Introduction
Morphine-induced hyperalgesia (MIH) is a type of
classic opioid-induced hyperalgesia (OIH) that is characterized by
increased sensitivity to noxious stimuli or even a painful response
to previously non-noxious stimuli (allodynia) induced by long-time
use of morphine (1). It was
suggested that paradoxical pain can be elicited by chronic opioid
exposure in humans and in animal models (2). Certain neuroplastic adaptations,
including increased expression of calcitonin gene related peptide,
substance P and various nociceptive receptors, were deemed to be
the possible mechanism underlying OIH (3,4). The
activation of mitogen-activated protein kinase (MAPK) in the
central and peripheral nervous systems was indicated to be a
possible signaling pathway in morphine-induced neuroplastic
adaptations by a series of findings from different laboratories
(4). Extracellular
signal-regulated protein kinase 1/2 (ERK1/2), a member of the MAPK
family, serves an important role in OIH by being activated by
self-phosphorylation and mediating the synthesis and expression of
downstream neuropeptides. The phosphorylated form of ERK1/2
(p-ERK1/2) can cause the activation of ERK1/2 and transfer the
electrical signal to the nucleus to cause cell damage (5–7).
Cannabinoid receptors, which comprise two subtypes,
including CB1 and CB2, belong to the G protein-coupled receptor
superfamily and are involved in the modulation of pain sensation
(8–10). CB1, which is mainly expressed in
the central nervous system (CNS), is considered to mediate the pain
sensation of the CNS (11).
CB1-knockout mice exhibited reduced locomotor activity and
hypoalgesia in hot plate and formalin tests (12). In addition, upregulation of CB1
receptor primarily within the ipsilateral superficial spinal cord
dorsal horn was revealed in sciatic nerve injury (chronic
constriction injury)-induced hyperalgesia model in rats, which was
partially attributed to ERK activation (13). A previous study suggested that the
CB1-selective cannabinoid receptor antagonist AM251 completely
reversed the peripheral antinociception induced by the µ-opioid
receptor agonist morphine but not by agonists of δ- or κ-opioid
receptors, which indicated that CB1 is involved in the analgesic
mechanism of morphine (14). The
CB2 receptor is expressed mainly in the immune system and in
hematopoietic cells (15). A
previous study indicated that co-administration of a selective CB2
agonist (AM 1241) attenuates chronic intraperitoneal morphine
exposure-mediated thermal hyperalgesia and tactile allodynia in
rats, which is partially due to attenuated immunoreactivity of the
spinal astrocyte and microglial marker and pro-inflammatory
mediators interleukin-1β and tumor necrosis factor-α (16). However, the exact association
between MIH and CB1/2 in CNS is poorly understood.
Electroacupuncture (EA) has been demonstrated to
effectively mitigate hyperalgesia induced by chronic constriction
injury (CCI) and cancer pain caused by intraplantar injection of
Walker 256 carcinoma cells in rats (17,18).
In addition, EA combined with a sub-threshold dose of morphine (2.5
mg/kg) enhanced the anti-inflammatory hyperalgesia effect compared
with that produced by each component alone in rats, which indicated
that there is a synergistic association between EA and morphine in
this regard (19). However,
whether EA can attenuate the hyperalgesia induced by chronic
morphine exposure is still unknown. Another study revealed that EA
inhibited zymosan-induced hypernociception in rats. The
CB1-selective antagonist AM251 and the CB2-selective antagonist
AM630 significantly reversed the antinociceptive and
anti-inflammatory effects of EA separately, suggesting that CB1 and
CB2 are involved in the mechanism of EA (20).
As mentioned above, ERK1/2, a classic member of the
MAPK family, is involved in the mechanism of OIH. However, ERK1/2
is also considered as a mediator between EA effects and CBs
(21). In the Freund's complete
adjuvant-induced hind paw pain model in rats, thermal hyperalgesia
and ERK phosphorylation in the ipsilateral dorsal horn of L4-5
segments were inhibited by EA stimulation (22). Notably, nocifensive behavior and
activation of ERK1/2 in the lumbar dorsal spinal cord were also
observed following intrathecal (IT) injection of a CB1 receptor
antagonist, namely AM251, which were both inhibited by IT injection
of a MAPK/ERK kinase inhibitor, namely U0126 (23). However, it is unknown whether
ERK1/2 is involved in mediating EA's effect through CB1/2 in the
spinal cord following MIH.
The present study hypothesized that EA could
ameliorate MIH and that this effect was partially mediated by CBs
via the ERK1/2 signaling pathway. The present study aimed to
evaluate the effect of EA on nociceptive behavior, as well as the
activation of ERK1/2 and to investigate the effect of CB1
activation or inhibition in regulating the effect of EA via the
ERK1/2 activated state in rats undergoing MIH.
Materials and methods
Animals
All experimental protocols were approved by the
Animal Experimental Ethics Committee of Tianjin Medical University
General Hospital (Tianjin, China). A total of 128 adult male
Sprague-Dawley rats, weighing 240±20 g each, were obtained from the
Laboratory Animal Center of the Military Medical Science Academy
(Beijing, China). For 1 week before the experiments, all animals
were housed in cages (5 rats per cage) at room temperature
(20–22°C) with 30–70% humidity on a 12 h light-dark cycle, were fed
a standard diet and had access to water.
IT morphine delivery
The rats were anesthetized with 3% sevoflurane plus
60% oxygen and catheterizations of the rat spinal subarachnoid
space were performed on anesthetized rats 3 days before morphine
administration, as described by Yaksh and Rudy (24). Briefly, rats were implanted with a
PE-10 polyethylene catheter (8 cm) the lumbar subarachnoid space.
Rats with no postoperative neurological deficits following surgery
were kept for the experiments. Animals showing neurological
dysfunction such as paralysis postoperatively were immediately
sacrificed using carbon dioxide. Upon surgery, rats were kept in
individual cages for 3 days before morphine administration.
EA
Rats received EA stimulation (16 rats/group) (2 Hz,
1.5 mA, 30 min) at 20 min after each administration, as described
by Yu et al (18). Briefly,
rats without any anesthetic drug received acupuncture with two
pairs of stainless acupuncture needles connected to two pairs of
electrodes. Each pair of needles was inserted perpendicularly ~6 mm
into the ipsilateral acupoints on the hind legs of the rats.
Acupoints were located according to the Zusanli (ST36) and
Yanglingquan (GB34) acupoints in humans. In rats, ST36 is located
at the proximal 1/5 point on the line from the depression lateral
to the patella ligament, while GB34 is located at the depression
anterior and inferior to the fibular head (19). A total of 2 non-acupoints were
located 0.5 cm horizontal and lateral to the ST36 and GB34
acupoints, respectively, at non-meridian points. A constant
electronic pulse (2 Hz, 1.5 mA) was administered by an
electroacupuncture stimulator (SDZ-II; Suzhou Medical Appliance
Factory, Suzhou, China) which was connected to the other end of the
electrodes. When the EA was starting, the current was stimulating
from one acupoint (ST36 or GB34) to another nonacupoint. Rats in
the control group (n=16) received the acupuncture needles at the
same points as the rats in the EA group but without EA treatment.
