Introduction
Alzheimer's disease (AD) is the most common
neurodegenerative cause of dementia and it is a progressive
neurodegenerative disorder of the central nervous system, which
leads to significant individual morbidity, mortality and economic
impact on the health care system (1,2). AD
may be characterized by a marked dsyregulation of the cholinergic
system, together with other neurotransmitter systems (including
glutamate and serotonin). Acetylcholinesterase (AChE) inhibitors
donepezil, galantamine along with rivastigmine and the
N-methyl-D-aspartate antagonist memantine are examples of the
current standard treatments, which do not inhibit the progression
of AD and offer marginal therapeutic advantages (3). Even though novel therapeutic methods
have appeared in recent years, there have been poor clinical
results, which makes cholinesterase inhibitors the primary approach
for AD treatment (3–5). The acetylcholine (Ach) signaling
pathway is additionally inhibited through the action of AChE
(ExPASy enzyme entry no. E.C. 3.1.1.7) as well as
butyrylcholinesterase (BChE; ExPASy enzyme entry no. E.C. 3.1.1.8)
(6–8), which regulates the levels of ACh by
hydrolysis (4). Controlled
inhibition of brain AChE and BChE may slow neurodegeneration in AD
(9–13).
The concentration of choline may be increased
through AChE in addition to BChE inhibition, by the application of
the
[1,1′-([1,1′-biphenyl]-4,4′-diyl)bis(3-(piperidin-1-yl)propan-1-one)
dihydrochloride; DL0410]-. DL0410 had been selected from
>100,000 compounds using high-throughput screening assays for
AChE in addition to BChE inhibitors (14). Subsequently, the in vitro
ability of DL0410 to inhibit AChE and BChE was determined with half
maximal inhibitory concentration (IC50) values of
0.286±0.004 and 3.962±0.099 µmol/l, respectively, which are
comparable with the donepezil as well as rivastigmine (15). The authors' previous study
demonstrated that DL0410 binds to the active-site groove of AChE
(16), whereas other previous
studies (14–18) confirmed its efficacy and safety.
Additionally, DL0410 was able to inhibit histamine receptor 3
(H3R) with IC50 values of 0.308±0.003 µmol/l.
H3Rs function as regulators that modulate the release of
ACh, dopamine, serotonin and norepinephrine (19). Consequently, H3R
antagonists have potential as a treatment for AD (19–22).
It was additionally demonstrated that DL0410 has the ability to
improve memory deficits in APP/PS1 transgenic mice and
Aβ1-42-induced amnesia in mice (15). Cholinesterase inhibition, Aβ plaque
inhibition, in addition to enhancement of synapse loss through the
regulation of synapse-associated protein expression may serve an
important role in beneficial effects of DL0410. As a result, DL0410
as multi-target-directed ligand may be considered as a candidate
drug for AD treatment (23).
In the present study, a sensitive, reproducible and
practical method was developed and used for the determination of
the pharmacokinetic properties of DL0410 in Sprague-Dawley (SD) rat
plasma and brain tissue for the first time. In addition, this is
the first study to the best of the authors the knowledge to
investigate the dose proportionality and bioavailability of DL0410.
The excretion of DL0410 was determined by liquid
chromatography-mass spectrometry (LC-MS) following liquid-liquid
extraction for biliary, urinary and fecal samples. Furthermore,
ultra-high performance liquid chromatography-quadrupole
time-of-flight mass spectrometry (UPLC-Q-TOF/MS) with electrospray
ionization (ESI) were additionally applied to identify the primary
metabolites of DL0410 in rat bile, urine and feces. The present
study aimed to provide preclinical pharmacokinetic information
regarding DL0410 and lay a foundation for evaluating the clinical
efficiency of DL0410 for oral administration.
Materials and methods
Reagents
DL0410 (purity >99%) was synthesized by the
Institute of the Materia Medica, Chinese Academy of Medical
Sciences (Beijing, China). Phenacetin (internal standard; IS) was
obtained from the National Institutes for Food and Drug Control
(Beijing, China). Acetonitrile (ACN; CH3CN; LC-MS-grade)
and methanol (MeOH; LC-MS grade) were purchased from J.T. Baker;
Thermo Fisher Scientific, Inc., (Waltham, MA, USA). Formicacid
[HCOOH; high (H)PLC-grade] was obtained from Tedia (Fairfield, CT,
USA). Recombinant CYP450s (CYP1A2, CYP2C9, CYP2D6 and CYP3A4) were
obtained from Corning Life Sciences (Tewksbury, MA, USA). Potassium
phosphate buffer (pH 7.4) was purchased from Beijing Solarbio
Science & Technology Co., Ltd. (Beijing, China). Ethyl acetate
(EtOAc) was purchased from Sinopharm Chemical Reagent Co., Ltd.
(Shanghai, China). Anticoagulation tubes with heparin were obtained
from Jiangsu Kangjian Healthcare Co., Ltd., (Taizhou, China). Pure
water was purchased from the Hangzhou Wahaha Company (Hangzhou,
China), with all other chemical reagents being of analytical grade
level or higher.
Experimental animals
A total of 90 rats were supplied by Beijing HFK
Bioscience Co., Ltd. (Beijing, China; cat. no. SCXK 2014-0004). All
experimental protocols involving 45 male and 45 female
Sprague-Dawley (SD) rats of 6–8 weeks old (210–230 g) were reviewed
and approved by the animal experimentation center of the Institute
of Materia Medica, Chinese Academy of Medical Sciences (Beijing,
China). The animals were allowed to acclimatize in the animal
facilities for ~7 days following arrival, complete with air
conditioning and an automatically controlled photoperiod of 12 h of
daylight. The temperature of the rearing room was maintained at
20–24°C and the relative humidity was 50–65%. Animals were given
free access to food and water for these 12 h prior to being used in
the experiments, during this time only the food was removed.
High performance LC-MS for plasma,
brain, bile, urine and fecal samples
The Agilent 1200 liquid chromatography-6100 mass
spectrometer (Agilent Technologies, Inc., Santa Clara, CA, USA) was
employed for the detection of DL0410 and phenacetin (IS). The
analytical column was Agilent Zorbax SB-C18 (100×2.1 mm; 3.5 µm;
Agilent Technologies, Inc.). The mobile phase was composed of
methanol-ACN-water (0.5% formic acid; 15:10:75, v/v) for plasma and
brain samples, and (15:9:76, v/v) for bile, urine, and fecal
samples. The injection volume was 10 µl and the flow rate was 0.3
ml/min. The column temperature was maintained at 35°C and the mass
spectrometer was used in the positive scan mode. The conditions of
ESI source were as follows: Drying gas flow was set at 10 l/min,
drying gas temperature was 350°C, nebulizer pressure was 35.0
pounds per square inch gauge and capillary voltage was 3,000 V. The
ESI was conducted using nitrogen to assist nebulization. The
typical compound parameters, fragmentor voltage and gain value,
were set at 90 V and 1.5, respectively. The MS detector was
operated in selective ion monitoring mode using the quantification
ions [M+H]+ at m/z 217.15 for DL0410 and m/z 180.22 for
IS.
