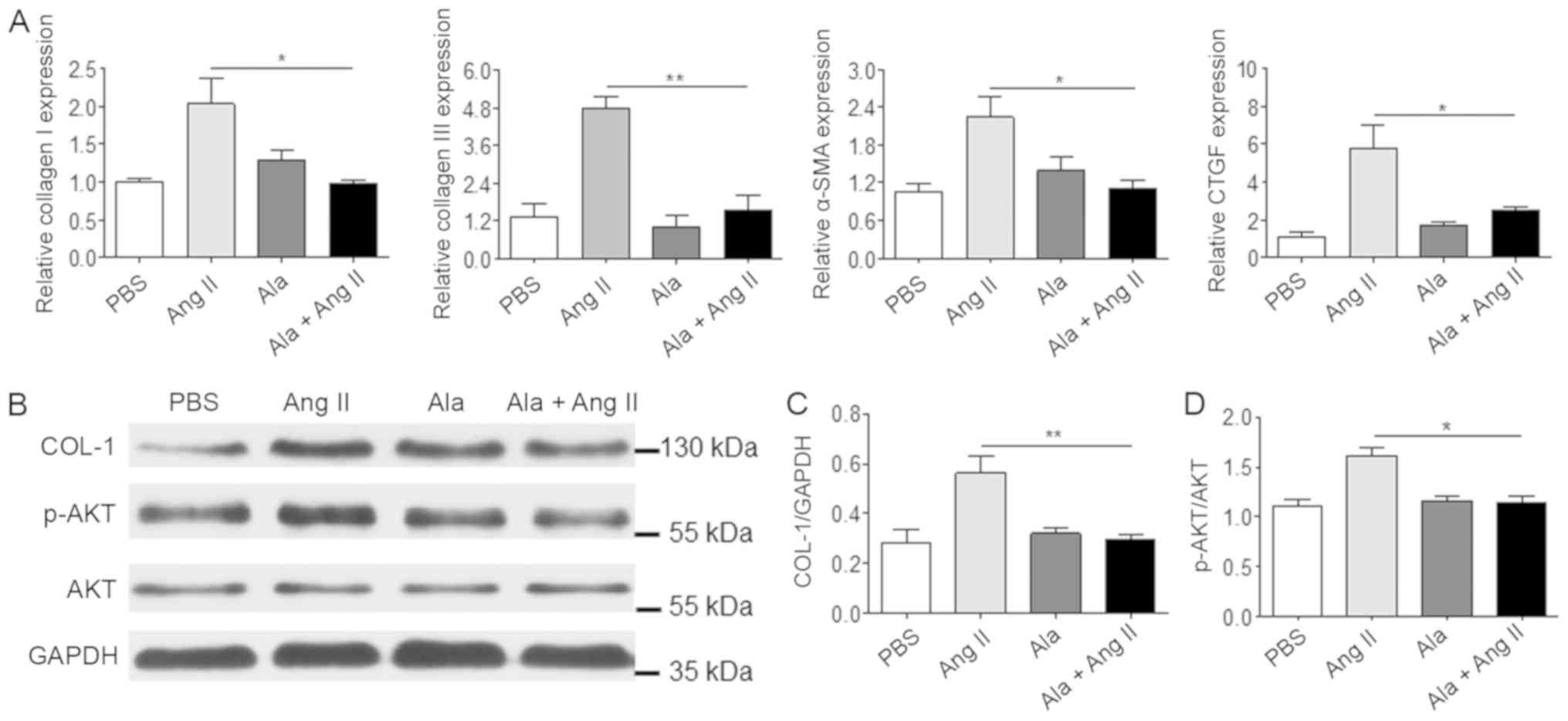 | Figure 5.Ala treatment decreases Ang
II-induced fibrotic marker protein expression in cardiac
fibroblasts. (A) mRNA levels of COL1A1, COL3A1, α-SMA and
CTGF were assessed by reverse transcription-quantitative
polymerase chain reaction. (B) Protein levels of COL-1 and p-Akt
were assessed by western blotting. (C) Quantification of relative
COL-I and (D) p-Akt levels, normalized to GAPDH and Akt,
respectively. Data are presented as the mean ± standard error of
the mean of triplicates and are representative of one experiment
out of three performed. *P<0.05, **P<0.01. α-SMA, α-smooth
muscle actin; Ala, alamandine; Akt, protein kinase B; Ang II,
angiotensin II; COL1A1/COL-1, collagen I; COL3A1, collagen type III
α 1 chain; CTGF, connective tissue growth factor; p,
phosphorylated. |
To determine whether the effects of Ala were
mediated by the MrgD receptor in CFs, CFs were treated with a
non-specific angiotensin AT2 receptor antagonist, PD123319, which
also blocks the MrgD receptor (16). The results demonstrated that
treatment with PD123319 significantly inhibited the effects of Ala
on Ang II-induced COL-1 protein expression and Akt phosphorylation
in CFs (Fig. 6).