These rats were kept in tubular acrylic holders for 30 min as
served as controls.
Experimental protocol
Experiment 1: Effects of EA on
MIH
The animals were randomly divided into 3 groups
(n=16 rats/group): The control group (C); the chronic morphine
group (M); and the morphine + EA at ST36-GB34 group (ME). Animals
in the M and ME groups were IT administrated twice with 15 µg (10
µl) morphine at 8 am and 6 pm daily for 8 days. Animals in the C
group were IT treated daily with 10 µl saline at the same time as
the M group for 8 days. The animals in the ME group received EA
stimulation (2 Hz, 1.5 mA, 30 min, two times/day) at the
Zusanli-Yanglingquan acupoints (ST36-GB34) 20 min after morphine or
saline administration every day. The mechanical withdrawal
threshold (MWT; n=8 rats/group) and thermal withdrawal latency
(TWL; n=8 rats/group) were determined at baseline (24 h before IT
administration, day-1) and at the same time following the second
treatment on days 1, 3, 5 and 7 after drug administration. On the
8th day after drug administration, randomized selecting of 6 rats
in each group to collect the L4-6 segments of the spinal
cord for determination of the levels of ERK1/2, p-ERK1/2 and CB1 in
the intumescentia lumbalis of the spinal cord (as shown in Fig. 1).
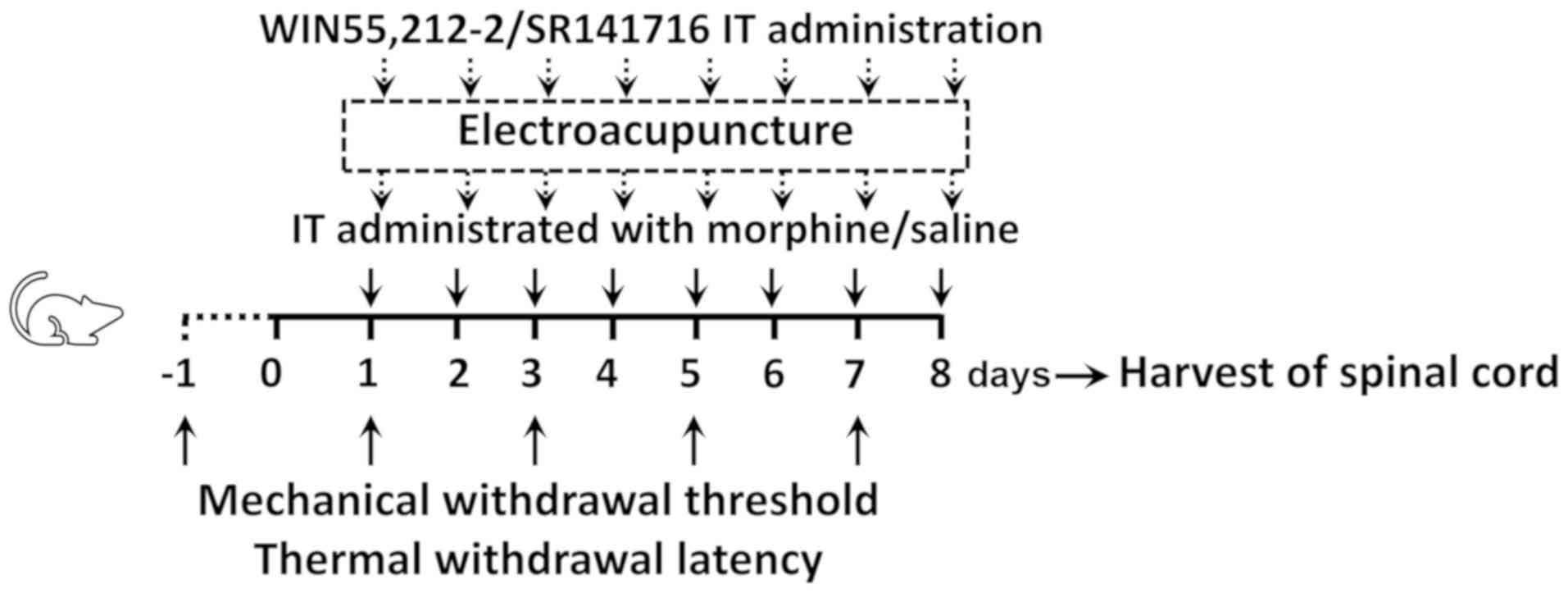 | Figure 1.Experimental design. Male
Sprague-Dawley rats (weighing 240±20 g each) randomly received C, M
or ME treatment. Animals in the M and ME groups were IT
administered with 15 µg (10 µl) morphine twice daily at 8 am and 6
pm for 8 days, while animals in the C group were IT treated with 10
µl saline at the same time as the M group daily for 8 days.
Furthermore, the animals in the ME group also received EA
stimulation (2 Hz, 1.5 mA, 30 min) at Zusanli-Yanglingquan
acupoints (ST36-GB34), 20 min after morphine or saline
administration every day. In order to verify the key role of CB1,
the CB1 agonist (WIN 55,212-2) and antagonist (SR141716) were IT
administered, respectively following morphine, while animals in the
other groups were injected with the same volume of saline.
Mechanical withdrawal threshold and thermal withdrawal latency were
determined at baseline (24 h before IT administration, day-1) and
at the same time after the second treatment on days 1, 3, 5 and 7
after administration. On the 8th day after administration, the
L4-6 segments of the spinal cord were collected for
determining the levels of ERK1/2, phosphorylated ERK1/2 and CB1 in
the intumescentia lumbalis of the spinal cord. IT, intrathecally;
EA, electroacupuncture; CB1, cannabinoid receptor 1; ERK1/2,
extracellular signal-regulated kinase 1/2; C, control; M, chronic
morphine; ME, morphine + EA at ST36-GB34. |
Experiment 2: Role of CB1 on the
protective effects of EA against MIH
The CB1 agonist WIN 55,212-2 and antagonist SR141716
were used in this experiment. The animals were randomly divided
into 5 groups (n=16 rats/group): i) The C group; ii) the M group;
ii) the ME group; iv) the morphine + EA treatment + CB1 agonist WIN
55,212-2 group (MEW); and v) the morphine + EA treatment + CB1
antagonist SR141716 group (MES). Saline, morphine and EA treatment
were administered as in experiment 1. WIN 55,212-2 (Cayman Chemical
Company, Ann Arbor, MI, USA; 30 µg) (25) and SR141716 (Cayman Chemical
Company; 30 µg) (26), which were
dissolved in 5% dimethyl sulfoxide (10 µl; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) were IT administrated to the MEW and MES
groups, respectively, upon morphine administration, followed by 10
µl normal saline. The animals in the C, M and EA groups received
the same volume of vehicle in identical conditions. MWT (n=8
rats/group) and TWL (n=8 rats/group) were determined at baseline
(24 h before IT administration) and at the same time after the
second treatment on days 1, 3, 5 and 7 post-administration. On day
8 after administration, randomized selecting of 6 rats in each
group to collect the L4-6 segments of the spinal cord to
detect the levels of ERK1/2, p-ERK1/2 and CB1 in the intumescentia
lumbalis of the spinal cord was performed (as shown in Fig. 1).