UPLC-Q-TOF-MS for metabolite
identification
Bile, urine and fecal metabolites were analyzed by
UPLC-Q-TOF-MS (XEVO G2; Waters Corporation, Milford, MA, USA) with
an Agilent Zorbax SB-C18 (100×2.1 mm; 3.5 µm) by gradient elution
using 0.2% formic acid in ACN (A) and 0.2% formic acid in water (B)
at a flow rate of 0.4 ml/min. The gradient profile was as follows:
0–2 min (A, 5%), 2–12 min (A, 2–20%), 12–14 min (A, 20–30%), 14–18
min (A, 30–100%), 18–21 min (A, 100%), 21–21.10 min (A, 100–5%) and
21–25 min (A, 5%), and was held for 2.5 min for the following run.
The injection volume was 10 µl and the temperature of the column
oven was set to 35°C. Ionization was performed in the positive ESI
mode. For MS detection, the optimum ESI conditions were as follows:
Source temperature of 100°C, desolvation temperature of 300°C,
desolvation gas flow of 900 l/h, cone gas flow of 50 l/h, capillary
voltage of 3.0 kV, sample cone voltage of 40 V, extraction cone of
1 eV and a scan range of 50–1,000 m/z.
Interaction of DL0410 with CYP450s in
vitro and in silico
Metabolism of DL0410 in recombinant CYP450s (CYP1A2,
CYP2C9, CYP2D6 and CYP3A4) was conducted initially by DL0410 (10
µM) in a typical incubation system, containing potassium phosphate
buffer (pH 7.4), NADPH-generating system and the appropriate
concentration recombinant CYP450s. Following pre-warming at 37°C
for 5 min, recombinant CYP450s (100 pmol/l) were added and
incubated at 37°C for 2 h. The reaction was terminated by adding
2-fold of ACN for protein precipitation and DL0410 extraction. The
mixture was centrifuged at 1,204 × g at room temperature for 10 min
and an aliquot of supernatant was transferred for LC-MS/MS
analysis. The CDOCKER protocol in Discovery Studio (DS) 2016
(Accelrys Software, Inc., San Diego, CA, USA) was utilized in the
present study to investigate the binding mode of DL0410 with
CYP450s, and the crystal structure of CYP450s were downloaded from
Protein Data Bank (PDB; identification no. 3QM4) (24). Prior to using the docking program,
the Prepare Protein tool, which is a plugin of DS 2016 was used to
perform a series of tasks, including inserting missing atoms in
incomplete residues, modeling missing loop regions, deleting
alternate conformations, removing waters, standardizing atom names
and protonating titratable residues.
Method validation
The LC-MS method for detecting the concentration of
DL0410 in the blood and brain samples was validated by examining
the specificity and sensitivity, linearity, precision and accuracy,
recovery, stability and the matrix effect. The precision and
accuracy for DL0410 were evaluated by quality control (QC) samples
(n=6) with low, medium and high concentrations (15.63, 62.50 and
500 ng/ml) on the same day and 5 independent days. A total of three
QC concentration levels in six replicates were used to examine the
extraction recovery, by recording the peak response of the
plasma/brain tissue and comparing it with the normal samples. The
short and long-term freezing and thawing stability of DL0410 in
plasma/brain tissue were evaluated by analysis of the QC samples at
three concentration levels. The short and long-term stability was
tested by storing the samples for 24 h at room temperature, 4°C and
−40°C for 4 weeks. The freezing-thawing stability was analyzed
following three freeze and thaw cycles, and the matrix effect was
additionally examined at three QC concentration levels by comparing
the peak area ratio of the neat standard in post-extracted samples
to those of neat standard solutions.
Experimental design and sample
collection
A total of 90 rats were used for plasma, brain,
bile, urine and feces sample collection. To collect the plasma
samples, 24 SD rats (12 males and 12 females) were separated into
four parallel designed groups at random. The drug was administered
orally in groups 1–3 (25, 50 and 100 mg/kg, respectively) and
intravenously (5 mg/kg) in group 4. Each group consisted of six
rats (three male and three female rats). The blood samples were
collected from the eye venous plexus (25). The blood samples were collected at
intervals of 0, 0.25, 0.33, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8, 12, 24,
36 and 48 h for oral administration, and 0.083, 0.25, 0.33, 0.5,
0.75, 1, 1.5, 2, 4, 6, 8, 12, 24, 36 and 48 h for intravenous
administration. Brain samples (n=6) of 54 SD rats (three males and
three females) were collected at 0, 0.25, 0.75, 1, 2, 4, 8, 12 and
24 h following oral administration of DL0410 (100 mg/kg). Normal
saline was subsequently used to immediately rinse the hippocampus
(HIP) and entorhinal cortex (EC), and blotted with filter paper.
Normal saline was used to homogenize the accurately weighed
tissues, which were subsequently stored at −40°C pending analysis.
Rats (n=6; sex ratio 3:3) were anesthetized with ether and the bile
duct was cannulated with tubing (Portex Nylon tubing; 0.75 mm
internal diameter; 0.94 mm external diameter). Bile samples were
collected via the bile cannula prior to drug administration as a
control and at 0–3, 3–6, 6–12, 12–24 and 24–36 h following
administration. Urine and feces were collected from six rats (sex
ratio 3:3). Urine was collected while the rats remained in isolated
metabolic cages at 0–2, 2–4, 4–6, 6–8, 8–12, 12–24, 24–48 and 48–72
h, and the volume was measured. Lastly, fecal samples were
collected at 0–12, 12–24, 24–36, 36–48 and 48–72 h. The fecal
samples were dried and crushed, and measured for volume. Bile,
urine and feces for the metabolite investigation were collected
between 2–8 h following oral administration of 100 mg/kg
DL0410.
Preparation of calibration standards
and QC samples
Primary stock solutions of DL0410 and IS were
separately prepared by forming a 1 mg/ml analyte/methanol solution,
and stored at −40°C. Stock solutions were diluted to working
solutions with methanol for use. Corresponding model working
solutions were spiked with 100 µl blank rat plasma and brain
tissue, and subsequently used as QC samples, at concentrations of
7.81 [lower limit of quantification (LLOQ)], 15.63, 62.50 and 500
ng/ml. To construct the plasma and brain sample calibration curves,
a series of working standard solution concentrations (7.81, 15.63,
31.25, 62.50, 125, 250, 500 and 1,000 ng/ml DL0410) were similarly
prepared. The seven-point concentration curve of biliary samples,
urinary samples and fecal samples were 15.63–1,000, 31.25–2,000 and
31.25–2,000 ng/ml, respectively.
Sample preparation
Rat plasma, brain, bile, urine and fecal samples
(100 µl) and 10 µl of IS solution (500 ng/ml) were mixed, with
1,000 µl ethyl acetate being added to extract the analytes.
Following vortexing at room temperature for 5 min, the mixture was
centrifuged at 1,304 × g for 10 min at room temperature. The
supernatant fluid was transferred to another tube and evaporated
under a light flow of nitrogen gas, until dry. The residue was
reconstituted in 75 µl methanol-ACN-water (15:10:75; v/v,
containing 0.5% formic acid) for plasma and brain samples, and 100
µl for biliary, urinary and fecal samples. Following
centrifugation, the supernatant was injected into the LC-MS system
for analysis.