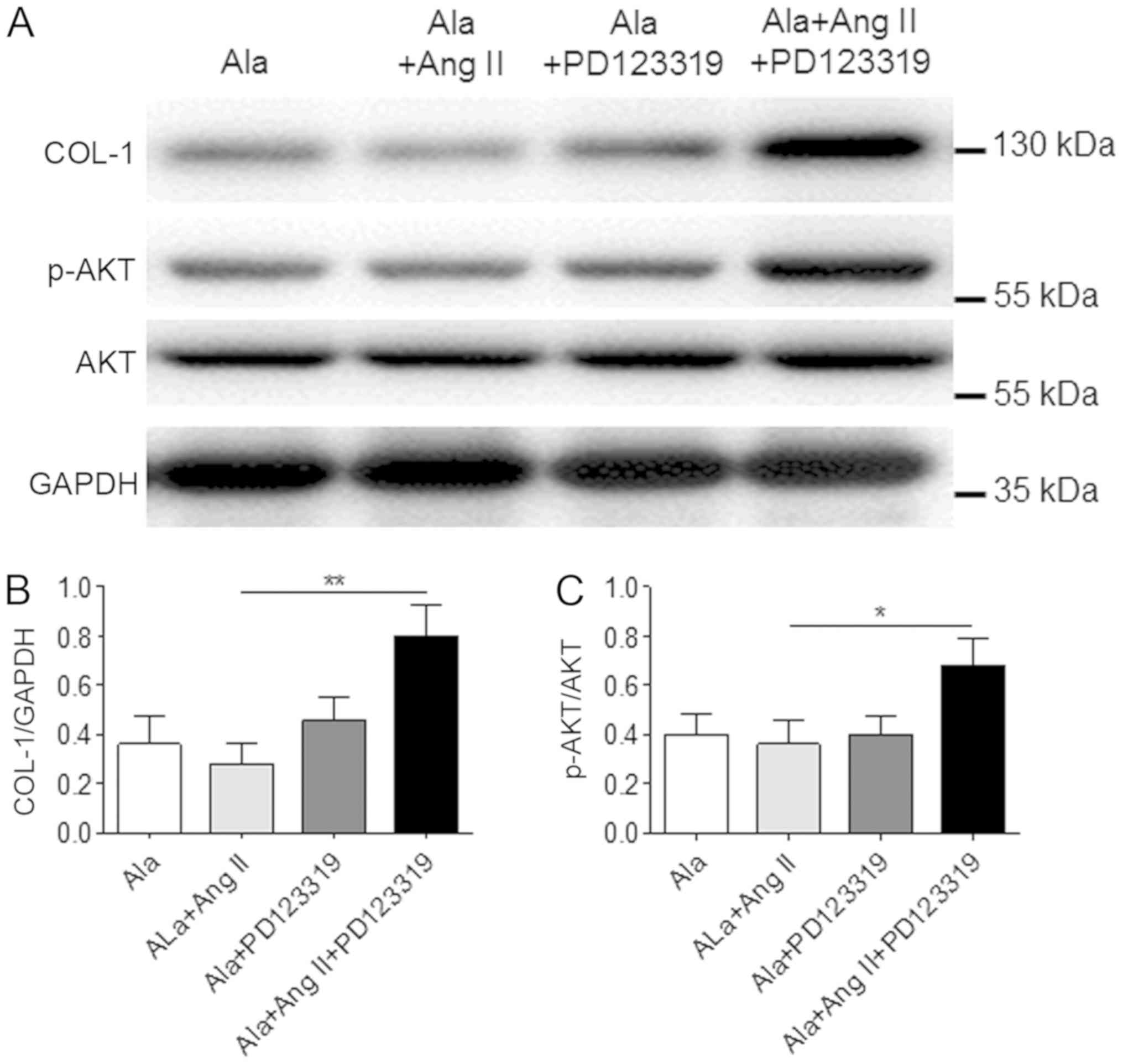 | Figure 6.A Mas-related G protein-coupled
receptor D antagonist inhibits the effects of Ala in cardiac
fibroblasts. (A) Cells were treated with Ala, Ala + Ang II, Ala +
PD123319 or Ala + Ang II + PD123319, and the protein expression
levels of COL-1 and p-Akt were assessed by western blotting. (B and
C) Quantification of relative COL-1 and p-Akt expression levels,
which were normalized to GAPDH, respectively. Data are presented as
the mean ± standard error of the mean of triplicates and are
representative of one experiment out of three performed.
