Introduction
Cardiac fibrosis is a common pathologic component of
various cardiovascular disorders, defined as the excessive
deposition of extracellular matrix (ECM) and the disturbance of
myocardial stiffness, which subsequently results in systolic and/or
diastolic dysfunction of the heart (1). Increased accumulation of myocardial
ECM also impairs the electrical conduction system and contributes
to arrhythmogenesis (2,3). Activation and transdifferentiation of
cardiac fibroblasts to myofibroblasts is a crucial event in cardiac
fibrosis, and is responsible for the excessive synthesis of ECM
(1). Therefore, improved
understanding of the pathogenesis of myofibroblast
transdifferentiation and the identification of novel therapeutic
targets may be of great therapeutic interest for the treatment of
cardiac fibrosis.
Transforming growth factor-β (TGF-β) is the most
widely known fibrogenic growth factor associated with cardiac
fibrosis, and promotes the transdifferentiation of cardiac
fibroblasts to myofibroblasts (4,5). In
response to cardiovascular insults, bioactive TGF-β is induced and
released from latent stores, subsequently binding to TGF-β
receptors, resulting in the activation of the canonical
Smad-dependent signaling pathway and the induction of a profibrotic
gene program (6). In addition,
TGF-β can stimulate myofibroblast transdifferentiation and promote
ECM synthesis via non-canonical pathways, including
mitogen-activated protein kinase (MAPK) and protein kinase B (AKT)
(7,8). These non-canonical pathways
coordinate with the Smad-dependent canonical pathway to induce
cardiac fibrosis (9). Furthermore,
negative regulators of AKT or p38 have been reported to inhibit
myofibroblast transdifferentiation and protect against cardiac
fibrosis (8,10). Thus, targeting TGF-β signaling, via
canonical or non-canonical pathways, may aid in developing
efficacious interventions against fibrosis.
MicroRNAs (miRNAs) are a class of small noncoding
RNA molecules that function as negative regulators of gene
expression by binding to the 3′-untranslated region (UTR) of target
mRNAs (11,12). Emerging evidence suggests that
miRNAs regulate the expression of key genes involved in fibrotic
diseases, particularly cardiac fibrosis (13,14).
Previous studies have demonstrated that miR-133a is
downregulated in transverse aortic constriction or
isoproterenol-induced fibrotic hearts, and that miR-133a
overexpression can reduce collagen deposition and improve cardiac
dysfunction (15). Pan et
al (16) observed that forced
expression of miR-101a/b suppressed the proliferation and
collagen production in rat neonatal cardiac fibroblasts.
Additionally, results from Nagpal et al (17) demonstrated that miR-125b was
important for the induction of cardiac fibrosis, and that the
inhibition of miR-125b may represent a novel therapeutic
approach for the treatment of cardiac fibrosis. These studies
indicated a central role for miRNAs in cardiac fibrosis.
miRNA-216a lies in the second intron of a
noncoding RNA (RP23-298H6.1–001) located on the mouse chromosome 11
(18). The majority of previous
studies into miR-216a have focused on tumors, identifying
miR-216a as a potential biomarker for certain types of
cancer (19,20). Xia et al (20) reported that miR-216a
contributed to hepatocarcinogenesis and tumor recurrence in
hepatocellular carcinoma. Recent studies, however, have suggested
that the functions of miR-216a extend beyond the regulation
of tumors, and that it serves important roles in other
pathophysiological processes. For example, Yang et al
(21,22) reported that miR-216a
promotes endothelial senescence and inflammation, and M1 macrophage
polarization via Smad3. Additionally, it was observed that miR-216a
levels were increased in mouse renal mesangial cells following
stimulation with TGF-β (23). The
present study hypothesized that miR-216a may be involved in
the pathogenesis of myofibroblast transdifferentiation and cardiac
fibrosis.
Materials and methods
Reagents
TGF-β (cat. no. ab50036) was purchased from Abcam.
AKT inhibitor MK2206 (cat. no. HY-10358) was purchased from
MedChemExpress LLC. The antagomir (5′-CACAGUUGCCAGCUGAGAUUA-3′) and
the agomir (5′-UAAUCUCAGCUGGCAACUGUG-3′) of miR-216a, their
negative controls (antagomir control, cat. no. miR3N0000001-4-5;
agomir control, cat. no. miR4N0000001-4-5), small interfering
(si)RNA against PTEN (siPten; 5′-TTCCGCCACTGAACATTGGAA-3′)
and negative control siRNA (cat. no. siN0000003-1-10) were
generated by Guangzhou RiboBio Co., Ltd. Alexa Fluor®
488-goat anti-rabbit immunoglobulin G (IgG) secondary antibody
(1:200; cat. no. A11008) for immunofluorescence detection was
obtained from Pierce (Thermo Fisher Scientific, Inc.). Primary
antibodies for total (T)-AKT (1:1,000; cat. no. 4691),
phosphorylated (P)-AKT (1:1,000; cat. no. 4060), T-glycogen
synthase kinase 3β (GSK3β; 1:1,000; cat. no. 9315), P-GSK3β
(1:1,000; cat. no. 9323P), T-Smad3 (1:1,000; cat. no. 9513s),
P-Smad3 (1:1,000; cat. no. 8769), PTEN (1:1,000; cat. no. 9559) and
GAPDH (1:1,000; cat. no. 2118) were purchased from Cell Signaling
Technology, Inc. The anti-TGF-β receptor II (TGFBR2; 1:1,000; cat.
no. ab61213) antibody was obtained from Abcam.