Statistical analysis
DAS 3.0 pharmacokinetic program (Chinese
Pharmacology Society, Shanghai, China) software was used to
calculate the pharmacokinetic parameters and a non-compartment
model analysis was employed. The absolute bioavailability
(Fabs%) was calculated as follows (26):
Fabs%=AUCpo × Div/AUCiv × Dpo ×100
AUCpo, area under the curve of oral administration;
AUCiv, area under the curve of intravenous injection; Dpo, dose of
oral administration; Div, dose of intravenous injection.
Standard deviations, accuracy [relative error (RE)]
and precision [relative standard deviation (RSD)] were calculated
using Microsoft Excel 2010 (Microsoft Corporation, Redmond, WA,
USA). Linear regression analysis, plasma concentration time curves
and cumulative excretion amount curves were constructed using
GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA,
USA).
Results
Validation of LC-MS method
The calibration curves demonstrated good linearity
for DL0410 in the 7.81–1,000 ng/ml range for the plasma and brain
samples, 15.63–2,000 ng/ml range for the biliary samples, and in
the 31.25–2,000 ng/ml range for the urinary and fecal samples. The
correlation coefficients (r) of DL0410 were 0.9998 in the plasma
samples, 0.9999 in the brain samples, 0.9948 in the biliary
samples, 0.9996 in the urinary samples and 0.9964 in the fecal
samples. LLOQ of plasma and brain samples was acquired with
sufficient precision and accuracy (Table I) (25).
 | Table I.Accuracy and precision of the samples
at LLOQ (7.812 ng/ml) and quality control with high, medium and low
concentration (500, 62.5 and 15.625 ng/ml, respectively; n=5). |
Table I.
Accuracy and precision of the samples
at LLOQ (7.812 ng/ml) and quality control with high, medium and low
concentration (500, 62.5 and 15.625 ng/ml, respectively; n=5).
| Analyte | Spiked concentration,
ng/ml | Mean measured
concentration, ng/ml | Relative error,
% | Intra-day RSD,
% | Inter-day RSD,
% |
|---|
| DL0410 in
plasma | 7.81 (LLOQ) | 8.12 | 3.91 | 4.41 | 8.78 |
|
| 15.63 | 15.19 | 5.27 | 3.71 | 4.91 |
|
| 62.50 | 63.10 | 3.68 | 4.46 | 2.50 |
|
| 500 | 534.20 | 6.84 | 5.62 | 3.95 |
| DL0410 in brain
sample | 7.81 (LLOQ) | 8.08 | 3.43 | 3.34 | 7.88 |
|
| 15.63 | 15.44 | −1.21 | 2.96 | 4.46 |
|
| 62.50 | 64.85 | 3.77 | 5.01 | 5.92 |
|
| 500 | 514.75 | 2.95 | 2.08 | 7.30 |
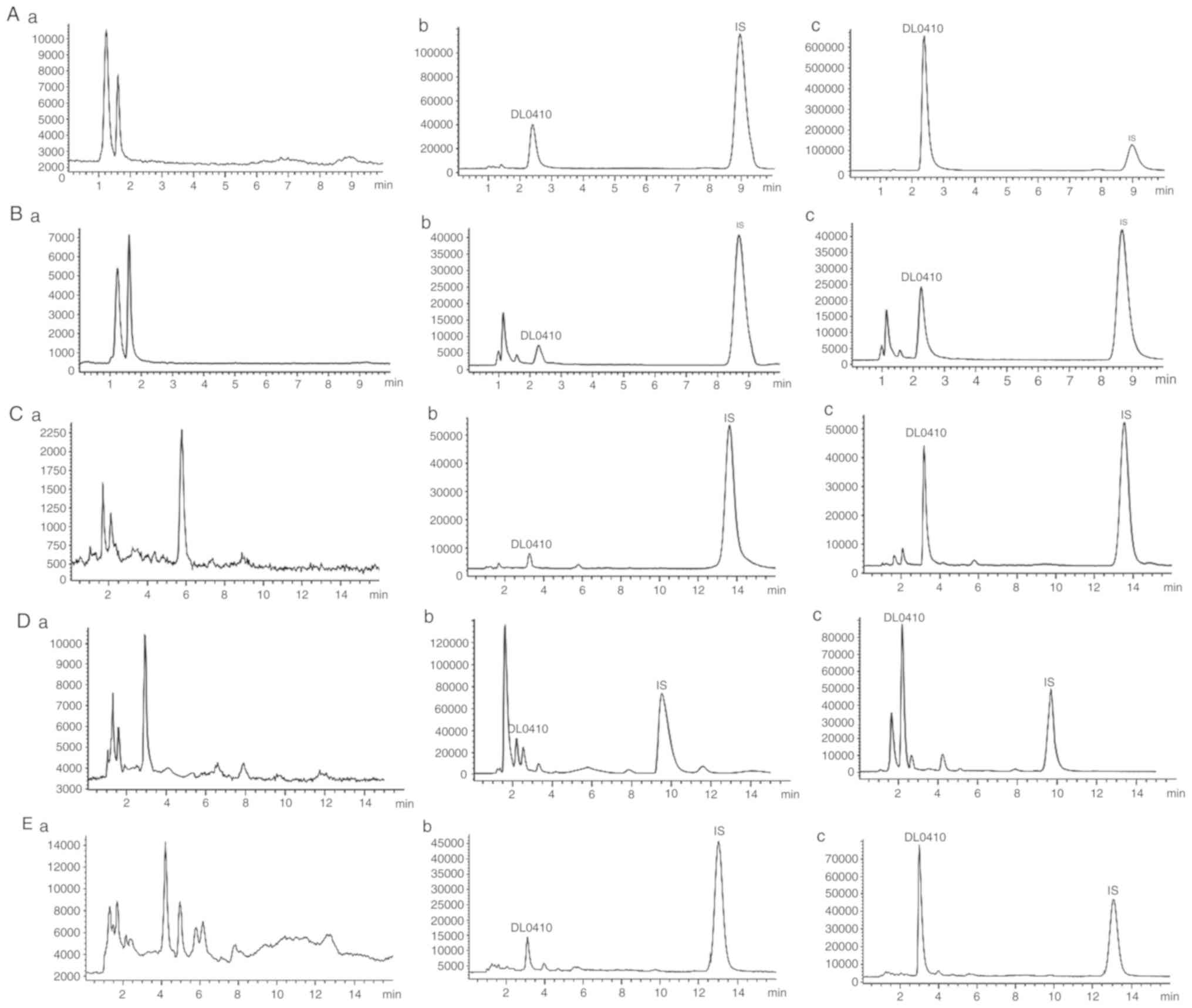
No endogenous interference with DL0410 or IS was
observed. Additionally, good separation was achieved for DL0410 and
IS. The representative chromatogram patterns of the blank samples,
samples at LLOQ and samples following oral administration are
presented in Fig. 1.
The RE and RSD values <10% (27) were within the tolerated limits for
the QCs with high, medium and low concentrations (Table I). The mean recoveries and matrix
effects for these three concentrations are presented in Table II. The RE determined from all of
the stability tests were within ±6.92% of each other (Table III). The results demonstrated
that the accuracy, precision, recovery and stability tests met the
criteria for quantitative determination in biological samples
(28).
| Table II.Recoveries and matrix effects of
DL0410 in rat plasma and brain samples (n=5). |
Table II.