Mechanical hyperalgesia
On days 1, 3, 5 and 7 post-administration, 8 rats
per group were chosen for the mechanical hyperalgesia test.
Mechanical hyperalgesia was assessed using an electronic von Frey
filament (BSEVF3; Harvard Apparatus, Holliston, MA, USA), as
described previously (18).
Animals were placed in individual wire cages (20×20×30 cm) and
allowed to acclimatize for 1 h before testing. Mechanical allodynia
was determined by calculating the mean value of 3 MWT measurements
with an interval of 5 min between each measurement. A cut-off
pressure of 60 g was used to prevent tissue damage.
Thermal hyperalgesia
On days 1, 3, 5 and 7 post-administration, another 8
rats per group (not the same rats that were used to do the
mechanical hyperalgesia test) were chosen to do the thermal
hyperalgesia test. Thermal hyperalgesia was determined with
Intelligence Hot plate equipment (YLS-6B; Zhenghua Biologic
Apparatus Facilities Ltd., Co., Hefei, China), as described
previously (18). Animals were
allowed to habituate to the environment for 1 h before testing.
Animals were placed on the hot plate (50°C) until a positive
response (a clear paw withdrawal) was observed. The time was then
recorded as the TWL. The mean TWL was obtained from the mean value
of the 3 measurements of TWL with an interval of 5 min between
each. A cut-off time of 30 sec was used to prevent tissue
damage.
Protein analysis by western
blotting
Tissues from the lumbar spinal cord (n=6 rats/group)
were quickly removed under anesthesia on day 8 after administration
and immediately frozen in liquid nitrogen at −196°C. Tissues were
homogenized in immunoprecipitation buffer (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing Protease Inhibitor
Cocktail (Sigma-Aldrich; Merck KGaA) and centrifuged at 15,000 × g
for 5 min at 4°C. Protein concentration was determined by the
bicinchoninic acid assay method (Pierce; Thermo Fisher Scientific,
Inc.). Equal quantities of protein (30 µg) were used to determine
the protein expression of ERK1/2, p-ERK1/2, CB1 and β-actin.
Samples were separated using SDS-PAGE (8–10% gradient gels) and
then transferred to nitrocellulose membranes. The membranes were
blocked with 5% non-fat milk for 1 h at room temperature and
incubated with anti-ERK1/2 (1:500; cat. no. 4696; Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-p-ERK1/2 antibody (1:500;
cat. no. 5726; Cell Signaling Technology, Inc.) or anti-CB1 (1:100;
cat. no. ab167366; Abcam, Cambridge, UK) or anti-β-actin antibodies
(1:5,000; cat no. A5441; Sigma-Aldrich; Merck KGaA) overnight at
4°C. The membranes were washed with 1X TBS-Tween-20 (TBST) buffer
for 30 min and incubated with an horseradish peroxidase conjugated
rabbit anti-mouse secondary antibody (1:2,000; cat. no. 58802; Cell
Signaling Technology, Inc.) for 2 h at room temperature. The
membranes were washed with TBST buffer for additional 30 min and
visualized using Immobilon Western Chemiluminescent HRP Substrate
(EMD Millipore, Billerica, MA, USA) for 1 min, followed by film
exposure for 30 sec to 2 min. The results were analyzed using
Quantity One analysis software (version 4.6.7; Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
All data are reported as the mean ± standard
deviation. An unpaired Student's t test was used if the values had
a Gaussian distribution, while Mann-Whitney test was used if values
did not have such a distribution, to analyze differences between 2
groups and one-way analysis of variance with Bonferroni comparison
was employed to analyze interactions among various groups.
P<0.05 was considered to indicate a statistically significant
difference. Significance testing was 2-tailed. Statistical analysis
was performed using GraphPad Prism software (version 5.0; GraphPad
Software, Inc., La Jolla, CA, USA) and SPSS statistical software
(version 16.0; SPSS, Inc., Chicago, IL, USA).
Results
EA at ST36-GB34 acupoints attenuates
IT MIH
Compared with the control animals (group C), IT
administration of morphine (group M) significantly decreased MWT
and TWL on days 5 and 7 (P<0.05; Fig. 2A and B), which indicated that the
animal model of IT morphine-induced hyperalgesia was successfully
established. In addition, compared with the M group, combined EA at
ST36-GB34 acupoints (group ME) induced a significant increase in
MWT and TWL on days 5 and 7 (P<0.05; Fig. 2A and B), which indicated that EA at
ST36-GB34 acupoints significantly reduced the mechanical and
thermal hyperalgesia induced by IT administration of morphine.
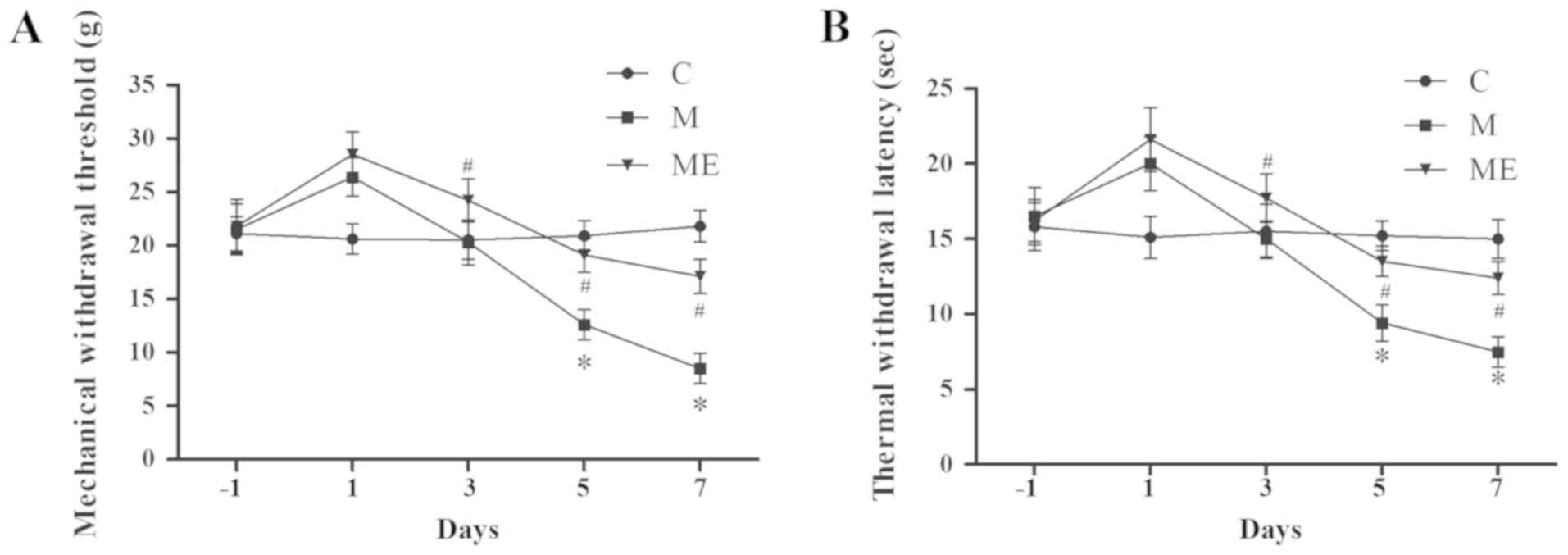 | Figure 2.Effects of EA on the behavioral tests
of morphine-induced hyperalgesia. On day-1 (1 day before IT
administration) and in days 1, 3, 5 and 7 after drug
administration, rats received IT normal saline, IT morphine and IT
morphine + EA at ST36-GB34. Next, (A) mechanical hyperalgesia and
(B) thermal hyperalgesia were evaluated by electronic von Frey
filament and hot plate, respectively. Data are expressed as the
mean ± standard deviation (n=8 rats/group for mechanical
hyperalgesia test and n=8 rats/group for thermal hyperalgesia
test). *P<0.05 vs. the C group, #P<0.05 vs. the M
group. IT, intrathecal; EA, electroacupuncture; C, control; M,
chronic morphine; ME, morphine + EA at ST36-GB34. |
Inhibition of the spinal cord ERK1/2
activation and increased expression of CB1 caused by EA at the
ST36-GB34 acupoints may be involved in the protective effects of EA
against MIH
Compared with the control group (C), repeated IT
treatment with morphine did not affect the protein levels of the
CB1 receptor in the spinal cord. However, in comparison with the
repeated administration of morphine group (M), repeated morphine
plus EA raised the expression of the CB1 receptor in the spinal
cord (P<0.05; Fig. 3A and B).