Cell culture and treatments
All of the animal experimental protocols were
approved by the Animal Care and Use Committee of Renmin Hospital of
Wuhan University (approval no. 20171003) and conducted in
accordance with the National Institutes of Health Guide for the
Care and Use of Laboratory Animals (24). A total of 40 male C57BL/6 mice
(age, 8–10 weeks; body weight, 23–28 g) were purchased from the
Institute of Laboratory Animal Science, Chinese Academy of Medical
Sciences. The animals were allowed free access to food/water in a
specific pathogen-free, environmentally controlled barrier
conditions (temperature, 20–25°C; humidity, 45–55%; 12-h light/dark
cycle) for 1 week prior to commencing the study. Adult mouse
cardiac fibroblasts were isolated as previously described (25). In brief, left ventricles were
harvested and digested in 0.125% trypsin and collagenase. The
culture was then collected and suspended in DMEM/F12 (HyClone; GE
Healthcare Life Sciences) medium with 10% fetal bovine serum
(HyClone; GE Healthcare Life Sciences) at 37°C for 90 min. The
adherent fibroblasts were prepared for the subsequent experiments
following synchronization for 12 h. The miR-216a antagomir
and agomir, and their negative controls were all diluted with
DMEM/F12 medium and then were mixed with Lipofectamine RNAiMAX
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) for 20 min at
room temperature. Then, when the cells had grown to 70–80%
confluency, they were incubated with the mixture at a final
concentration of 50 nM at 37°C for 24 h, followed with TGF-β
stimulation for an additional 24 h. To inhibit AKT activity,
cardiac fibroblasts were pretreated with MK2206 (1 µM) for 24 h
(26). PTEN knockdown was
performed using siPten, and the efficiency of the knockdown
was assessed via western blotting. Briefly, the siPten and
its negative control were diluted with DMEM/F12 medium and then
mixed with Lipofectamine RNAiMAX reagent for 20 min at room
temperature. Then, the cells (at 40–50% confluency) were incubated
with the mixture at a final concentration of 50 nM at 37°C for 4 h,
followed by miR-216a antagomir transfection for 24 h and TGF-β
stimulation for an additional 24 h as aforementioned.
Western blotting
Western blotting was performed as previously
described (27,28). Briefly, cultured cardiac
fibroblasts were lysed in RIPA lysis buffer (50 mM Tris-HCl, 0.5%
NP-40, 250 mM NaCl, 5 mM EDTA and 50 mM NaF) and the protein
concentration was evaluated using a Rapid Gold BCA Protein Assay
kit from Pierce (cat. no. A53225; Thermo Fisher Scientific, Inc.).
Total proteins (50 µg) were loaded, separated via 10% SDS-PAGE and
electrically transferred to PVDF membranes (cat. no. IPFL00010; EMD
Millipore). Non-specific binding was blocked with 5% non-fat milk
at room temperature for 1 h. Then, the proteins were incubated with
the indicated antibodies at 4°C overnight, followed by incubation
with secondary antibodies (IRDye® 800CW conjugated goat
anti-mouse IgG; 1:1,000; cat. no. 925-32210; LI-COR Biosciences) at
room temperature for 1 h in the dark. Proteins were scanned and
quantified using an Odyssey Infrared Imaging System (Odyssey
version 3.0 Software; LI-COR Biosciences) in a blinded manner, and
target proteins were normalized to GAPDH or the corresponding total
proteins.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from fibroblasts using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and transcribed to cDNA using a Maxima First Strand cDNA
Synthesis kit (Roche) according to the manufacturer's protocols.
Levels of miR-216a were detected using a
BulgeLoop™ miRNA RT-qPCR System (Guangzhou RiboBio Co.,
Ltd.). The thermocycling conditions were as follows: 95°C for 10,
then 40 cycles of 95°C for 2 sec, 60°C for 20 sec and 70°C for 10
sec. The data were analyzed using the 2−∆∆Cq method as
previously described (29). Total
mRNA levels were normalized to GAPDH, and miR-216a levels
were normalized to U6. The primer sequences were as follows: Mouse
collagen 1 (Col 1), forward, 5′-AGGCTTCAGTGGTTTGGATG-3′ and
reverse, 5′-CACCAACAGCACCATCGTTA-3′; mouse collagen 3 (Col
3), forward, 5′-CCCAACCCAGAGATCCCATT-3′ and reverse,
5′-GAAGCACAGGAGCAGGTGTAGA-3′; mouse connective tissue growth factor
(Ctgf), forward, 5′-TGTGTGATGAGCCCAAGGAC-3′ and reverse,
5′-AGTTGGCTCGCATCATAGTTG-3′; mouse fibronectin (Fn),
forward, 5′-CCGGTGGCTGTCAGTCAGA-3′ and reverse,
5′-CCGTTCCCACTGCTGATTTATC-3′; mouse miR-216a, forward,
5′-CATGATCAGCTGGGCCAAGACACAGTTGCCAGCTG-3′ and reverse,
5′-TAATCTCAGCTGGCAA-3′; mouse GAPDH, forward,
5′-CGTGCCGCCTGGAGAAACC-3′ and reverse,
5′-TGGAAGAGTGGGAGTTGCTGTTG-3′, and U6, forward,
5′-CTCGCTTCGGCAGCACA-3′ and reverse,
5′-AACGCTTCACGAATTTGCGT-3′.
Immunofluorescence staining
Immunofluorescence staining was performed to detect
the expression of the myofibroblast transdifferentiation biomarker
α-smooth muscle actin (α-SMA) in mouse cardiac fibroblasts, as
previously described (30). In
brief, cardiac coverslips were fixed with 4% paraformaldehyde for
15 min at room temperature and then permeabilized with 0.2% Triton
X-100, followed by incubation with 10% goat serum (GeneTex, Inc.)
for 1 h at room temperature. The cells were then incubated with
anti-α-SMA (1:100; cat. no. ab5694; Abcam) at 4°C overnight,
followed by incubation with the secondary antibody for 1 h at room
temperature. The nuclei were stained with DAPI at room temperature
for 30 sec. Images were captured using an Olympus DX51 fluorescence
microscope (magnification, ×400; Olympus Corporation) and
quantified using Image-Pro Plus 6.0 (Media Cybernetics, Inc.). A
total of 10–15 fields of view were observed per coverslip.
Bioinformatic prediction
The online database TargetScanMouse (Release 7.1,
http://www.targetscan.org/mmu_71/) was
employed for target prediction and analysis of miR-216a
(31).