Recoveries and matrix effects of
DL0410 in rat plasma and brain samples (n=5).
| Analyte | Spiked
concentration, ng/ml | Recovery, % | Recovery RSD,
% | Matrix effects,
% | Matrix effects RSD,
% |
|---|
| DL0410 in
plasma | 15.63 | 99.44 | 2.19 | 97.99 | 3.66 |
|
| 62.50 | 93.66 | 8.37 | 100.84 | 6.73 |
|
| 500 | 105.64 | 2.51 | 105.58 | 1.77 |
| DL0410 in brain
sample | 15.63 | 75.31 | 2.76 | 102.56 | 4.11 |
|
| 62.50 | 87.42 | 5.30 | 102.30 | 3.58 |
|
| 500 | 90.15 | 2.86 | 93.78 | 2.50 |
| Table III.Stability of DL0410 in rat plasma and
brain samples (n=5). |
Table III.
Stability of DL0410 in rat plasma and
brain samples (n=5).
|
|
| DL0410 in
plasma | DL0410 in brain
sample |
|---|
|
|
|
|
|
|---|
| Condition | Spiked
concentration, ng/ml | Measured
concentration, ng/ml | RE, | Measured
concentration, (ng/ml) | RE, % |
|---|
| Three freeze/thaw
cycles | 15.63 | 14.67±0.74 | −6.14 | 15.35±0.76 | −1.74 |
|
| 62.50 | 66.78±1.01 | 6.86 | 61.42±3.80 | −1.73 |
|
| 500 | 500.99±5.93 | −0.88 | 498.70±34.39 | −1.07 |
| Short-term | 15.63 | 15.37±0.47 | −1.61 | 15.434±0.74 | −1.27 |
| (25°C for 4 h) | 62.50 | 64.65±18.81 | 3.43 | 61.03±3.48 | −2.35 |
|
| 500 | 461.97±16.65 | −7.61 | 498.70±13.25 | −0.26 |
| Autosampler | 15.63 | 15.47±0.66 | −1.02 | 15.10±1.08 | −3.36 |
| (25°C for 24
h) | 62.50 | 64.03±2.15 | 2.44 | 66.04±2.87 | 5.67 |
|
| 500 | 491.76±24.80 | −1.47 | 467.64±18.90 | −6.46 |
| Long-term | 15.63 | 16.70±0.17 | 6.92 | 15.70±0.53 | 0.49 |
| (−40°C for 4
weeks) | 62.50 | 60.38±1.52 | −3.4 | 61.73±5.29 | −1.28 |
|
| 500.00 | 526.50±10.94 | 5.24 | 489.78±19.89 | −2.05 |
Method application in a
pharmacokinetic study
The established LC-MS method was successfully
applied to measure the DL0410 concentration in the rat plasma
samples following oral and intravenous administration. The time
profiles of the plasma concentration of DL0410 following oral
administration at 25, 50 and 100 mg/kg and following intravenous
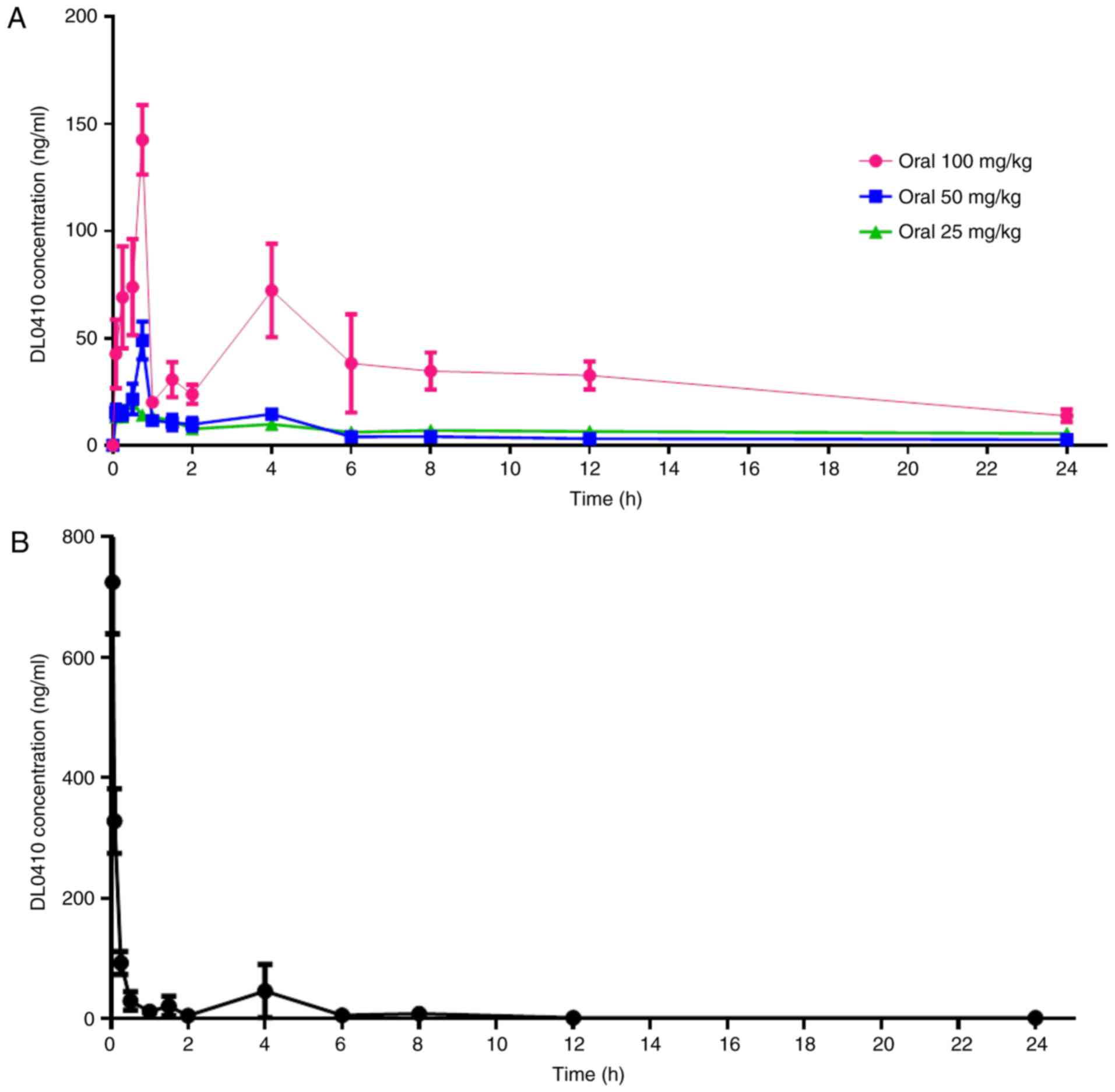
administration at a dose of 5 mg/kg are presented in Fig. 2. The primary plasma pharmacokinetic
data are summarized in Table IV.