Furthermore, there was also a significant increase in p-ERK1/2
(P<0.05; Fig. 3C and D) but not
in ERK1/2 (P>0.05; Fig. 3C and
D) levels in the spinal cord of animals in the chronic morphine
(M) group compared with the C group. EA at ST36-GB34 acupoints (ME)
induced a significant decrease in p-ERK1/2 (P<0.05; Fig. 3C and D) but not ERK1/2 (P>0.05;
Fig. 3C and D) levels compared
with the ME group, which indicated that EA stimulation at acupoints
attenuated the level of ERK1/2 activation caused by chronic IT
administration of morphine.
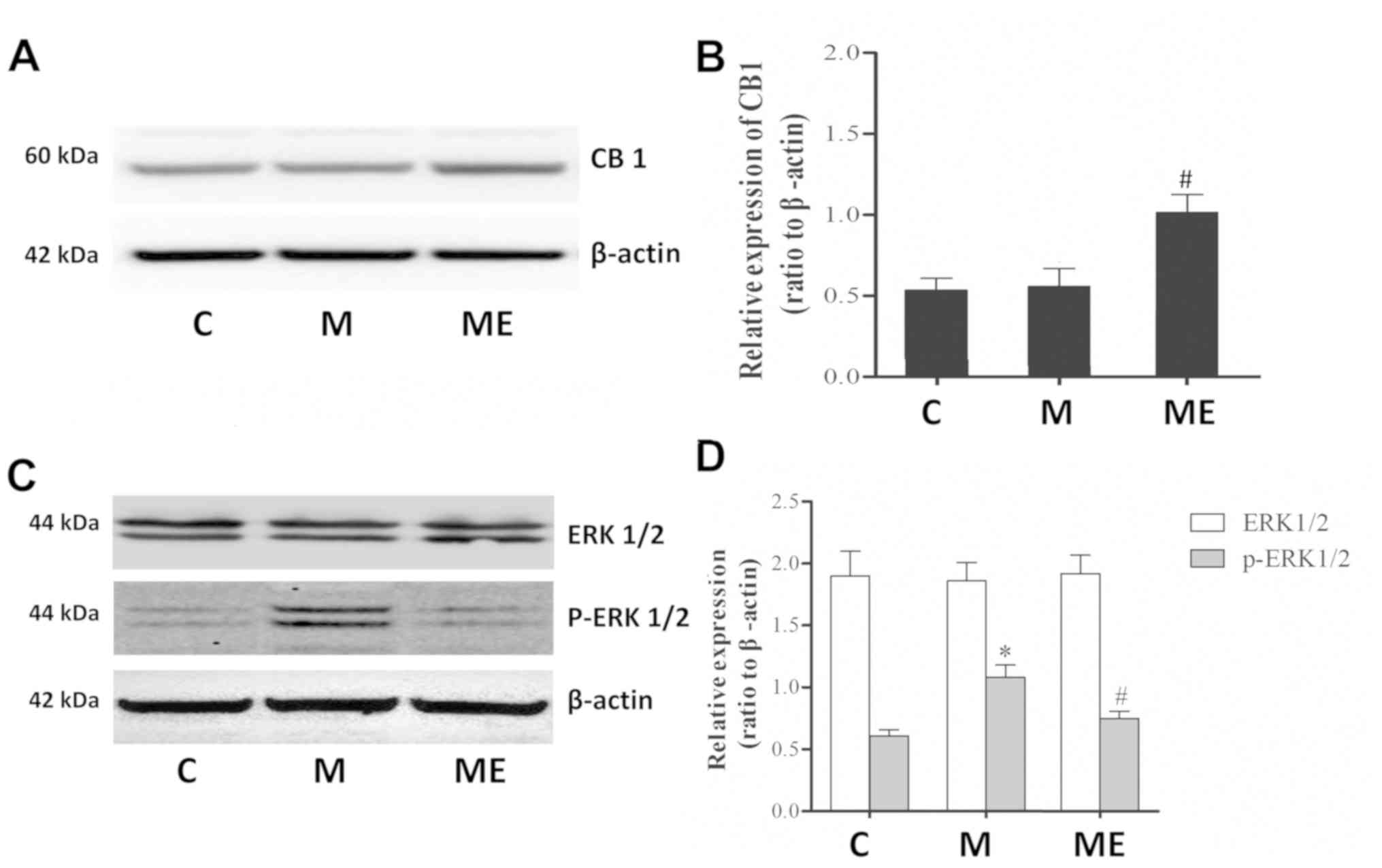 | Figure 3.Effects of EA on CB1, ERK1/2 and
p-ERK1/2 expression in morphine-induced hyperalgesia. Spinal cord
tissue from different groups was collected 8 days after intrathecal
treatment with saline, morphine and EA. (A) CB1, (C) ERK1/2 and
p-ERK1/2 levels were detected by western blotting. Quantitative
analysis of (B) CB1, (D) ERK1/2 and p-ERK1/2 are shown as the ratio
of protein relative density to β-actin. Data are expressed as the
mean ± standard deviation (n=6 rats/group). *P<0.05 vs. the C
group, #P<0.05 vs. the M group. EA,
electroacupuncture; CB1, cannabinoid receptor 1; ERK1/2,
extracellular signal-regulated kinase 1/2; p, phosphorylated; C,
control; M, chronic morphine; ME, morphine + EA at ST36-GB34. |
IT administration of WIN
55,212-2/SR141716 enhances/attenuates the inhibitory effects of EA
on MIH at the ST36-GB34 acupoints
As mentioned above, IT administration of morphine
significantly decreased MWT and TWL (P<0.05 in group M vs. group
C; Fig. 4A and B). EA at the
ST36-GB34 acupoints induced a significant increase in MWT and TWL
(P<0.05 in the ME group vs. the M group; Fig. 4A and B). However, compared with the
ME group, IT administration of WIN 55,212-2 (group MEW)
significantly increased MWT and TWL on days 3–7 (P<0.05 for MWT
on day 3 and TWL on day 7; P<0.01 for MWT on days 5 and 7 and
for TWL on days 3 and 5; Fig. 4A and
B). Compared with the ME group, IT administration of SR141716
(group MES) significantly decreased MWT and TWL on days 3 to 7
(P<0.01; Fig. 4A and B). These
results demonstrated that activation of CB1 significantly enhanced
the inhibitory effect of EA on IT morphine-induced mechanical and
thermal hyperalgesia at the ST36-GB34 acupoints, while inhibition
of CB1 attenuated the inhibitory effect of EA.