Statistical analysis
Results were presented as the mean ± standard error
of the mean. Unpaired Student's t-tests (two-tailed) were used to
compare differences between two groups. One-way ANOVA followed by a
Tukey's post hoc test was performed to determine differences across
multiple groups. All data were analyzed using SPSS 22.0 software
(IBM Corporation) P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-216a inhibition attenuates
TGF-β-induced myofibroblast transdifferentiation
Alterations in the expression of miR-216a
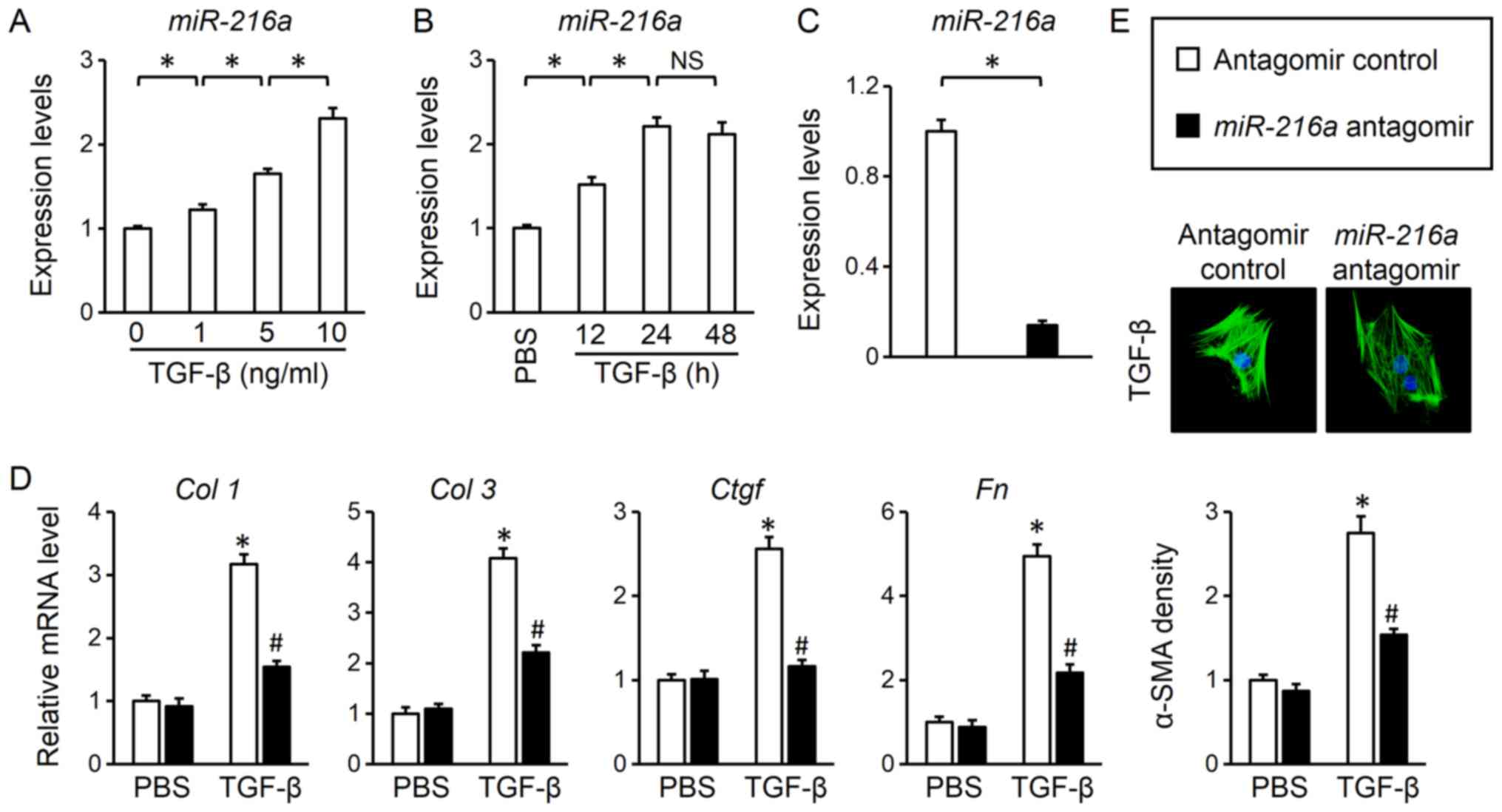
were evaluated in TGF-β-treated adult mouse cardiac fibroblasts. As
presented in Fig. 1A, TGF-β
incubation increased miR-216a levels in a dose-dependent
manner. In addition, miR-216a levels were increased in a
time-dependent manner, albeit with no significant difference
between 24 and 48 h (Fig. 1B).
Therefore, a 24-h treatment period with 10 ng/ml of TGF-β was
selected for further experiments. An antagomir was used to inhibit
miR-216a expression in cultured adult mouse cardiac
fibroblasts; the efficiency of transfection was demonstrated via
RT-qPCR analysis (Fig. 1C). As
presented in Fig. 1D,
miR-216 inhibition suppressed collagen synthesis in response
to TGF-β stimulation, as determined by the significantly decreased
mRNA levels of fibrotic markers, Col 1, Col 3, Ctgf and
Fn compared with the control. Increased α-SMA expression in
response to TGF-β is a hallmark of myofibroblast
transdifferentiation (27). Of
note, it was observed that miR-216a inhibition also
significantly reduced the TGF-β-induced expression of α-SMA and
myofibroblast transdifferentiation (Fig. 1E). Thus, the results indicated that
miR-216a was upregulated following TGF-β treatment, and that
miR-216a inhibition attenuated TGF-β-induced myofibroblast
transdifferentiation.