For oral administration, during the 45 min following dose
administration, the DL0410 plasma concentrations increased rapidly
to maximum serum concentration (Cmax; 195.79±46.76 ng/ml
for 100 mg/kg; 56.15±4.01 ng/ml for 50 mg/kg; and 21.48±3.46 ng/ml
for 25 mg/kg; Fig. 2A).
Specifically, there were two peaks observed in Fig. 2. Following intravenous
administration, the plasma concentration of DL0410 decreased
(Fig. 2B). The fold increase in
oral dosage (25 vs. 50 vs. 100 mg/kg) did not lead to a
fold-increase in AUC0-t (978.14±229.53 µg/l × h vs.
238.87±63.03 µg/l × h vs. 135.40±22.41 µg/l × h; Table IV). The data demonstrated that
Cmax and AUC0-t of DL0410 did not increase in
a dose-dependent manner at doses between 25 and 100 mg/kg. The
half-life (t1/2) extended with the increasing dosage
between 6.05 and 8.23 h. The oral bioavailability of DL0410 in rats
was 10.78 and 12.19% at the low and medium doses (25 and 50 mg/kg),
respectively, and increased to 24.18% at 100 mg/kg. The
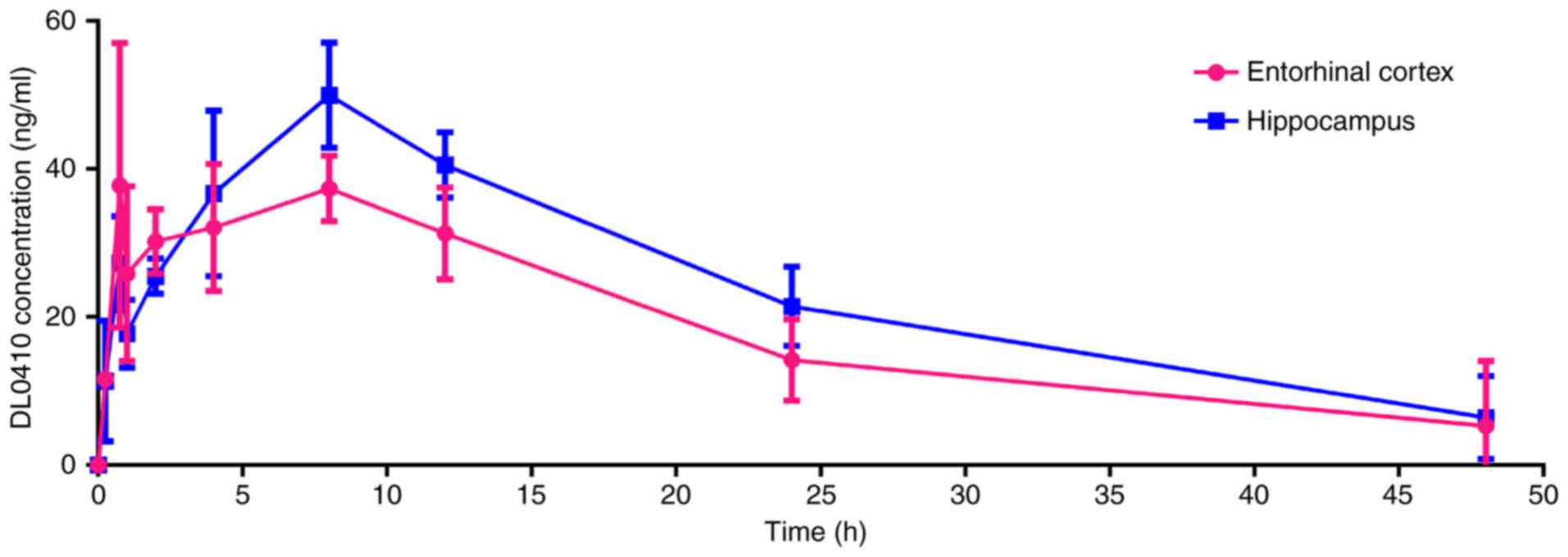
concentration-time profile of DL0410 in the EC and HIP following
oral administration at a dose of 100 mg/kg to rats is presented in
Fig. 3. Time to maximum
concentration (Tmax) of DL0410 in the two regions was ~6
h and the values of Cmax were 50.27±6.92 ng/ml and
46.23±13.17 ng/ml for HIP and EC, respectively.
| Table IV.Pharmacokinetic parameters of DL0410
following intravenous (5 mg/kg) and oral (100, 50 and 25 mg/kg)
administration in rats (n=6). |
Table IV.
Pharmacokinetic parameters of DL0410
following intravenous (5 mg/kg) and oral (100, 50 and 25 mg/kg)
administration in rats (n=6).
|
|
|
| Oral dose |
|---|
|
|
|
|
|
|---|
| Parameter | Unit | Intravenous dose 5