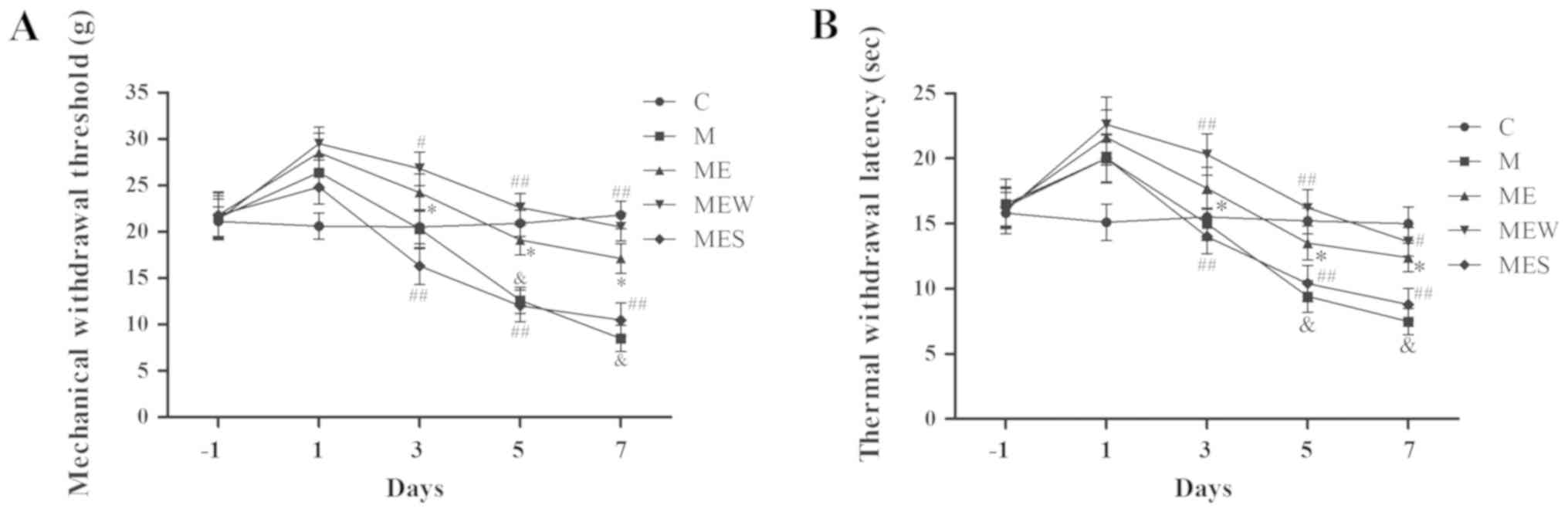 | Figure 4.Effects of EA upon administration of
WIN 55,212-2 or SR141716 on the behavioral tests of
morphine-induced hyperalgesia. On day-1 (1 day before IT
administration) and on days 1, 3, 5 and 7 after drug administration
in rats that received IT normal saline, IT morphine, IT morphine +
EA at ST36-GB34, IT morphine + EA treatment + CB1 agonist WIN
55,212-2 or IT morphine + EA treatment + CB1 antagonist SR141716,
(A) mechanical hyperalgesia and (B) thermal hyperalgesia were
assessed by electronic von Frey filament and hot plate,
respectively. Data are expressed as the mean ± standard deviation
(n=8 rats/group for mechanical hyperalgesia test and n=8 rats/group
for thermal hyperalgesia test). *P<0.05 vs. the C group,
#P<0.05 vs. the M group, ##P<0.01 vs.
the M group, &P<0.05 vs. the ME group. IT,
intrathecal; EA, electroacupuncture; CB1, cannabinoid receptor 1;
C, control; M, chronic morphine; ME, morphine + EA at ST36-GB34;
MEW, the morphine + EA treatment + CB1 agonist WIN 55,212-2 group;
MES, the morphine + EA treatment + CB1 antagonist SR141716
group. |
Inhibition of ERK1/2 activation
induced CB1 overexpression may enhance the inhibitory effects of EA
on MIH at the ST36-GB34 acupoints
There were significant increases in p-ERK1/2 levels
in the spinal cord of the animals in the chronic morphine (M) group
compared with those in the C group (P<0.05 M vs. C; Fig. 5C and D). These increases were
significantly attenuated by EA at the ST36-GB34 acupoints (group
ME; P<0.05; Fig. 5C and D).
Compared with the ME group, IT administration of the CB1 agonist
WIN 55,212-2 combined with EA significantly increased the CB1
levels but decreased the p-ERK1/2 levels in the spinal cord of rats
with IT MIH (P<0.01; Fig. 5).
On the contrary, there was a significant decrease in CB1 protein
level and a significant increase in p-ERK1/2 level in the spinal
cord of rats that received IT administration of the CB1 antagonist
SR141716 combined with EA (P<0.05 compared with the ME group;
Fig. 5A). There was no significant
difference in total ERK1/2 levels across all groups (P>0.05;
Fig. 5C and D). These results
indicated that EA at the ST36-GB34 acupoints may have a protective
effect against MIH through upregulating CB1 and downregulating
ERK1/2 activation.