miR-216a activation exacerbates
TGF-β-induced myofibroblast transdifferentiation
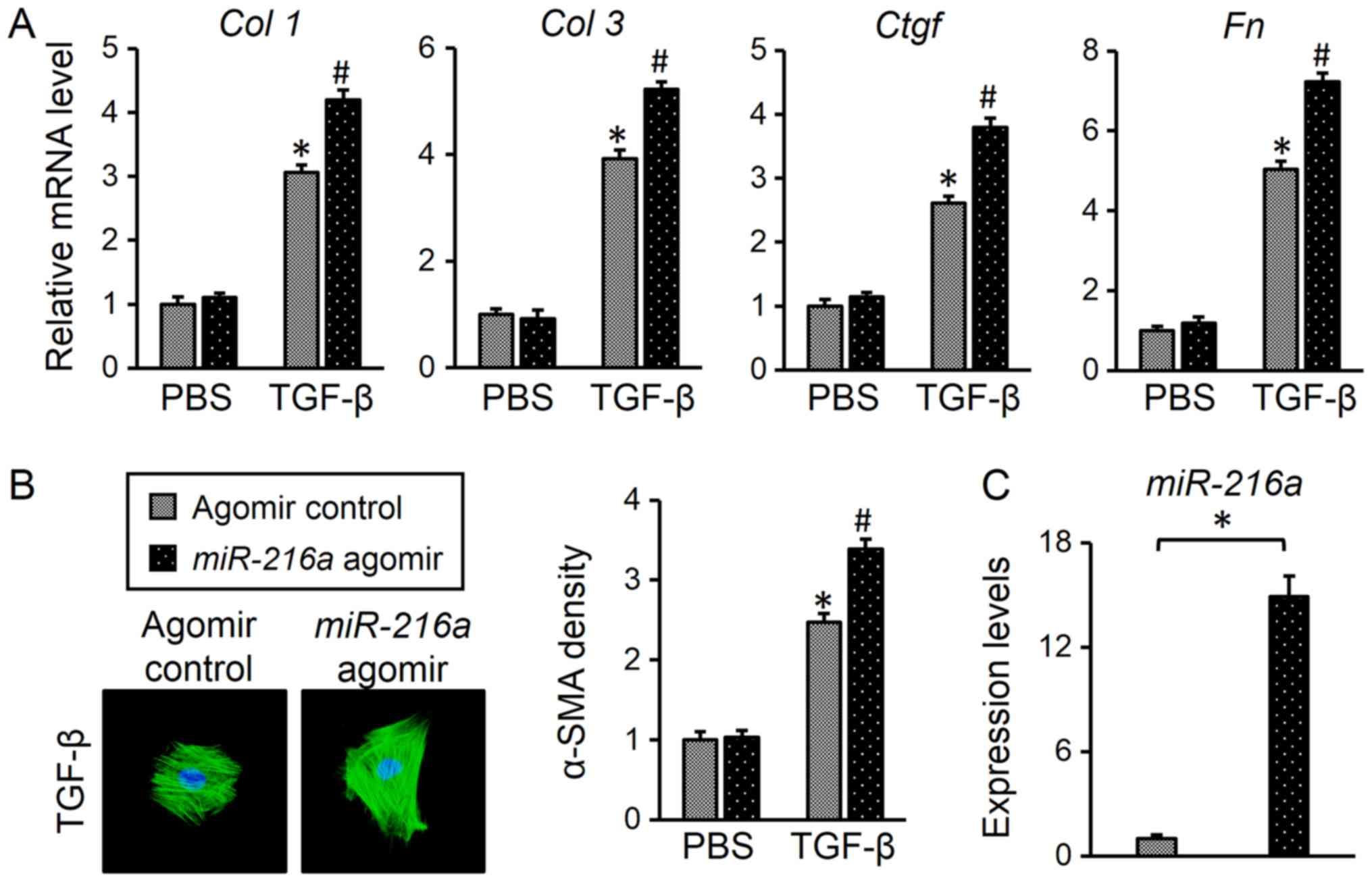
Next, the present study investigated whether
miR-216a overexpression promoted myofibroblast
transdifferentiation in response to TGF-β. Cardiac fibroblasts were
transfected with a miR-216a agomir (Fig. 2C), and it was observed that
miR-216a upregulation enhanced the TGF-β-induced increase in
the expression of fibrotic markers compared with the control
(Fig. 2A). In addition, it was
determined that the miR-216a agomir further promoted α-SMA
expression in response to TGF-β compared with the control (Fig. 2B). Collectively, these findings
suggested that miR-216a is required for the induction of
myofibroblast transdifferentiation.
AKT/GSK3β is involved in the
regulation of myofibroblast transdifferentiation by miR-216a
A previous study reported that TGF-β/Smad is the
most common signaling pathway responsible for myofibroblast
transdifferentiation, and that Smad3 is a critical and necessary
mediator in this process (6). In
the present study, however, the results demonstrated that
miR-216a inhibition did not significantly affect the
phosphorylation of Smad3 (Fig. 3A and
B). TGFBR2 is the primary receptor of TGF-β and mediates its
profibrotic effect. Furthermore, bioinformatics analysis using
TargetScan indicated that the TGFBR2 gene may be a target
for miR-216a (data not shown). Therefore, the expression
levels of TGFBR2 were analyzed via western blotting; however, the
protein levels of TGFBR2 were not altered by the up- or
downregulation of miR-216a, with or without TGF-β treatment
(Fig. 3A, C, E and F).