mg/kg | 100 mg/kg | 50 mg/kg | 25 mg/kg |
|---|
|
AUC(0-t) | µg/l × h | 251.10±183.05 | 978.14±229.53 | 238.87±63.03 | 135.40±22.41 |
|
MRT(0-t) | h | 19.91±1.88 | 12.17±1.45 | 6.73±1.20 | 4.51±0.89 |
|
t1/2z | h | 0.08±0.02 | 8.23±4.02 | 7.54±2.97 | 6.05±0.34 |
|
Tmax | h | 0.03 | 0.75±0.27 | 0.65±0.23 | 0.63±0.12 |
|
Cmax | µg/l | 724.85±85.92 | 195.79±46.76 | 56.15±4.01 | 21.48±3.46 |
|
Fabs% |
| – | 24.18±0.03 | 12.19±0.03 | 10.78±0.02 |
Excretion study
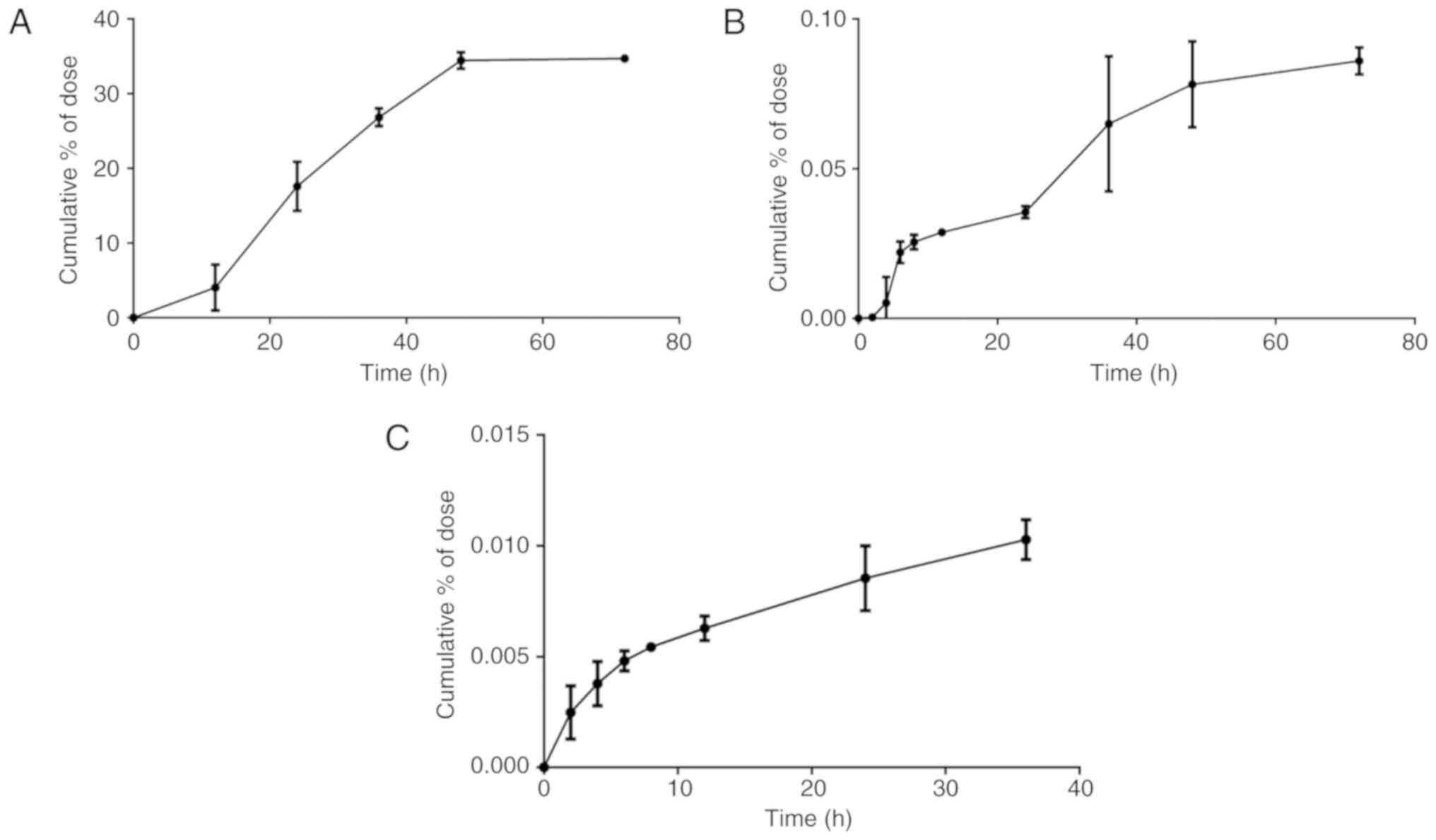
The cumulative DL0410 excretion results in rat bile,
urine and feces, following oral administration with DL0410 (100
mg/kg), are presented in Fig. 4.
The cumulative urinary excretion of DL0410 was very small, only
0.086% of the dose in 72 h. The accumulated excretion of DL0410 in
bile for 36 h was even lower, with 0.010% of the dose being
excreted. The cumulative fecal excretion of DL0410 accounted for
34.71%.
Metabolism of DL0410 identified in
vivo and in vitro
Following a full scan and subsequent product ion
scan identification of the bile, urine and fecal samples following
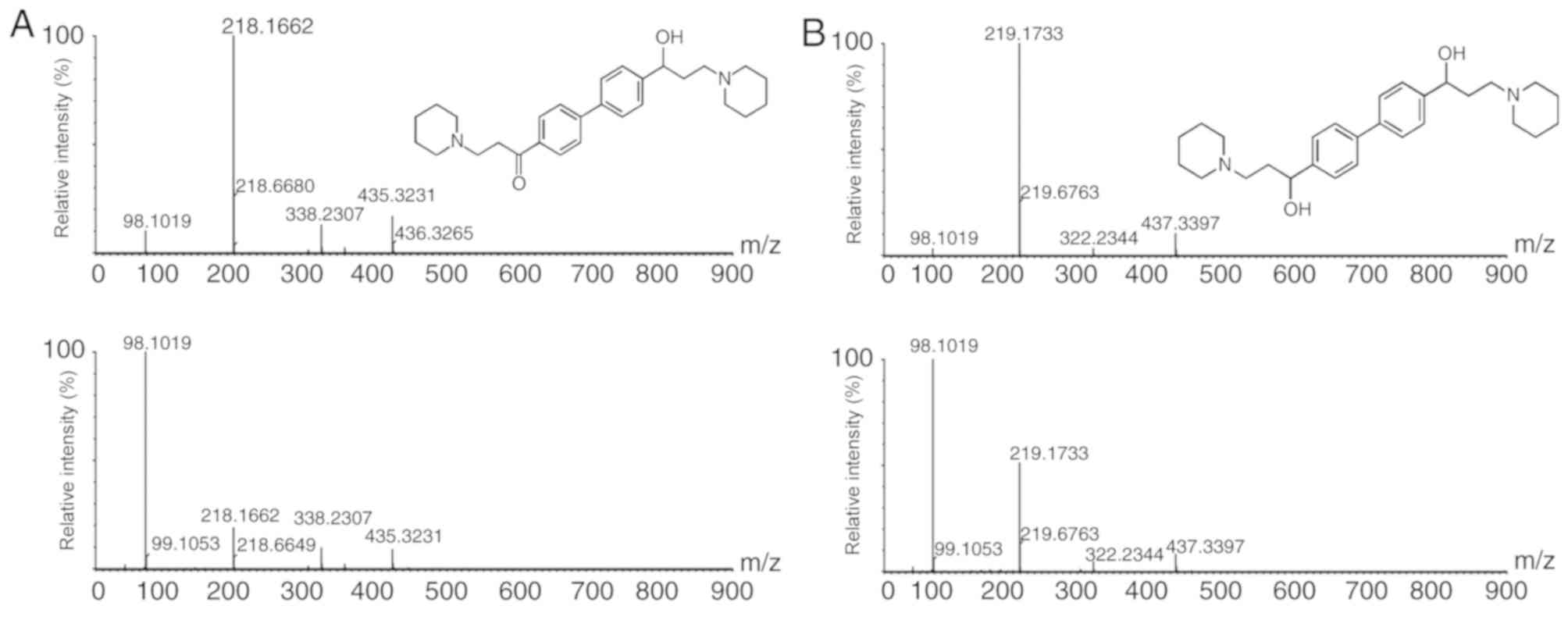
DL0410 administration, two common DL0410 metabolites at m/z
435.3231 and m/z 437.3397 were identified (Fig. 5). Recombinant human CYP450 enzymes
(CYP1A2, CYP 2C9, CYP2D6 and CYP3A4) were used to evaluate the
contribution of CYP450 enzymes to the reduction reaction. As a
result, CYP2D6 was identified to be involved in the reduction of
the carbonyl group.
Docking study of CYP2D6 and
DL0410
The molecular docking assessment between CYP2D6 and
DL0410 was performed using the CDOCKER module in the DS 2016
package. The crystal structure of CYP2D6 was obtained from the PDB
(identification no. 3QM4) (26).