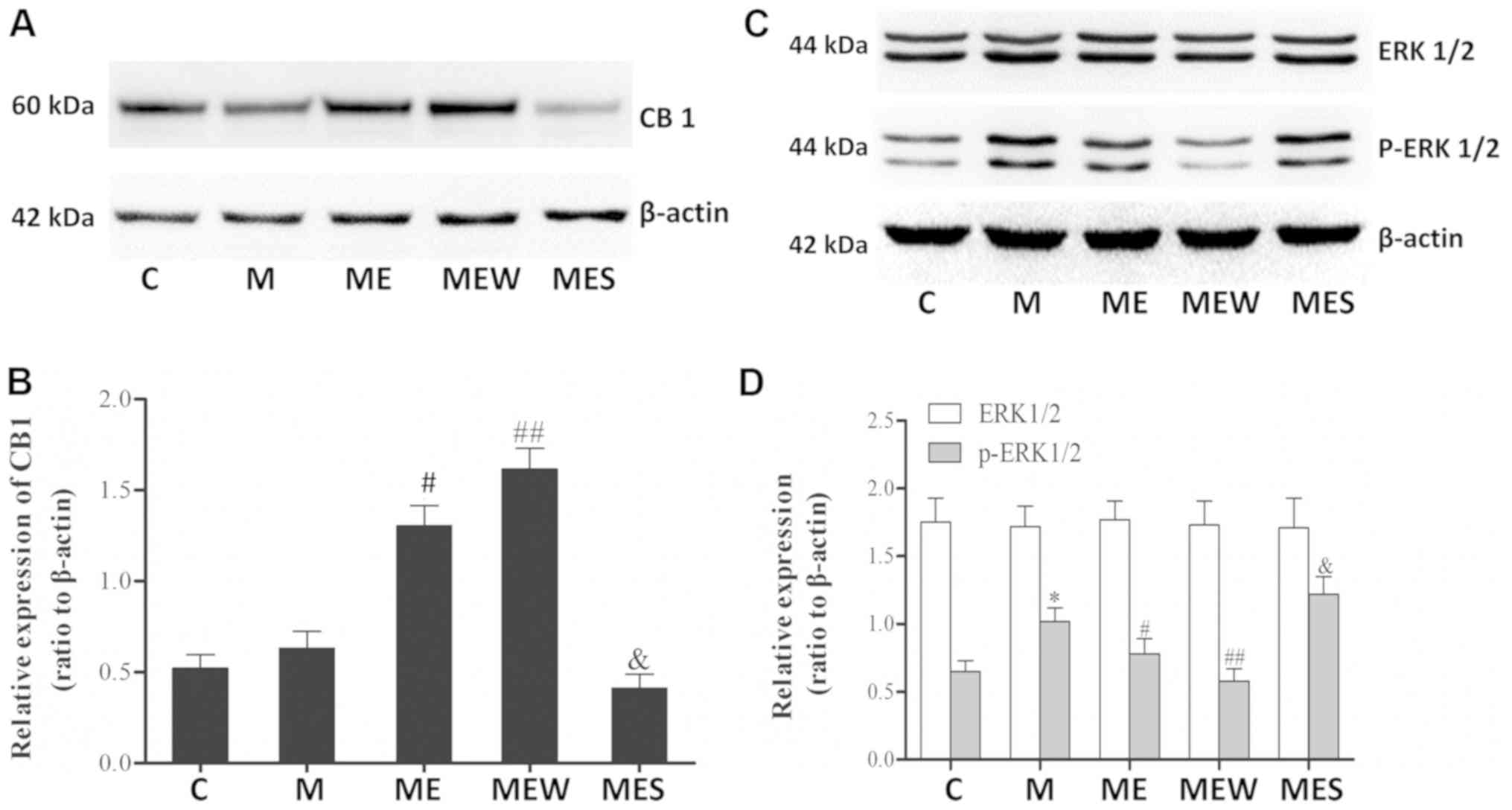 | Figure 5.Effects of EA upon administration of
WIN 55,212-2 or SR141716 on CB1, ERK1/2 and p-ERK1/2 expression in
morphine-induced hyperalgesia. Spinal cord tissue from different
groups was collected 8 days after intrathecal treatment with
saline, morphine, EA, WIN 55,212-2 and SR141716. (A) CB1, (C)
ERK1/2 and p-ERK1/2 were detected by western blotting. Quantitative
analysis of (B) CB1, (D) ERK1/2 and p-ERK1/2 are shown as the ratio
of protein relative density to β-actin. Data are expressed as the
mean ± standard deviation (n=6 rats/group). *P<0.05 vs. the C
group, #P<0.05 vs. the M group,
##P<0.01 vs. the M group, &P<0.05
vs. the ME group. EA, electroacupuncture; CB1, cannabinoid receptor
1; ERK1/2, extracellular signal-regulated kinase 1/2; p,
phosphorylated; C, control; M, chronic morphine; ME, morphine + EA
at ST36-GB34; MEW, the morphine + EA treatment + CB1 agonist WIN
55,212-2 group; MES, the morphine + EA treatment + CB1 antagonist
SR141716 group. |
Discussion
The present study used a rat model of chronic MIH to
investigate the antinociceptive mechanism of EA and to explore the
effect of CB1 in this mechanism. The results of the present study
indicated that 2-Hz EA at the ST36-GB34 acupoints attenuated MIH,
which was accompanied by an increase in CB1 levels and a decrease
in p-ERK1/2 levels. The present results also revealed that IT
administration of the CB1 agonist WIN 55,212-2 combined with EA at
the above acupoints enhanced the antinociceptive effect of EA on
MIH and induced an increase in CB1 levels and a decrease in
p-ERK1/2 levels compared with administration of EA alone, while the
CB1 antagonist SR141716 had the opposite effect. These data
indicated that EA at the ST36-GB34 acupoints may have a protective
effect against MIH through upregulating CB1 and downregulating
ERK1/2 activation.
As a type of Chinese traditional therapy, EA is used
to treat various diseases. There is increasing evidence that EA may
have clinical potential in attenuating certain types of chronic
pain, including adjuvant arthritis, CCI and cancer-associated pain
(17,18,27).
Frequency is regarded as an important parameter in EA treatment,
with 2, 15 and 100 Hz being the most commonly used frequencies for
analgesic therapy. Among these frequencies, 2-Hz EA has been
demonstrated to have a better analgesic effect for neuropathic
pain; thus, this frequency was selected in the present study
(18). The data from the present
study revealed that 2-Hz EA at the ST36-GB34 acupoints but not at
non-acupoints attenuated mechanical and thermal hyperalgesia caused
by IT administration of morphine, which is similar to the findings
of previous studies regarding the antinociceptive effect of EA at
the ST36 and GB34 acupoints (17,18,28).
Activation of ERK1/2 within spinal neurons by
various peripheral noxious stimulation has been reported to be
involved in generating pain hypersensitivity (29). Activation of ERK1/2 induced
short-term functional changes by non-transcriptional processing and
long-term neuronal plastic changes by increasing the gene
transcription of hyperalgesia-associated downstream neuropeptides
(30). Furthermore, there are
accumulating data about the roles of ERK in mediating the neuronal
plasticity that contributes to MIH (4). Previous evidence has suggested that
activation of ERK in the spinal cord is implicated in the formation
of MIH (7,31). It was reported that IT injection of
morphine for 7 days induced a remarkable increase in p-ERK1/2
levels in the spinal cord of rats, which contributed to morphine
tolerance and associated hyperalgesia (7,32).