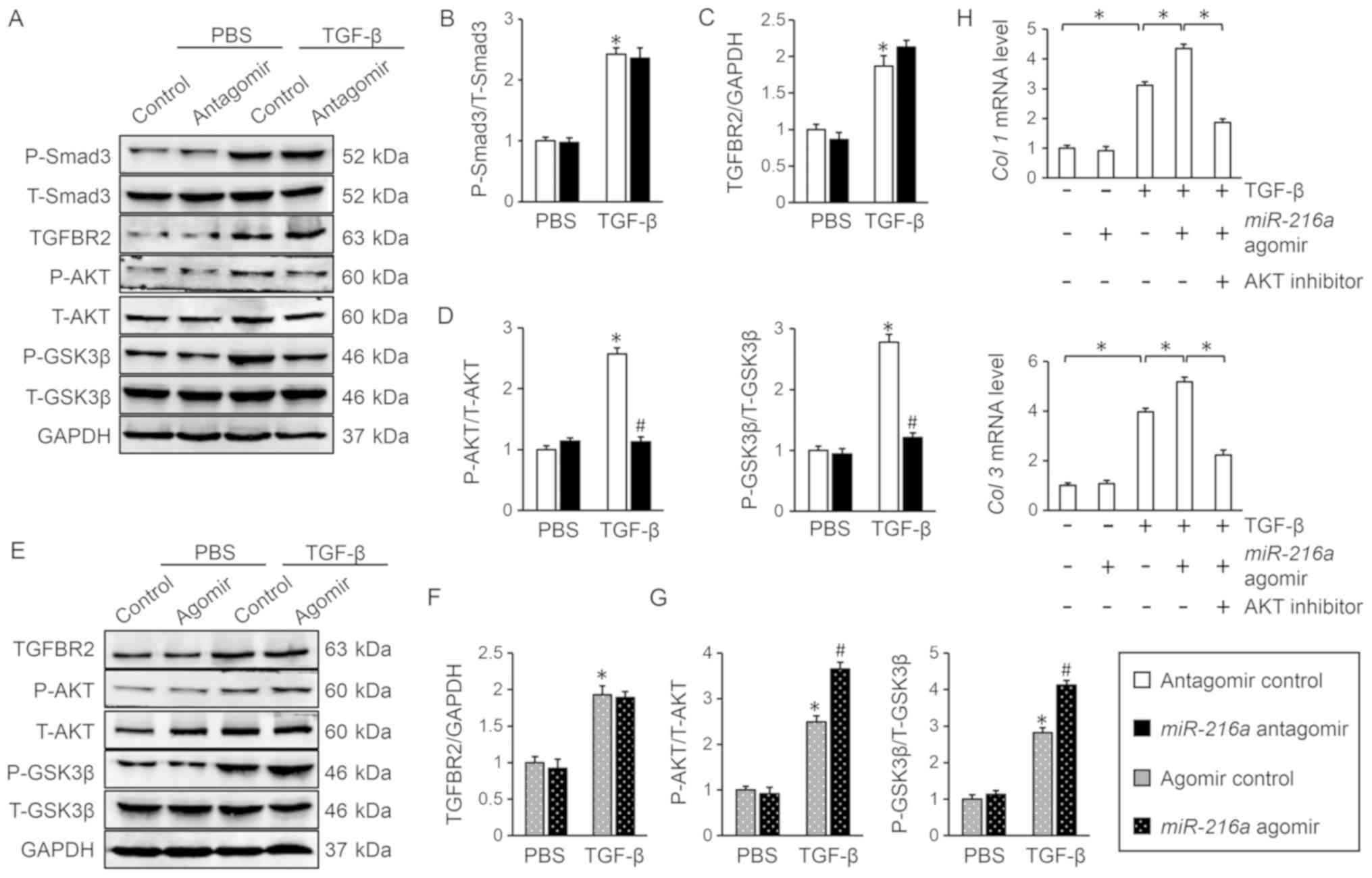 | Figure 3.AKT/GSK3β is involved in the
regulation of myofibroblast transdifferentiation by
miR-216a. (A-D) Representative western blot images and
quantification following transfection with miR-216a
antagomir, in the presence or absence of TGF-β (n=6). *P<0.05
vs. antagomir control + PBS; #P<0.05 vs. antagomir
control + TGF-β. (E-G) Representative western blot images and
quantification following transfection with miR-216a agomir,
in the presence or absence of TGF-β (n=6). *P<0.05 vs. agomir
control + PBS; #P<0.05 vs. agomir control + TGF-β.
(H) Relative mRNA levels of Col 1 and Col 3 in the
presence of AKT inhibitor MK2206 (n=6). *P<0.05, with
comparisons indicated by brackets. Data are presented as the mean ±
standard error of the mean. GSK3β, glycogen synthase kinase 3β;
miR-216a, microRNA-216a; TGF-β, transforming growth
factor-β; Col, collagen; P-, phosphorylated; T-, total;
TGFB2R, TGF-β receptor II. |
In addition to the canonical Smad-dependent
signaling, TGF-β also activates Smad-independent pathways,
including the AKT pathway (8).
Previous studies have reported that AKT and its downstream target
GSK3β also contribute to the regulation of myofibroblast
transdifferentiation (8,32). Therefore, the phosphorylation
status of AKT and GSK3β was examined in the present study, and the
results demonstrated that the miR-216a antagomir
significantly inhibited TGF-β-induced AKT/GSK3β activation compared
with the control (Fig. 3D); by
contrast, the miR-216a agomir further promoted AKT/GSK3β
phosphorylation following treatment with TGF-β (Fig. 3G). Additionally, inhibition of AKT
with MK2206 significantly attenuated the miR-216a
agomir-mediated increase in myofibroblast transdifferentiation in
response to TGF-β stimulation, as determined by the decreased mRNA
levels of Col 1 and Col 3 (Fig. 3H). Collectively, these results
indicated that miR-216a promoted TGF-β-induced myofibroblast
transdifferentiation via activating AKT/GSK3β.
miR-216a activates AKT via inhibition
of PTEN
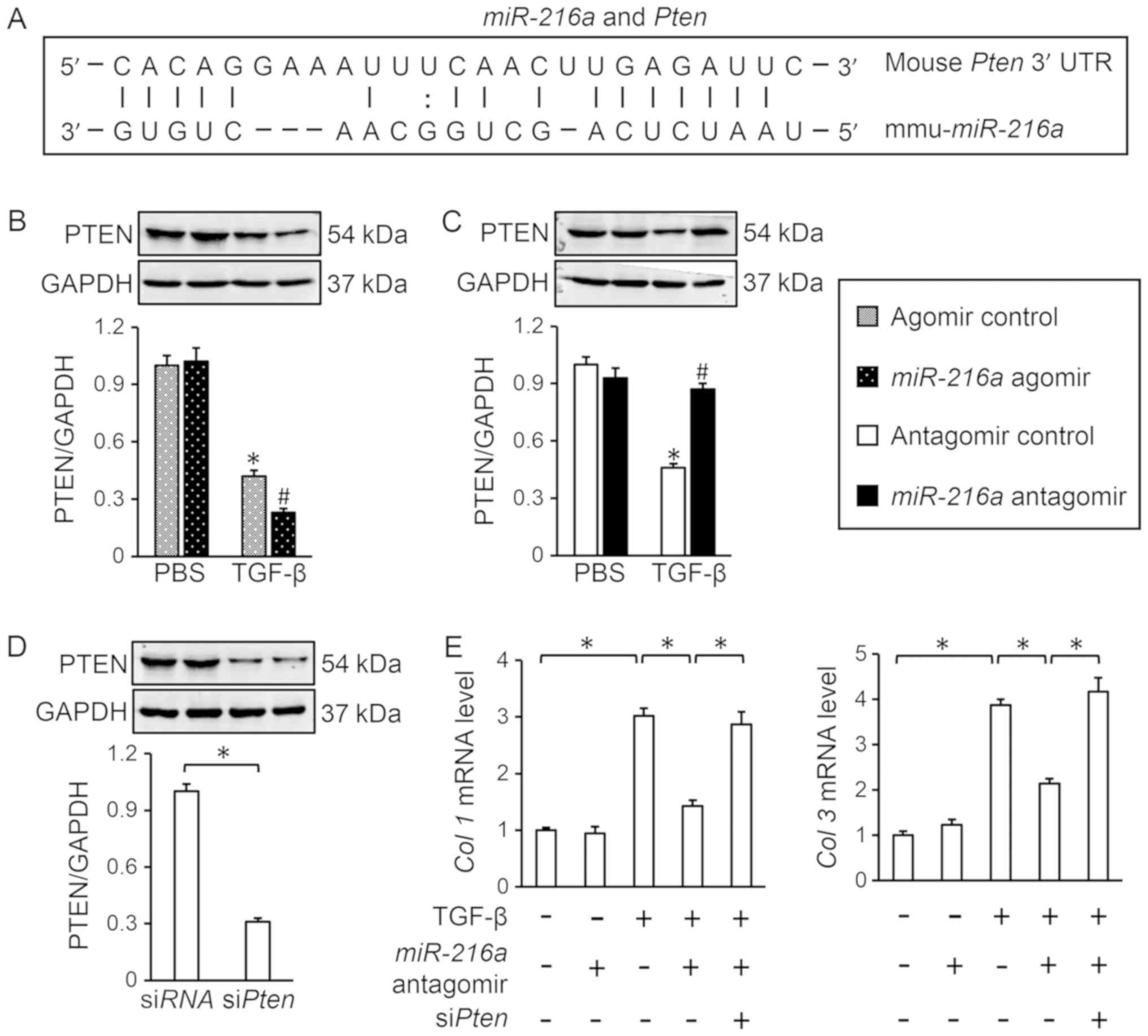
Finally, the possible mechanisms underlying the
miR-216a-mediated activation of AKT were investigated.
miRNAs exert biological regulation on various pathophysiological
procedures via complementary binding to cognate mRNA transcripts,
and subsequent degradation of the targeted transcripts (12). Among the miR-216a target
genes predicted using TargetScan was Pten, a negative
regulator of AKT signaling (Fig.
4A) (33). It was observed
that the miR-216a agomir further decreased PTEN expression
in response to TGF-β in cardiac fibroblasts (Fig. 4B), while miR-216a antagomir
significantly attenuated TGF-β-induced PTEN reduction compared with
the control (Fig. 4C). To
determine the role of PTEN in AKT activation by miR-216a,
the expression of PTEN was knocked down in cardiac fibroblasts
using siPten (Fig. 4D).
RT-qPCR analysis revealed that PTEN knockdown abolished the
miR-216a antagomir-mediated effects on TGF-β-induced
myofibroblast transdifferentiation, as determined by the mRNA
levels of Col 1 and Col 3 (Fig. 4E). Thus, the present data suggested
that miR-216a may activate AKT by inhibiting PTEN.
Discussion
Myofibroblast transdifferentiation enhances collagen
synthesis and is responsible for the occurrence of cardiac
fibrosis; however, there is no available strategy to effectively
suppress this pathological process (1,27).
In the present study, it was observed that miR-216a