The water molecules in the protein were removed, and the protein
was prepared by adding hydrogen and correcting the incomplete
residues using the Prepare Protein tool; the protein was
subsequently refined. The structure of DL0410 was prepared and was
followed by hydrogen addition, conversion into a 3D structure, pH
based ionization and charge neutralization. The binding site of
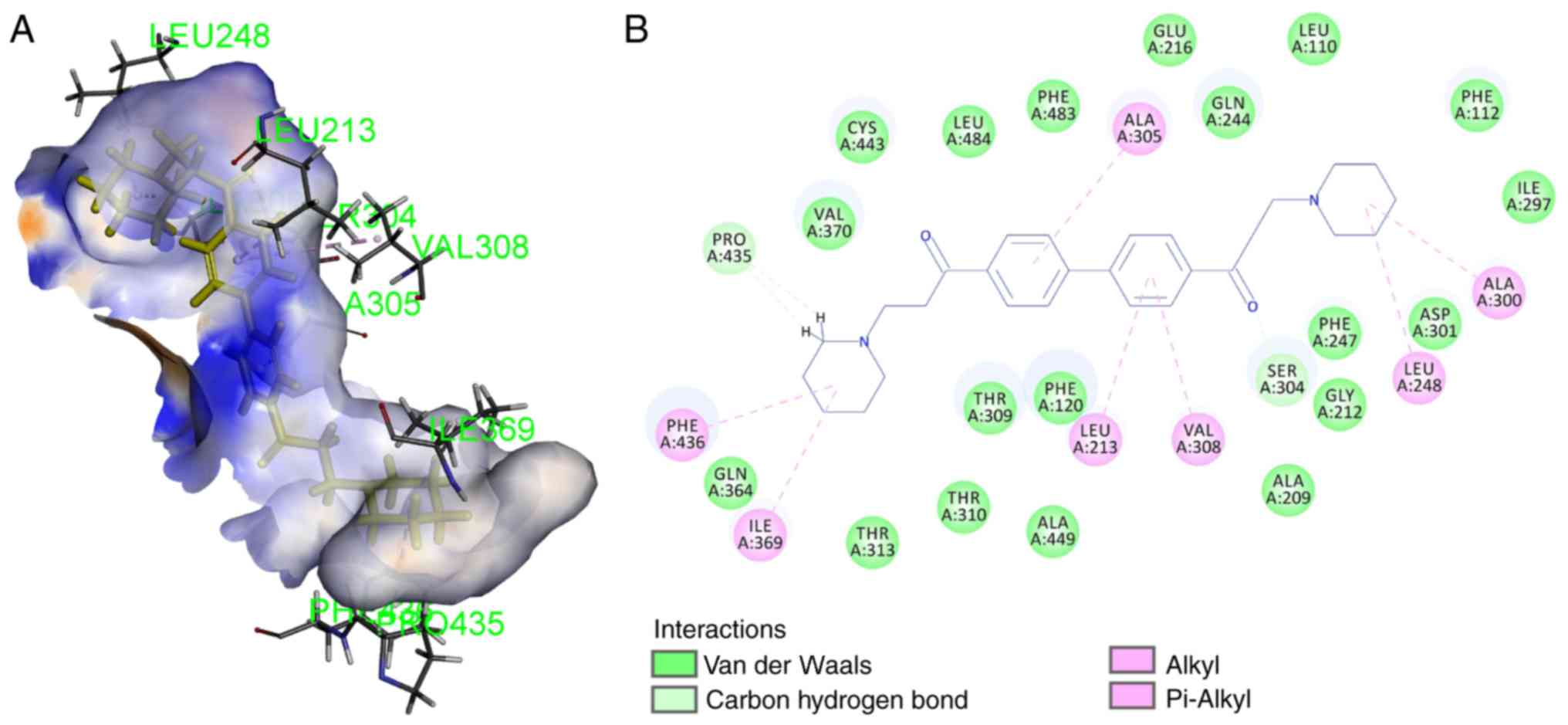
CYP2D6 was defined by the ligand prinomastat. To validate the
docking approach for the protein structure used, prinomastat was
redocked to the active site. The root-mean-square deviation was
0.6229, which indicated the reliability of this docking model. The
results demonstrated that DL0410 has a moderate interaction with
CYP2D6 by residues of ILE213, LEU248, ALA200, VAL308, ILE369,
PRO435 and PHE436 (Fig. 6).
-CDOCKER ENERGY and -CDOCKER INTERACTION ENERGY (a higher value
indicates a more favorable binding) were 24.3048 and 59.4411,
respectively, which suggests DL0410 has a good affinity with CYP2D6
in silico.
Discussion
DL0410, a novel cholinesterase inhibitor and H3R
antagonist, is currently under preclinical development for use in
AD treatment. It is crucial to investigate the pharmacokinetic
profile of DL0410 for its demonstrated effectiveness in AD
treatment. The present study aimed to evaluate the bioavailability
and dose proportionality of DL0410 in rats with a validated method
of LC-MS.
Following oral and intravenous administration of
DL0410, respectively, pharmacokinetic parameters were acquired. The
results demonstrated that DL0410 exhibited a fast phase of
absorption, reaching Cmax at Tmax of nearly
45 min following the three orally administered doses. There were
double peaks in the concentration-time profiles of oral
administration of DL0410. The possible mechanisms are primarily
associated with enterohepatic circulation, gastric emptying delay,
the difference of absorption in various intestinal segments and
recirculation (29). Nevertheless,
AUC0-t values of these three DL0410 dosages were not
proportionately increased. Furthermore, the t1/2
extended with the increasing dosage. Therefore, the results
supported non-linear plasma pharmacokinetics of DL0410 across the
investigated dosage range in rats. This is possibly due to the
saturability of drug metabolic enzymes and transporters. Therefore,
t1/2 and other pharmacokinetic parameters are no longer
constant and AUC and Cmax are not proportionately
increased (30).
Oral DL0410 demonstrated a low level of
bioavailability, which is a result of a number of factors,
including low permeability, first-pass DL0410 metabolism in the
liver, small and large intestinal tracts in addition to low aqueous
solubility. Whether DL0410 was able to distribute into the EC in
addition to the HIP, which are the most important brain regions for
learning and memory, was additionally assessed. The detectable
level in the brain demonstrated that DL0410 was able to cross the
blood-brain barrier and disperse into the two regions.
The cumulative excretion percentages in urine, feces
and bile following oral administration were evaluated. Fecal
excretion was the dominant route of elimination for DL0410. The
marked difference in excretion levels may be attributed to the low
oral bioavailability. DL0410 is poorly absorbed into blood and is
instead quickly excreted through the feces. Furthermore, the
present study suggested that DL0410 was excreted primarily as
metabolites. Based on the results from the present study on rat
bile, urine and fecal metabolites of DL0410 following oral
administration, the common metabolites at m/z 435.3231 and m/z
437.3397 were assigned to carbonyl reduction metabolites. CYP2D6
was identified for the involvement of this reduction reaction in
vitro and in silico.
In conclusion, a rapid, highly selective and
reliable LC-MS method for the determination of DL0410 in biological
matrices was developed and applied to investigate the
pharmacokinetics, brain, distribution and excretion of DL0410 in
rats. Furthermore, metabolism of DL0410 in urine was identified by
UPLC-Q-TOF-MS. The present results provide useful insight for the
continued evaluation of DL0410 as a therapeutic agent for AD.
Acknowledgements
Not applicable.