*P<0.05, **P<0.01. Ala, alamandine; Akt, protein kinase B;
Ang II, angiotensin II; COL-1, collagen I; p, phosphorylated. |
Discussion
The RAS comprises numerous peptides and enzymes,
including Ang I, Ang II, ACE1, ACE2, Ang A, Ang (1–7) and
Ala (13,17,18).
ACE1 cleaves the C-terminal of dipeptide of Ang I to form Ang II,
and Ang A is thought to be generated from Ang II via the
decarboxylation of aspartate (17,19).
Ang II and Ang A promote vasoconstriction by binding to the AT1
receptor (20). Ala is the only
one of these factors that may be derived from Ang A and Ang
(1–7) (21),
and has been reported to exhibit anti-hypertensive effects in SHRs
and to reduces the effects of fibrosis in isoproterenol-treated
rats (13); however, whether Ala
can protect against the development of fibrosis in SHRs remains
unknown.
The pathological mechanisms underlying cardiac
fibrosis are complicated and not completely understood. Compared
with cardiac hypertrophy, fibrosis can occur in pressure
over-loaded ventricles, as well as in normotensive dilated
ventricles and atria (3).
Therefore, nonhemodynamic factors, including cardiac fibroblast
cell proliferation and increased collage synthesis, have been
proposed to serve a role in the development of fibrosis (22). In the present study, compared with
the frequently used, short-term model of cardiac fibrosis, the
therapeutic effects of Ala in a chronic, long-term model of cardiac
fibrosis were investigated using 50 week-old SHRs. This model is
notably similar to clinical patients with cardiac fibrosis than
short-term models, including Ang II or isoproterenol-induced
models. In addition, the present study aimed to identify the
therapeutic, rather than protective, effects of Ala on cardiac
fibrosis, which have been assessed previously, as well as other RAS
signaling inhibitors (12,21,23,24).
The results of the present study indicated that Ala exhibited
better therapeutic effects against cardiac fibrosis than Hyd, a
vasodilator used for treatment of hypertension; these treatments
revealed a similar reduction in BP. Ala significantly decreased the
expression levels of fibrotic markers compared with in the control
group; however, the differences in the expression of Col-1 and CTGF
did not exhibit significant differences compared with the Hyd
group. Based on our in vitro data, it was proposed that the
anti-fibrotic effects of Ala mainly occur in CFs. The results of
the present study indicated that reductions in elevated BP may not
be sufficient for the control of chronic cardiac fibrosis in SHRs.
Hyd treatment resulted in a similar reduction of BP, but less
improvement of cardiac fibrosis compared with Ala treatment. This
suggested that Ala performed additional alleviation of cardiac
fibrosis independent of BP reduction, which further suggests that
this novel component of the RAS system may serve an important role
in the reduction of chronic cardiac fibrosis in our in vivo
model.
A role for Ang II in fibrosis has been suggested by
several studies [reviewed in (22)]. Two types of RAS system inhibitors,
including ACE inhibitors and antagonists of the Ang II receptor
type 1 (AT1) receptor, have been reported to inhibit the effects of
Ang II on cardiomyocytes and cardiac fibroblast cells (8), and to prevent cardiac hypertrophy and
fibrosis (9,25). However, Ang II reactivation or
aldosterone escape mediated by the potent profibrotic factor,
aldosterone, during long-term RAS inhibition therapy can attenuate
the clinical benefits of a RAS blockade, particularly on cardiac
fibrosis (26). The results of the
present study revealed that Ala exerts a significant antifibrotic
effect on cardiac tissue in the chronic hypertension rat model,
suggesting that it may have potential as a supplementary treatment
for classical RAS inhibition by ACE inhibitors or AT1 receptor
blockers. Furthermore, the results suggest that Ala may be able to
reverse chronic hypertension-induced cardiac fibrosis in those with
a genetic predisposition this disease; thus, Ala may be considered
as a potential novel therapeutic target in clinical practice.