Inhibition of ERK1/2 activation by IT injection of the ERK1/2
inhibitor U0126 or knockdown of spinal ERK1/2 by antisense
oligonucleotides attenuated withdrawal-induced mechanical allodynia
in rats (31,33). Consistent with previous studies,
the results of the present study indicated that the p-ERK1/2 levels
in the spinal cord significantly increased by IT injection of
morphine (15 µg, twice a day) for 7 days in rats with MIH. Recent
studies suggested that EA at acupoints attenuated hyperalgesia
caused by peripheral noxious stimulation and decreased the
activation of ERK1/2 (18,28). The results of the present study
revealed that, accompanied by attenuated mechanical and thermal
hyperalgesia, EA at the ST36-GB34 acupoints induced a decrease in
p-ERK1/2 levels in the spinal cord of rats that received IT
morphine for 7 days. These data demonstrated that inhibition of
ERK1/2 activation is at least partially involved in the EA
treatment of MIH.
CB1 is highly expressed in regions involved in pain
transmission and modulation, including the majority (76–83%) of
nociceptive neurons of dorsal root ganglions, the dorsal horn of
the spinal cord, the thalamus and the periaqueductal grey (34,35).
In the spinal cord, results revealed that CB1 levels were a
slightly increased by IT morphine and EA administration could
greatly increase the expression of CB1 under IT morphine
administration. Upregulation of CB1 partially participated in the
antinociceptive effect of EA and CB1 may serve a major role in this
process at the level of the spinal cord, which is in agreement with
a previous study (36).
As aforementioned, upregulation of the CB1 receptor
in the spinal cord was observed in a sciatic nerve injury-induced
hyperalgesia model in rats (13).
Another study suggested that the CB1 antagonist AM251 completely
reversed the peripheral antinociception induced by morphine, which
demonstrated that CB1 is involved in the analgesic mechanism of
morphine (14). Consistent with
earlier studies (13,14), the results of the present study
revealed that chronic IT injection of morphine significantly
increases CB1 protein levels along with the onset of hyperalgesia.
In a pain model of L5 spinal nerve ligation,
intraperitoneal injection of the CB1 agonist WIN 55,212-2
significantly attenuated mechanical hyperalgesia and thermal
allodynia, while co-administration of the CB1 antagonist SR141716
but not the CB2 antagonist SR144528 prevented this effect,
suggesting that this effect of WIN 55,212-2 is mediated via the CB1
receptor (37). Additionally, CB1
was also considered to be involved in the mechanism of EA
treatment. The CB1 selective antagonist AM251 significantly
reversed the antinociceptive and anti-inflammatory effects of EA in
a rat model of zymosan-induced hypernociception (20). The results of the present study
revealed that IT injection of the CB1 agonist WIN 55,212-2 enhanced
the antinociceptive effect of EA and induced a significant increase
in CB1 protein levels in a rat model of MIH, while IT injection of
the CB1 antagonist SR141716 induced the opposite results. These
data demonstrated that the CB1 receptor system was partially
involved in the mechanism of EA treatment. Various studies have
suggested that the ERK signaling pathway is involved in the
antinociceptive mechanism of the CB1 receptor system (23,38).
Katsuyama et al (23)
demonstrated that IT injection of the CB1 antagonist AM251 induced
a remarkable activation of ERK1/2 in the spinal cord along with
nocifensive behavior in mice, while the CB1 agonist ACEA and the
MAPK/ERK inhibitor U0126 reversed these results. A previous study
suggested that the ERK1/2 signaling pathway may be involved in EA
pretreatment-induced cerebral ischemic tolerance via the
cannabinoid CB1 receptor in rats (21). The results of the present study
revealed that the CB1 agonist WIN 55,212-2 combined with EA
decreased the p-ERK1/2 levels compared with EA treatment alone,
while the CB1 antagonist SR141716 induced the opposite results.
These data demonstrated that the enhancement produced by the CB1
agonist WIN 55,212-2 on the effect of EA in attenuating MIH was
partially mediated by inhibition of ERK1/2 activation. However, the
results of the current study revealed that EA treatment alone
increased the CB1 protein levels and decreased the p-ERK1/2 levels
in rats with MIH, which indicated that other mechanisms probably
participated in the inhibition of ERK1/2 activation.
In conclusion, the present study suggests that EA
produces an antinociceptive effect on IT injection of
morphine-induced hyperalgesia partially through the inhibition of
ERK1/2 activation. Activation of the CB1 receptor induced an
enhancement of the EA-mediated antinociceptive effect on MIH
partially through regulation of the spinal CB1-ERK1/2 signaling
pathway. The current study may contribute to the understanding of
the antinociceptive mechanism of EA and developing novel methods
for the treatment of MIH.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Fund of China (grant no. 81671888 awarded to Dr
Yonghao Yu; and grant nos. 81772043 and 81471842 awarded to Dr
Keliang Xie).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ, GW, KX and YoY conceived and designed the study.
YZ, YaY, YuY, YC and CW performed the experiments. YZ and YaY wrote
the manuscript. KX and YoY reviewed and edited the manuscript. All
authors read and approved the manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Animal Experimental Ethics Committee of Tianjin Medical University
General Hospital (Tianjin, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Angst MS and Clark JD: Opioid-induced
hyperalgesia: A qualitative systematic review. Anesthesiology.
104:570–587. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ossipov MH, Lai J, Vanderah TW and Porreca
F: Induction of pain facilitation by sustained opioid exposure:
Relationship to opioid antinociceptive tolerance. Life Sci.
73:783–800. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ossipov MH, Lai J, King T, Vanderah TW and
Porreca F: Underlying mechanisms of pronociceptive consequences of
prolonged morphine exposure. Biopolymers. 80:319–324. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Y and Sommer C: The role of
mitogen-activated protein kinase (MAPK) in morphine tolerance and
dependence. Mol Neurobiol. 40:101–107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma W, Zheng WH, Powell K, Jhamandas K and
Quirion R: Chronic morphine exposure increases the phosphorylation
of MAP kinases and the transcription factor CREB in dorsal root
ganglion neurons: An in vitro and in vivo study. Eur J Neurosci.
14:1091–1104. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Durham PL and Russo AF: Serotonergic
repression of mitogen-activated protein kinase control of the
calcitonin gene-related peptide enhancer. Mol Endocrinol.
12:1002–1009. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Z, Ma W, Chabot JG and Quirion R:
Cell-type specific activation of p38 and ERK mediates calcitonin
gene-related peptide involvement in tolerance to morphine-induced
analgesia. FASEB J. 23:2576–2586. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zogopoulos P, Vasileiou I, Patsouris E and
Theocharis SE: The role of endocannabinoids in pain modulation.
Fundam Clin Pharmacol. 27:64–80. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matsuda LA, Lolait SJ, Brownstein MJ,
Young AC and Bonner TI: Structure of a cannabinoid receptor and
functional expression of the cloned cDNA. Nature. 346:561–564.