Funding
The present study was financially supported by
grants from the Research Special Fund for the Public Welfare
Industry of Health (grant no. 200802041), and the National Great
Science and Technology Projects (grant nos. 2013ZX09402203 and
2014ZX09507003002).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
AL and GD designed the experiment and revised the
manuscript. XP, YZ, JS and DK performed the experiments. SW and LW
acquired the data. XP analyzed the data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All experimental protocols involving Sprague-Dawley
rats were reviewed and approved by the animal experimentation
center of the Institute of Materia Medica, Chinese Academy of
Medical Sciences (Beijing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Abdalla A: Tau protein as a target for
Alzheimer's disease management. Saudi Pharm J. 23:405–406. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mucke L: Neuroscience: Alzheimer's
disease. Nature. 461:895–897. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tayeb HO, Yang HD, Price BH and Tarazi FI:
Pharmacotherapies for Alzheimer's disease: Beyond cholinesterase
inhibitors. Pharmacol Ther. 134:8–25. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martinelli-Boneschi F, Giacalone G,
Magnani G, Biella G, Coppi E, Santangelo R, Brambilla P, Esposito
F, Lupoli S, Clerici F, et al: Pharmacogenomics in Alzheimer's
disease: A genome-wide association study of response to
cholinesterase inhibitors. Neurobiol Aging. 34:1711.e7–13. 2013.
View Article : Google Scholar
|
|
5
|
Citron M: Alzheimer's disease: Strategies
for disease modification. Nat Rev Drug Discov. 9:387–398. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mushtaq G, Greig NH, Khan JA and Kamal MA:
Status of acetylcholinesterase and butyrylcholinesterase in
Alzheimer's disease and type 2 diabetes mellitus. CNS Neurol Disord
Drug Targets. 13:1432–1439. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ferreira-Vieira TH, Guimaraes IM, Silva FR
and Ribeiro FM: Alzheimer's disease: Targeting the cholinergic
system. Curr Neuropharmacol. 14:101–115. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deb PK, Sharma A, Piplani P and
Akkinepally RR: Molecular docking and receptor-specific 3D-QSAR
studies of acetylcholinesterase inhibitors. Mol Divers. 16:803–823.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Silman I and Sussman JL:
Acetylcholinesterase: How is structure related to function? Chem
Biol Interact. 175:3–10. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Decroocq C, Stauffert F, Pamlard O,
Oulaïdi F, Gallienne E, Martin OR, Guillou C and Compain P:
Iminosugars as a new class of cholinesterase inhibitors. Bioorg Med
Chem Lett. 25:830–833. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Saeed A, Mahesar PA, Zaib S, Khan MS,
Matin A, Shahid M and Iqbal J: Synthesis, cytotoxicity and
molecular modelling studies of new phenylcinnamide derivatives as
potent inhibitors of cholinesterases. Eur J Med Chem. 78:43–53.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Krátký M, Štěpánková Š, Vorčáková K and
Vinšová J: Salicylanilide diethyl phosphates as cholinesterases
inhibitors. Bioorg Chem. 58:48–52. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tao LX, Huang XT, Chen YT, Tang XC and
Zhang HY: Acetylcholinesterase-independent protective effects of
huperzine A against iron overload-induced oxidative damage and
aberrant iron metabolism signaling in rat cortical neurons. Acta
Pharmacol Sin. 37:1391–1400. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou D, Zhou W, Song JK, Feng ZY, Yang RY,
Wu S, Wang L, Liu AL and Du GH: DL0410, a novel dual cholinesterase
inhibitor, protects mouse brains against Aβ-induced neuronal damage
via the Akt/JNK signaling pathway. Acta Pharmacol Sin.
37:1401–1412. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang RY, Zhao G, Wang DM, Pang XC, Wang
SB, Fang JS, Li C, Liu AL, Wu S and Du GH: DL0410 can reverse
cognitive impairment, synaptic loss and reduce plaque load in
APP/PS1 transgenic mice. Pharmacol Biochem Behav. 139:15–26. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lian W, Fang J, Xu L, Zhou W, Kang D,
Xiong W, Jia H, Liu AL and Du GH: DL0410 ameliorates memory and
cognitive impairments induced by scopolamine via increasing
cholinergic neurotransmission in mice. Molecules. 22(pii):
E4102017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fang J, Yang R, Gao L, Zhou D, Yang S, Liu
AL and Du GH: Predictions of BuChE inhibitors using support vector
machine and naive bayesian classification techniques in drug
discovery. J Chem Inf Model. 53:3009–3020. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lian W, Jia H, Xu L, Zhou W, Kang D, Liu A
and Du G: Multi-protection of DL0410 in ameliorating cognitive
defects in D-galactose induced aging mice. Front Aging Neurosci.
9:4092017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Clapham J and Kilpatrick GJ: Histamine H3
receptors modulate the release of [3H]-acetylcholine from slices of
rat entorhinal cortex: Evidence for the possible existence of H3
receptor subtypes. Br J Pharmacol. 107:919–923. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Arrang JM, Garbarg M and Schwartz JC:
Auto-inhibition of brain histamine release mediated by a novel
class (H3) of histamine receptor. Nature. 302:832–837. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Blandina P, Giorgetti M, Bartolini L,
Cecchi M, Timmerman H, Leurs R, Pepeu G and Giovannini MG:
Inhibition of cortical acetylcholine release and cognitive
performance by histamine H3 receptor activation in rats. Br J
Pharmacol. 119:1656–1664. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bongers G, Bakker RA and Leurs R:
Molecular aspects of the histamine H3 receptor. Biochem Pharmacol.
73:1195–1204. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fang J, Li Y, Liu R, Pang X, Li C, Yang R,
He Y, Lian W, Liu AL and Du GH: Discovery of multitarget-directed
ligands against Alzheimer's disease through systematic prediction
of chemical-protein interactions. J Chem Inf Model. 55:149–164.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang A, Savas U, Hsu MH, Stout CD and
Johnson EF: Crystal structure of human cytochrome P450 2D6 with
prinomastat bound. J Biol Chem. 287:10834–10843. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang D, Huang C, Xin W, Ma X, Zhang W,
Zhang T and Du G: Preclinical pharmacokinetic evaluation and
metabolites identification of methyl salicylate-2-O-β-d-lactoside
in rats using LC-MS/MS and Q-TOF-MS methods. J Pharm Biomed Anal.
109:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tian S, He G, Song J, Wang S, Xin W, Zhang
D and Du G: Pharmacokinetic study of baicalein after oral
administration in monkeys. Fitoterapia. 83:532–540. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bressolle F, Bromet-Petit M and Audran M:
Validation of liquid chromatographic and gas chromatographic
methods. Applications to pharmacokinetics. J Chromatogr B Biomed
Appl. 686:3–10. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie X, Li Y, Gao D, Zhang Y and Ren Y:
Quantitative determination of euphol in rat plasma by LC-MS/MS and
its application to a pharmacokinetic study. Biomed Chromatogr.
28:1229–1234. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Godfrey KR, Arundel PA, Dong Z and Bryant
R: Modelling the double peak phenomenon in pharmacokinetics. Comput
Methods Programs Biomed. 104:62–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang QJ, Si L, Tang H, Sveigaard HH, Chow
EC and Pang KS: PBPK modeling to unravel nonlinear pharmacokinetics
of verapamil to estimate the fractional clearance for verapamil
N-demethylation in the recirculating rat liver preparation. Drug
Metab Dispos. 43:631–645. 2015. View Article : Google Scholar : PubMed/NCBI
|