To further investigate the mechanism by which Ala
prevents cardiac fibrosis, the effects of Ala on collagen synthesis
and Akt activation in primary CFs from normal rats was
investigated. Akt signaling is involved in cardiac fibrosis, and
activated Akt is required for Ang II- and uremia-induced cardiac
fibrosis (27,28). Ang II acts as a profibrotic factor
for cardiac fibrosis via the AT1 receptor; the Ang II-mediated
anti-apoptotic effects on cardiac fibroblast cells are also Akt
dependent (29). In the present
study, Ang II treatment was observed to promote a significant
increase in the phosphorylation of Akt in CFs, whereas this was
inhibited by Ala. In addition, the known AT2 receptor antagonist,
PD123319 which also binds to MrgD receptor (16), suppresses the effects of Ala on
collagen synthesis and Akt activation in primary CFs. Ala has no
affinity for the AT2 receptor (13); the in vitro data of the
present study demonstrated that PD123319 can suppress the
inhibition of Akt phosphorylation induced by Ala treatment, which
suggests that the AT2 receptor may not be involved in Ang
II-mediated Akt activation.
A previous study reported that PD123319 is not a
specific antagonist for the AT2 receptor, but can bind to MrgD
receptors (13). AT2 is highly
expressed in the developing fetus, but its expression is low in the
adult cardiovascular system; however, increased AT2 expression is
observed in conditions of inflammation, hypertension and
atherosclerosis (30). This
protein has vasoactive properties in human radial arteries
(31). MrgD has been reported as
the main receptor for Ala, which exhibits no affinity to MAS or AT2
(6); thus, it was proposed that
Ala inhibits Ang II-induced Akt activation mainly via interactions
with MrgD in the present study. MrgD is expressed predominantly in
dorsal root ganglion neurons, which are key for pain perception
(32,33). This protein is also stably
expressed in an oncogenic fibroblast cell line, and in this
context, overexpression of MrgD promotes cell growth (34). The known ligand of MrgD, β-alanine,
has also been demonstrated to enhance spheroid formation induced by
MrgD and tumorigenesis (18,34).
The potential role of MrgD during cardiac remodeling is not fully
understood; however, in the present study, the MrgD ligand Ala
exhibited antiproliferative activity, unlike the effects of
β-alanine on MrgD in fibroblasts (34). Therefore, the findings of the
present study suggest that MrgD may induce a variety of signal
transduction pathways upon interaction with different ligands, and
this protein may represent a therapeutic target for cardiac
remodeling.
Acknowledgements
We thank Dr Wei Sun and Ms. Ming Qiu of Nanjing
Medical University (Nanjing, China) for the technical help and
writing assistance.
Funding
The present study was supported by the Kangda
College of Nanjing Medical University (grant no. 2015012) and the
National Natural Science Foundation of China (grant no.
81400315).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LW and PL designed the study and wrote the
manuscript. CL and XC performed and analyzed the experiments. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal procedures were approved by the
Experimental Animal Care and Use Committee of Nanjing Medical
University and were conducted in accordance with National
Institutes of Health guide for the Care and Use of Laboratory
Animals (35) and comply with the
ARRIVE guidelines (http://www.nc3rs.org.uk/arrive-guidelines) and the
AVMA euthanasia guidelines 2013 (36).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fujiu K and Nagai R: Fibroblast-mediated
pathways in cardiac hypertrophy. J Mol Cell Cardiol. 70:64–73.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weber KT and Brilla CG: Pathological
hypertrophy and cardiac interstitium. Fibrosis and
renin-angiotensin-aldosterone system. Circulation. 83:1849–1865.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Young MJ and Funder JW: The
renin-angiotensin-aldosterone system in experimental
mineralocorticoid-salt-induced cardiac fibrosis. Am J Physiol.