expression was upregulated in TGF-β-treated cardiac fibroblasts,
which in turn activated the AKT/GSK3β signaling pathway and induced
myofibroblast transdifferentiation. Furthermore, it was revealed
that AKT inhibition abolished the miR-216a agomir-mediated
acceleration of myofibroblast transdifferentiation, and that
miR-216a activated AKT via the inhibition of PTEN. The
results indicated that miR-216a is involved in myofibroblast
transdifferentiation, and that targeting of miR-216a may aid
the development of efficacious interventions to treat cardiac
fibrosis.
In response to mechanical or neurohumoral
stimulation, cardiac fibroblasts transdifferentiate into
myofibroblasts that produce large amounts of ECM and trigger the
fibrotic process (1).
Additionally, myofibroblasts secrete various factors that
accelerate cardiac remodeling via autocrine and paracrine pathways
(34). Nagpal et al
(17) demonstrated that inhibiting
the fibroblast-to-myofibroblast transition is required for the
treatment of human cardiac fibrosis. miRNAs are now considered to
be important regulators of gene expression in various
pathophysiological processes (11). Previous studies have reported that
miRNAs are specifically involved in the regulation of cardiac
fibrosis (15–17). The present data demonstrated that
an miR-216a agomir enhanced TGF-β-induced myofibroblast
transdifferentiation, whereas an miR-216a antagomir
inhibited this process and decreased ECM synthesis. It was
previously reported that miR-216a was upregulated in
TGF-β-treated mouse glomerular mesangial cells, leading to
glomerular mesangial cell survival and hypertrophy (18). Additionally, it was observed that
miR-216a mediated TGF-β-induced collagen expression in
kidney cells (23). The heart
comprises numerous types of cells; previous studies have identified
important roles for various other cell types in the regulation of
cardiac fibrosis, in addition to cardiac fibroblasts. For example,
activation of M2 macrophages is associated with cardiac fibrosis
(35,36). Yang et al (22) demonstrated that miR-216a
promoted M1 macrophage polarization via Smad3 activation. The
present study demonstrated that transfection with an
miR-216a antagomir did not affect Smad3 phosphorylation, but
it increased the phosphorylation of AKT/GSK3β. These results
suggested that the functional effects of miR-216a are cell
type-dependent, and that upregulation of miR-216a in cardiac
fibroblasts may lead to cardiac fibrosis. These data collectively
provided rationale for the treatment of myofibroblast
transdifferentiation and cardiac fibrosis via the targeting of
miR-216a in cardiac fibroblasts.
AKT/GSK3β signaling serves an important role in the
pathological fibrotic response (8). AKT is phosphorylated and activated in
response to fibrotic stimulation, and AKT inhibition alleviates
pressure overload-induced cardiac fibrosis (37). Activated AKT phosphorylates and
inactivates GSK3β, which is also an important regulator of cardiac
fibrosis (32). Lal et al
(32) previously demonstrated that
GSK3β physically interacted with Smad3, inhibiting its
transcriptional activity. Specific deletion of GSK3β in cardiac
fibroblasts induced a profibrotic myofibroblast phenotype and
contributed to the pathogenesis of cardiac fibrosis post-myocardial
infarction (32). In the present
study, it was revealed that an miR-216a agomir further
enhanced TGF-β-induced AKT/GSK3β phosphorylation; conversely, an
miR-216a antagomir inhibited AKT/GSK3β activation.