1990. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Munro S, Thomas KL and Abu-Shaar M:
Molecular characterization of a peripheral receptor for
cannabinoids. Nature. 365:61–65. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matsuda LA, Bonner TI and Lolait SJ:
Localization of cannabinoid receptor mRNA in rat brain. J Comp
Neurol. 327:535–550. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zimmer A, Zimmer AM, Hohmann AG, Herkenham
M and Bonner TI: Increased mortality, hypoactivity, and hypoalgesia
in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci USA.
96:5780–5785. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lim G, Sung B, Ji RR and Mao J:
Upregulation of spinal cannabinoid-1-receptors following nerve
injury enhances the effects of Win 55,212-2 on neuropathic pain
behaviors in rats. Pain. 105:275–283. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
da Fonseca Pacheco D, Klein A, de Castro
Perez A, da Fonseca Pacheco CM, de Francischi JN and Duarte ID: The
mu-opioid receptor agonist morphine, but not agonists at delta- or
kappa-opioid receptors, induces peripheral antinociception mediated
by cannabinoid receptors. Br J Pharmacol. 154:1143–1149. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pacher P and Mechoulam R: Is lipid
signaling through cannabinoid 2 receptors part of a protective
system. Prog Lipid Res. 50:193–211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tumati S, Largent-Milnes TM, Keresztes A,
Ren J, Roeske WR, Vanderah TW and Varga EV: Repeated morphine
treatment-mediated hyperalgesia, allodynia and spinal glial
activation are blocked by co-administration of a selective
cannabinoid receptor type-2 agonist. J Neuroimmunol. 244:23–31.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Z, Wang C, Gu G, Li H, Zhao H, Wang
K, Han F and Wang G: The effects of electroacupuncture at the ST36
(Zusanli) acupoint on cancer pain and transient receptor potential
vanilloid subfamily 1 expression in Walker 256 tumor-bearing rats.
Anesth Analg. 114:879–885. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu J, Zhao C and Luo X: The effects of
electroacupuncture on the extracellular signal-regulated kinase
1/2/P2X3 signal pathway in the spinal cord of rats with chronic
constriction injury. Anesth Analg. 116:239–246. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yin CS, Jeong HS, Park HJ, Baik Y, Yoon
MH, Choi CB and Koh HG: A proposed transpositional acupoint system
in a mouse and rat model. Res Vet Sci. 84:159–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gondim DV, Araújo JC, Cavalcante AL, Havt
A, Quetz Jda S, Brito GA, Ribeiro Rde A and Lima Vale M: CB1 and
CB2 contribute to antinociceptive and anti-inflammatory effects of
electroacupuncture on experimental arthritis of the rat
temporomandibular joint. Can J Physiol Pharmacol. 90:1479–1489.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Du J, Wang Q, Hu B, Peng Z, Zhao Y, Ma L,
Xiong L, Lu Y, Zhu X and Chen S: Involvement of ERK 1/2 activation
in electroacupuncture pretreatment via cannabinoid CB1 receptor in
rats. Brain Res. 1360:1–7. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jang JY, Kim HN, Koo ST, Shin HK, Choe ES
and Choi BT: Synergistic antinociceptive effects of
N-methyl-D-aspartate receptor antagonist and electroacupuncture in
the complete Freund's adjuvant-induced pain model. Int J Mol Med.
28:669–675. 2011.PubMed/NCBI
|
|
23
|
Katsuyama S, Mizoguchi H, Komatsu T,
Nagaoka K, Sakurada S and Sakurada T: The cannabinoid 1 receptor
antagonist AM251 produces nocifensive behavior via activation of
ERK signaling pathway. Neuropharmacology. 59:534–541. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yaksh TL and Rudy TA: Chronic
catheterization of the spinal subarachnoid space. Physiol Behav.
17:1031–1036. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cui JH, Kim WM, Lee HG, Kim YO, Kim CM and
Yoon MH: Antinociceptive effect of intrathecal cannabinoid receptor
agonist WIN 55,212-2 in a rat bone tumor pain model. Neurosci Lett.
493:67–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Johanek LM, Heitmiller DR, Turner M, Nader
N, Hodges J and Simone DA: Cannabinoids attenuate capsaicin-evoked
hyperalgesia through spinal and peripheral mechanisms. Pain.
93:303–315. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shou Y, Yang Y, Xu MS, Zhao YQ, Ge LB and
Zhang BM: Electroacupuncture inhibition of hyperalgesia in rats
with adjuvant arthritis: Involvement of cannabinoid receptor 1 and
dopamine receptor subtypes in striatum. Evid Based Complement
Alternat Med. 2013:3934602013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Park JY, Park JJ, Jeon S, Doo AR, Kim SN,
Lee H, Chae Y, Maixner W, Lee H and Park HJ: From peripheral to
central: The role of ERK signaling pathway in acupuncture
analgesia. J Pain. 15:535–549. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ji RR, Baba H, Brenner GJ and Woolf CJ:
Nociceptive-specific activation of ERK in spinal neurons
contributes to pain hypersensitivity. Nat Neurosci. 2:1114–1119.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Obata K and Noguchi K: MAPK activation in
nociceptive neurons and pain hypersensitivity. Life Sci.
74:2643–2653. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cao JL, Liu HL, Wang JK and Zeng YM: Cross
talk between nitric oxide and ERK1/2 signaling pathway in the
spinal cord mediates naloxone-precipitated withdrawal in
morphine-dependent rats. Neuropharmacology. 51:315–326. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen Y, Geis C and Sommer C: Activation of
TRPV1 contributes to morphine tolerance: Involvement of the
mitogen-activated protein kinase signaling pathway. J Neurosci.
28:5836–5845. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cao JL, He JH, Ding HL and Zeng YM:
Activation of the spinal ERK signaling pathway contributes
naloxone-precipitated withdrawal in morphine-dependent rats. Pain.
118:336–349. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rani Sagar D, Burston JJ, Woodhams SG and
Chapman V: Dynamic changes to the endocannabinoid system in models
of chronic pain. Philos Trans R Soc Lond B Biol Sci. 367:3300–3311.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Svízenská I, Dubový P and Sulcová A:
Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution,
ligands and functional involvement in nervous system structures-a
short review. Pharmacol Biochem Behav. 90:501–511. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wei H, Yao X, Yang L, Wang S, Guo F, Zhou
H, Marsicano G, Wang Q and Xiong L: Glycogen synthase kinase-3β is
involved in electroacupuncture pretreatment via the cannabinoid CB1
receptor in ischemic stroke. Mol Neurobiol. 49:326–336. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bridges D, Ahmad K and Rice AS: The
synthetic cannabinoid WIN55,212-2 attenuates hyperalgesia and
allodynia in a rat model of neuropathic pain. Br J Pharmacol.
133:586–594. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ribeiro R, Wen J, Li S and Zhang Y:
Involvement of ERK1/2, cPLA2 and NF-κB in microglia suppression by
cannabinoid receptor agonists and antagonists. Prostaglandins Other
Lipid Mediat. 100-101:1–14. 2013. View Article : Google Scholar : PubMed/NCBI
|