271:E883–E888. 1996.PubMed/NCBI
|
|
4
|
MacKenna D, Summerour SR and Villarreal
FJ: Role of mechanical factors in modulating cardiac fibroblast
function and extracellular matrix synthesis. Cardiovasc Res.
46:257–263. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Davis J and Molkentin JD: Myofibroblasts:
Trust your heart and let fate decide. J Mol Cell Cardiol. 70:9–18.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bader M, Peters J, Baltatu O, Muller DN,
Luft FC and Ganten D: Tissue renin-angiotensin systems: New
insights from experimental animal models in hypertension research.
J Mol Med (Berl). 79:76–102. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ruiz-Ortega M, Lorenzo O, Rupérez M,
Esteban V, Suzuki Y, Mezzano S, Plaza JJ and Egido J: Role of the
renin-angiotensin system in vascular diseases: Expanding the field.
Hypertension. 38:1382–1387. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Villarreal FJ, Kim NN, Ungab GD, Printz MP
and Dillmann WH: Identification of functional angiotensin II
receptors on rat cardiac fibroblasts. Circulation. 88:2849–2861.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim S, Ohta K, Hamaguchi A, Yukimura T,
Miura K and Iwao H: Angiotensin II induces cardiac phenotypic
modulation and remodeling in vivo in rats. Hypertension.
25:1252–1259. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kawano H, Do YS, Kawano Y, Starnes V, Barr
M, Law RE and Hsueh WA: Angiotensin II has multiple profibrotic
effects in human cardiac fibroblasts. Circulation. 101:1130–1137.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
González A, López B, Querejeta R and Diez
J: Regulation of myocardial fibrillar collagen by angiotensin II. A
role in hypertensive heart disease? J Mol Cell Cardiol.
34:1585–1593. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Simko F, Pechanova O, Pelouch V,
Krajcirovicova K, Mullerova M, Bednarova K, Adamcova M and Paulis
L: Effect of melatonin, captopril, spironolactone and simvastatin
on blood pressure and left ventricular remodelling in spontaneously
hypertensive rats. J Hypertens. (Suppl 27):S5–S10. 2009. View Article : Google Scholar
|
|
13
|
Lautner RQ, Villela DC, Fraga-Silva RA,
Silva N, Verano-Braga T, Costa-Fraga F, Jankowski J, Jankowski V,
Sousa F, Alzamora A, et al: Discovery and characterization of
alamandine: A novel component of the renin-angiotensin system. Circ
Res. 112:1104–1111. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Habiyakare B, Alsaadon H, Mathai ML, Hayes
A and Zulli A: Reduction of angiotensin A and alamandine
vasoactivity in the rabbit model of atherogenesis: Differential
effects of alamandine and Ang(1–7). Int J Exp Pathol. 95:290–295.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Soares ER, Barbosa CM, Campagnole-Santos
MJ, Santos RAS and Alzamora AC: Hypotensive effect induced by
microinjection of Alamandine, a derivative of angiotensin-(1–7),
into caudal ventrolateral medulla of 2K1C hypertensive rats.
Peptides. 96:67–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jankowski V, Vanholder R, van der Giet M,
Tölle M, Karadogan S, Gobom J, Furkert J, Oksche A, Krause E, Tran
TN, et al: Mass-spectrometric identification of a novel angiotensin
peptide in human plasma. Arterioscler Thromb Vasc Biol. 27:297–302.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shinohara T, Harada M, Ogi K, Maruyama M,
Fujii R, Tanaka H, Fukusumi S, Komatsu H, Hosoya M, Noguchi Y, et
al: Identification of a G protein-coupled receptor specifically
responsive to beta-alanine. J Biol Chem. 279:23559–23564. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang R, Smolders I, Vanderheyden P,
Demaegdt H, Van Eeckhaut A, Vauquelin G, Lukaszuk A, Tourwé D, Chai
SY, Albiston AL, et al: Pressor and renal hemodynamic effects of
the novel angiotensin A peptide are angiotensin II type 1A receptor
dependent. Hypertension. 57:956–964. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Coutinho DC, Foureaux G, Rodrigues KD,
Salles RL, Moraes PL, Murça TM, De Maria ML, Gomes ER, Santos RA,
Guatimosim S and Ferreira AJ: Cardiovascular effects of angiotensin
A: A novel peptide of the renin-angiotensin system. J Renin
Angiotensin Aldosterone Syst. 15:480–486. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qaradakhi T, Apostolopoulos V and Zulli A:
Angiotensin (1–7) and Alamandine: Similarities and differences.