Inhibition of AKT attenuated the miR-216a agomir-mediated
promotion of myofibroblast transdifferentiation. Of note, an
alteration in Smad3 phosphorylation was not observed, suggesting
that the AKT/GSK3β pathway also contributes to fibrotic regulation,
independent of Smad3. TGFBR2 has been identified as the primary
receptor of TGF-β, and it delivers its profibrotic signal from the
cell membrane into the cytoplasm (38); however, it was revealed that TGFBR2
protein levels were unchanged following transfection with the
agomir or antagomir of miR-216a, with or without TGF-β.
Collectively, these data indicated that the effects of
miR-216a on myofibroblast transdifferentiation may be
specifically mediated by the downstream, non-canonical AKT/GSK3β
signaling pathway independent of TGFBR2. PTEN is the main negative
regulator of AKT signaling, and its inhibition results in the
accumulation of phosphatidyl (3,4,5)-trisphosphate, mimicking the effect of
PI3K activation and inducing the activation of downstream AKT
signaling (33). In the present
study, it was observed that miR-216a was predicted to bind
the 3′-UTR of Pten, thus potentially leading to the
downregulation of PTEN protein levels and subsequent activation of
AKT. Functional experiments confirmed that transfection with the
miR-216a agomir decreased PTEN levels in combination with
TGF-β, whereas the miR-216a antagomir alleviated the
TGF-β-induced downregulation of PTEN. Pten knockdown
attenuated the beneficial effects of the miR-216a antagomir.
Nie et al (39) recently
reported that PTEN downregulation by miR-217 enhances the
proliferation of fibroblasts and accelerates collagen synthesis. In
the present study, it was demonstrated that downregulation of PTEN
by miR-216a promoted fibrotic progress via activation of the
downstream AKT/GSK3β pathway.
In conclusion, it was revealed that miR-216a
exacerbated TGF-β-induced myofibroblast transdifferentiation via
the PTEN-dependent activation of AKT/GSK3β signaling. The present
study identified roles and underlying mechanisms of miR-216a
in myofibroblast transdifferentiation, suggesting that
miR-216a may be a novel therapeutic target for the treatment
of cardiac fibrosis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Nature
Science Foundation of Hubei Province (grant no. 2014CFA061), the
Foundation Research Funds for the Central Research Funds for the
Central Universities, China (grant nos. 2042016kf0082 and
2042017kf0158) and the Major Program of Technological Innovation of
Hubei Province (grant no. 2016ACA153).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CQ and BY contributed to the conception and design
of the experiments. XL, TY, LW, SL and XZ performed the
experiments. CQ, GW, JL and SS analyzed the experimental results
and interpreted the data. CQ and BY drafted and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All of the animal experimental protocols were
approved by the Animal Care and Use Committee of Renmin Hospital of
Wuhan University (approval no. 20171003).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kong P, Christia P and Frangogiannis NG:
The pathogenesis of cardiac fibrosis. Cell Mol Life Sci.
71:549–574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nguyen TP, Qu Z and Weiss JN: Cardiac
fibrosis and arrhythmogenesis: The road to repair is paved with
perils. J Mol Cell Cardiol. 70:83–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Coronel R, Wilders R, Verkerk AO,
Wiegerinck RF, Benoist D and Bernus O: Electrophysiological changes
in heart failure and their implications for arrhythmogenesis.
Biochim Biophys Acta. 1832:2432–2441. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Teekakirikul P, Eminaga S, Toka O, Alcalai
R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, et al:
Cardiac fibrosis in mice with hypertrophic cardiomyopathy is
mediated by non-myocyte proliferation and requires Tgf-β. J Clin
Invest. 120:3520–3529. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
TGF-β: The master regulator of fibrosis. Nat Rev Nephrol.
12:325–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Khalil H, Kanisicak O, Prasad V, Correll
RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, et al:
Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac
fibrosis. J Clin Invest. 127:3770–3783. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Molkentin JD, Bugg D, Ghearing N, Dorn LE,
Kim P, Sargent MA, Gunaje J, Otsu K and Davis J:
Fibroblast-specific genetic manipulation of p38 mitogen-activated
protein kinase in vivo reveals its central regulatory role in
fibrosis. Circulation. 136:549–561. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma ZG, Yuan YP, Zhang X, Xu SC, Wang SS
and Tang QZ: Piperine attenuates pathological cardiac fibrosis via
PPAR-γ/AKT pathways. EBioMedicine. 18:179–187. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vivar R, Humeres C, Ayala P, Olmedo I,
Catalán M, Garcia L, Lavandero S and Diaz-Araya G: TGF-β1 prevents
simulated ischemia/reperfusion-induced cardiac fibroblast apoptosis
by activation of both canonical and non-canonical signaling
pathways. Biochim Biophys Acta. 1832:754–762. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zaidi SH, Huang Q, Momen A, Riazi A and
Husain M: Growth differentiation factor 5 regulates cardiac repair
after myocardial infarction. J Am Coll Cardiol. 55:135–143. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang X, Tsitsiou E, Herrick SE and
Lindsay MA: MicroRNAs and the regulation of fibrosis. FEBS J.
277:2015–2021. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dai Y, Khaidakov M, Wang X, Ding Z, Su W,
Price E, Palade P, Chen M and Mehta JL: MicroRNAs involved in the
regulation of postischemic cardiac fibrosis. Hypertension.
61:751–756. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matkovich SJ, Wang W, Tu Y, Eschenbacher
WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM and Dorn GN II:
MicroRNA-133a protects against myocardial fibrosis and modulates
electrical repolarization without affecting hypertrophy in
pressure-overloaded adult hearts. Circ Res. 106:166–175. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pan Z, Sun X, Shan H, Wang N, Wang J, Ren
J, Feng S, Xie L, Lu C, Yuan Y, et al: MicroRNA-101 inhibited
postinfarct cardiac fibrosis and improved left ventricular
compliance via the FBJ osteosarcoma oncogene/transforming growth
factor-β1 pathway. Circulation. 126:840–850. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nagpal V, Rai R, Place AT, Murphy SB,
Verma SK, Ghosh AK and Vaughan DE: MiR-125b is critical for
fibroblast-to-myofibroblast transition and cardiac fibrosis.