Pharmacol Res. 111:820–826. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun Y, Ramires FJ, Zhou G, Ganjam VK and
Weber KT: Fibrous tissue and angiotensin II. J Mol Cell Cardiol.
29:2001–2012. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Varo N, Etayo JC, Zalba G, Beaumont J,
Iraburu MJ, Montiel C, Gil MJ, Monreal I and Díez J: Losartan
inhibits the post-transcriptional synthesis of collagen type I and
reverses left ventricular fibrosis in spontaneously hypertensive
rats. J Hypertens. 17:107–114. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brown L, Duce B, Miric G and Sernia C:
Reversal of cardiac fibrosis in deoxycorticosterone acetate-salt
hypertensive rats by inhibition of the renin-angiotensin system. J
Am Soc Nephrol. 10 (Suppl 11):S143–S148. 1999.PubMed/NCBI
|
|
25
|
Crawford DC, Chobanian AV and Brecher P:
Angiotensin II induces fibronectin expression associated with
cardiac fibrosis in the rat. Circ Res. 74:727–739. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Athyros VG, Mikhailidis DP, Kakafika AI,
Tziomalos K and Karagiannis A: Angiotensin II reactivation and
aldosterone escape phenomena in renin-angiotensin-aldosterone
system blockade: Is oral renin inhibition the solution? Expert Opin
Pharmacother. 8:529–535. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao QD, Viswanadhapalli S, Williams P,
Shi Q, Tan C, Yi X, Bhandari B and Abboud HE: NADPH oxidase 4
induces cardiac fibrosis and hypertrophy through activating
Akt/mTOR and NFkB signaling pathways. Circulation. 131:643–655.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin CY, Hsu YJ, Hsu SC, Chen Y, Lee HS,
Lin SH, Huang SM, Tsai CS and Shih CC: CB1 cannabinoid receptor
antagonist attenuates left ventricular hypertrophy and Akt-mediated
cardiac fibrosis in experimental uremia. J Mol Cell Cardiol.
85:249–261. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tian B, Liu J, Bitterman P and Bache RJ:
Angiotensin II modulates nitric oxide-induced cardiac fibroblast
apoptosis by activation of AKT/PKB. Am J Physiol Heart Circ
Physiol. 285:H1105–1112. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Arumugam S, Sreedhar R, Thandavarayan RA,
Karuppagounder V, Krishnamurthy P, Suzuki K, Nakamura M and
Watanabe K: Angiotensin receptor blockers: Focus on cardiac and
renal injury. Trends Cardiovasc Med. 26:221–228. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zulli A, Hare DL, Buxton BF and Widdop RE:
Vasoactive role for angiotensin II type 2 receptors in human radial
artery. Int J Immunopathol Pharmacol. 27:79–85. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dong X, Han S, Zylka MJ, Simon MI and
Anderson DJ: A diverse family of GPCRs expressed in specific
subsets of nociceptive sensory neurons. Cell. 106:619–632. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lembo PM, Grazzini E, Groblewski T,
O'Donnell D, Roy MO, Zhang J, Hoffert C, Cao J, Schmidt R,
Pelletier M, et al: Proenkephalin A gene products activate a new
family of sensory neuron-specific GPCRs. Nat Neurosci. 5:201–209.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nishimura S, Uno M, Kaneta Y, Fukuchi K,
Nishigohri H, Hasegawa J, Komori H, Takeda S, Enomoto K, Nara F and
Agatsuma T: MRGD, a MAS-related G-protein coupled receptor,
promotes tumorigenisis and is highly expressed in lung cancer. PLoS
One. 7:e386182012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Council NR: Guide for the care and use of
laboratory animals. The National Academies Press; Washington, DC:
1996, PubMed/NCBI
|
|
36
|
Leary S, Underwood W, Anthony R, Cartner
S, Corey D, Grandin T, Greenacre C, Gwaltney-Brant S, McCrackin MA,
Meyer R, et al: AVMA guidelines for the euthanasia of animals: 2013
Edition. American Veterinary Medical Association. 2013.
|