Circulation. 133:291–301. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kato M, Putta S, Wang M, Yuan H, Lanting
L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, et al:
TGF-beta activates Akt kinase through a microRNA-dependent
amplifying circuit targeting PTEN. Nat Cell Biol. 11:881–889. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hou BH, Jian ZX, Cui P, Li SJ, Tian RQ and
Ou JR: miR-216a may inhibit pancreatic tumor growth by targeting
JAK2. FEBS Lett. 589:2224–2232. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xia H, Ooi LL and Hui KM:
MicroRNA-216a/217-induced epithelial-mesenchymal transition targets
PTEN and SMAD7 to promote drug resistance and recurrence of liver
cancer. Hepatology. 58:629–641. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang S, Mi X, Chen Y, Feng C, Hou Z, Hui R
and Zhang W: MicroRNA-216a induces endothelial senescence and
inflammation via Smad3/IκBα pathway. J Cell Mol Med. 22:2739–2749.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang S, Li J, Chen Y, Zhang S, Feng C, Hou
Z, Cai J, Wang Y, Hui R, Lv B and Zhang W: MicroRNA-216a promotes
M1 macrophages polarization and atherosclerosis progression by
activating telomerase via the Smad3/NF-κB pathway. Biochim Biophys
Acta Mol Basis Dis. Jan 26–2018.(Epub ahead of print). doi:
10.1016/j.bbadis.2018.06.016. View Article : Google Scholar :
|
|
23
|
Kato M, Wang L, Putta S, Wang M, Yuan H,
Sun G, Lanting L, Todorov I, Rossi JJ and Natarajan R:
Post-transcriptional up-regulation of Tsc-22 by Ybx1, a target of
miR-216a, mediates TGF-{beta}-induced collagen expression in kidney
cells. J Biol Chem. 285:34004–34015. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
National Research Council (US) Institute
for Laboratory Animal Research, . Guide for the care and use of
laboratory animals. National Academies Press; 1996
|
|
25
|
O'Connell TD, Rodrigo MC and Simpson PC:
Isolation and culture of adult mouse cardiac myocytes. Methods Mol
Biol. 357:271–296. 2007.PubMed/NCBI
|
|
26
|
Geyer FC, Li A, Papanastasiou AD, Smith A,
Selenica P, Burke KA, Edelweiss M, Wen HC, Piscuoglio S, Schultheis
AM, et al: Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT
pathway genes as drivers of breast adenomyoepitheliomas. Nat
Commun. 9:18162018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang X, Ma ZG, Yuan YP, Xu SC, Wei WY,
Song P, Kong CY, Deng W and Tang QZ: Rosmarinic acid attenuates
cardiac fibrosis following long-term pressure overload via
AMPKα/Smad3 signaling. Cell Death Dis. 9:1022018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma ZG, Yuan YP, Xu SC, Wei WY, Xu CR,
Zhang X, Wu QQ, Liao HH, Ni J and Tang QZ: CTRP3 attenuates cardiac
dysfunction, inflammation, oxidative stress and cell death in
diabetic cardiomyopathy in rats. Diabetologia. 60:1126–1137. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ma ZG, Dai J, Yuan YP, Bian ZY, Xu SC, Jin
YG, Zhang X and Tang QZ: T-bet deficiency attenuates cardiac
remodelling in rats. Basic Res Cardiol. 113:192018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. Aug 12–2015.(Epub ahead of print). doi: 10.7554/eLife.05005.
2015. View Article : Google Scholar
|
|
32
|
Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi
RJ, Guo Y, Yu D, Tsai EJ, Woodgett J, Gao E and Force T: Cardiac
fibroblast glycogen synthase kinase-3β regulates ventricular
remodeling and dysfunction in ischemic heart. Circulation.
130:419–430. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shojaee S, Chan LN, Buchner M, Cazzaniga
V, Cosgun KN, Geng H, Qiu YH, von Minden MD, Ernst T, Hochhaus A,
et al: PTEN opposes negative selection and enables oncogenic
transformation of pre-B cells. Nat Med. 22:379–387. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee AA, Dillmann WH, McCulloch AD and
Villarreal FJ: Angiotensin II stimulates the autocrine production
of transforming growth factor-beta 1 in adult rat cardiac
fibroblasts. J Mol Cell Cardiol. 27:2347–2357. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shirakawa K, Endo J, Kataoka M, Katsumata
Y, Yoshida N, Yamamoto T, Isobe S, Moriyama H, Goto S, Kitakata H,
et al: IL (Interleukin)-10-STAT3-galectin-3 axis is essential for
osteopontin-producing reparative macrophage polarization after
myocardial infarction. Circulation. 138:2021–2035. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Horckmans M, Ring L, Duchene J, Santovito
D, Schloss MJ, Drechsler M, Weber C, Soehnlein O and Steffens S:
Neutrophils orchestrate post-myocardial infarction healing by
polarizing macrophages towards a reparative phenotype. Eur Heart J.
38:187–197. 2017.PubMed/NCBI
|
|
37
|
Ma ZG, Zhang X, Yuan YP, Jin YG, Li N,
Kong CY, Song P and Tang QZ: A77 1726 (leflunomide) blocks and
reverses cardiac hypertrophy and fibrosis in mice. Clin Sci (Lond).
132:685–699. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bottinger EP, Jakubczak JL, Roberts IS,
Mumy M, Hemmati P, Bagnall K, Merlino G and Wakefield LM:
Expression of a dominant-negative mutant TGF-beta type II receptor
in transgenic mice reveals essential roles for TGF-beta in
regulation of growth and differentiation in the exocrine pancreas.
EMBO J. 16:2621–2633. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nie X, Fan J, Li H, Yin Z, Zhao Y, Dai B,
Dong N, Chen C and Wang DW: miR-217 promotes cardiac hypertrophy
and dysfunction by targeting PTEN. Mol Ther Nucleic Acids.
12:254–266. 2018. View Article : Google Scholar : PubMed/NCBI
|